

Abstract
Ebola Virus (EBOV) is a highly pathogenic member of the family Filoviridae of viruses that causes severe hemorrhagic fever. Infection proceeds through fusion of the host cell and viral membranes, a process that is mediated by the viral envelope glycoprotein (GP). Following endosomal uptake, a key step in viral entry is the proteolytic cleavage of GP by host endosomal cysteine proteases. Cleavage exposes a binding site for the host cell receptor Niemann-Pick C1 (NPC1) and may induce conformational changes in GP leading to membrane fusion. However, the precise details of the structural changes in GP associated with proteolysis and the role of these changes in viral entry have not been established. Here, we have employed synthetic antibody technology to identify antibodies targeting EBOV GP prior to and following proteolysis (i.e. in the “uncleaved” [GPUNCL] and “cleaved” [GPCL] forms). We identified antibodies with distinct recognition profiles: FabCL bound preferentially to GPCL (EC50 = 1.7 nM), whereas FabUNCL bound specifically to GPUNCL (EC50 = 75 nM). Neutralization assays with GP-containing pseudotyped viruses indicated that these antibodies inhibited GPCL or GPUNCL mediated viral entry with specificity matching their recognition profiles (IC50: 87 nM for IgGCL; 1 μM for FabUNCL). Competition ELISAs indicate that FabCL binds an epitope distinct from that of KZ52, a well-characterized EBOV GP antibody, and from that of the luminal domain of NPC1. The binding epitope of FabUNCL was also distinct from that of KZ52, suggesting that FabUNCL binds a novel neutralization epitope on GPUNCL. Furthermore, the neutralizing ability of FabCL suggests that there are targets on GPCL available for neutralization. This work showcases the applicability of synthetic antibody technology to the study of viral membrane fusion, and provides new tools for dissecting intermediates of EBOV entry.
Keywords: Ebola Virus, Filovirus, Viral Membrane Fusion, Synthetic Antibodies, Antibody Engineering, Phage Display
Introduction
Ebola virus (EBOV) is a member of the family Filoviridae of negative-stranded, enveloped viruses (“filoviruses”) that cause severe hemorrhagic fever[1,2]. Three EBOV species (Zaire, Sudan, and Bundibugyo) have been associated with large outbreaks that have up to 90% human case fatality rates[3, 4]. Despite the increasing re-emergence of EBOV and outbreaks of new species[5,6], there are currently no treatments or vaccines approved for use in humans in the United States. As with other enveloped viruses, EBOV entry into host cells requires fusion of the viral and host cell membranes, a process that is mediated by the envelope glycoprotein (GP)[7–9]. In its native (“prefusion”) form, GP contains three copies each of the two subunits, GP1 (the surface subunit) and GP2 (the transmembrane subunit) (Figure 1)[10,11]. GP1, the larger of the two subunits (130 kDa), is responsible for cell attachment. It also contains a heavily glycoslyated mucin-like domain (muc) that projects from its surface and may play a role in immune evasion[11]. GP2 is much smaller (24 kDa) and contains two heptad repeat regions (N-terminal and C-terminal, NHR and CHR, respectively) that form a highly stable six-helix bundle in the post-fusion conformation. Folding of the six-helix bundle during viral entry provides the driving force for fusion between the viral and host cell the viral membrane by its transmembrane domain[10,11]. Upon viral attachment and uptake, priming and triggering events in endosomal compartments release constraints on prefusion GP, resulting in deployment of the fusion machinery[12].
Figure 1.
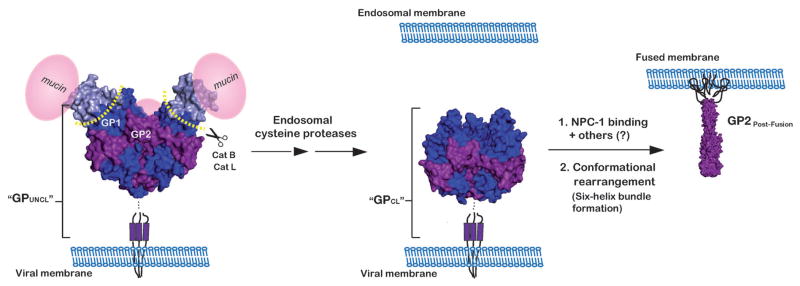
Schematic of the EBOV glycoprotein, GP, throughout its proposed structural transitions during entry into host cells. EBOV GP contains three copies each of a 130 kDa surface subunit, GP1 (blue), and a 24 kDa transmembrane subunit, GP2 (purple). GP1 contains a heavily glycosylated carboxyl-terminal mucin-like domain (represented in pink spheres, “mucin”). GP2 contains a distinct hydrophobic patch at the N-terminus (the fusion peptide) and N-terminal and C-terminal heptad repeat regions (NHRs and CHRs, respectively). After the virus is taken up into the endocytic pathway, endosomal cysteine proteases (cathepsins B and L (Cat B/Cat L) cleave and remove the glycan cap and mucin-like domains of GP1. In response to an unknown trigger, the cleaved GP next undergoes a large conformational change, resulting in the formation of a stable six-helix bundle. Formation of this six-helix bundle is thought to provide the energy required for fusion of the viral and host cell membranes. Here, GP is depicted in its prefusion trimer (PDB ID 3CSY, ref 10), both before and after cathepsin cleavage and in its post-fusion six-helix bundle (GP2Post-Fusion) (PDB ID 1EBO, ref 54). The antigens used for the selection (GPUNCL and GPCL) are soluble forms (i.e. lacking the transmembrane domain) of GP prior to and following proteolysis (respectively). The model for GPCL was generated by modifying the PDB file for GPUNCL (PBD ID 3CSY) to reflect the residues that are removed during cathepsin cleavage (see ref 43).
A key step in the entry process is the proteolytic cleavage of GP by host cysteine proteases present in the endosome. Zaire EBOV appears to be dependent on the cysteine proteases cathepsin B and cathepsin L (Cat B/Cat L) for entry[12–14]; however, other filoviruses vary in their dependence on these two proteases[15]. The proteolytic cleavage event removes most of GP1 (leaving only a small 17 kDa fragment) and is necessary, but not sufficient, to trigger viral membrane fusion[16]. GP cleavage appears to play at least two roles in entry. First, cleavage is thought to unmask a binding site for the endosomal cholesterol transporter Niemann-Pick C1 (NPC1), which was recently shown to be a critical intracellular receptor for filovirus entry[17–19]. Second, proteolytic cleavage may prime GP2 for conformational change by removing constraints imposed by GP1[13,20]. In analogy to other enveloped viruses, the next step of EBOV entry involves a dramatic conformational change in the proteolytically cleaved GP, leading to projection of the GP2 N-terminal fusion loop into the host cell membrane. GP2 is then thought to collapse into the stable post-fusion six-helix bundle, supplying the energy needed to overcome barriers associated with membrane fusion[7,8] (Figure 1). Despite recent progress, many questions remain regarding EBOV viral entry. Structural changes in GP associated with endosomal proteolytic cleavage are incompletely defined, and our understanding of these changes derives from in vitro experiments - no probes are currently available to detect cleaved forms of GP generated within the endosomes of intact cells.
Monoclonal antibodies are essential reagents for understanding viral membrane fusion and identifying epitopes for immunotherapy or vaccine development. In the well-studied systems of HIV-1 and influenza, conformation- or strain-specific antibodies targeting the viral envelope glycoproteins have been used to discern which conformations are most relevant to membrane fusion and how such conformations could be mimicked by designed immunogens[21–27]. Furthermore, antibodies that have high specificity for epitopes or conformational intermediates critical to the viral membrane fusion pathway typically have high neutralization potency and therefore immunotherapeutic promise. B-cell repertoires from HIV-1 or influenza survivors have been a fruitful source of neutralizing antibodies for these purposes, isolated by phage display or other methods[28–31]. However, there are limited natural sources of human EBOV antibodies targeting fusion-relevant forms of GP because survivors typically have low antibody titers, and most antibodies that arise from natural infection react preferentially with a soluble form of GP (sGP) that is secreted by the virus but is not relevant to membrane fusion[32–34]. At present, only two neutralizing antibodies targeting GP have been structurally characterized[35–37]. Other antibodies that target various epitopes of GP have been reported, but none of these harbors a human framework.
Here we describe the isolation of new GP-targeting antibodies from synthetic antibody repertoires. Synthetic antibody technology is a powerful approach to identification and characterization of monoclonal antibodies. Structural and bioinformatic analysis of existing antibody-antigen structures provides insight into which residues have optimal physicochemical characteristics for molecular recognition. In the most extreme case, antibody libraries in which complementarity-determining regions (CDRs) vary between only two residues – Tyr and Ser – are sufficient to support specific and high affinity antigen interactions against some targets[38,39]. The in vitro nature of the selection process, and the fact that synthetic repertoires are predicated on principles of protein recognition rather than immune response, permits identification of antibody fragments against targets that have resisted traditional antibody isolation methods. Furthermore, stringency of the selections can be highly controlled, and it is therefore possible to identify antibodies with membranes. In the prefusion conformation, GP1 is closely associated with GP2 and tethered to it via a disulfide bond. GP2 unique or enhanced specificity profiles (for example, see refs 40–42).
Results and Discussion
Identification of Synthetic Antibodies that Recognize Distinct Forms of GP
We sought to identify antibody fragments with orthogonal binding and neutralization specificities for the prefusion and proteolytically cleaved intermediate forms of EBOV GP. We employed a synthetic antigen binding fragment (Fab) library since such libraries have demonstrated utility against non-immunogenic targets. “Library F” contains binomial Tyr/Ser randomization in non-structural positions of CDR-H1 and CDR-H2, and additional variation at CDR-L3 and CDR-H3 that encodes the nine residues Tyr/Ser/Gly/Ala/Phe/Trp/His/Pro/Val in a 5/4/4/2/1/1/1/1/1 ratio (Figure 2A). This amino acid distribution mimics the observed distribution in natural CDR segments[38]. Antibodies against over 100 diverse antigens with high affinities and specificities have been identified using Library F (Sidhu, unpublished). Full details for Library F synthesis and characterization will be published elsewhere; the diversity was 3 × 1010 unique members.
Figure 2.

A) Amino acid sequences for Library F, FabCL, and FabUNCL. Invariant residues in Library F are highlighted in grey. Oligos of varying length were used during library construction in the LCDR3 (3–7 X residues) and HCDR3 (1–17 X residues) regions. X encodes nine residues Tyr/Ser/Gly/Ala/Phe/Trp/His/Pro/Val in a 5/4/4/2/1/1/1/1/1 ratio. B&C) ELISA binding profiles for FabCL (B) and FabUNCL (C) against GPUNCL and GPCL. BSA was included as a negative control protein. The half-maximal binding titers (EC50) were as follows: B) 1.7 nM for FabCL against GPCL and 13 nM against GPUNCL; C) 75 nM for FabUNCL against GPUNCL.
Previous work has demonstrated that the soluble form of EBOV GP lacking the transmembrane region and mucin-like domain serves as a structural mimic for the prefusion form (“GPUNCL”)[12,19]. In addition, treatment of GPUNCL with thermolysin mimics proteolysis by Cat L and Cat B (“GPCL”)[14,16,43]. We therefore used soluble GPUNCL and GPCL ectodomains as surrogates for the membrane-bound prefusion and proteolytic intermediate forms, respectively, in phage display selections to isolate Fabs from Library F. After 4 or 5 rounds of selection against GPUNCL or GPCL, respectively, clones displaying at least 10-fold preference for GPUNCL or GPCL over a negative control protein were tested for discrimination between the two forms of GP. We identified two Fab-phage clones with unique recognition properties on-phage; surprisingly both were isolated from selection against GPCL ectodomain. These two clones exhibited the best specificity for GPUNCL or GPCL out of a panel of 12 that were characterized extensively from the selection. FabCL bound preferentially to GPCL and FabUNCL bound preferentially to GPUNCL. Both clones displayed moderate levels of cross-reactivity for the different forms of GP, but nonetheless had distinct preferences (Figure 2B and 2C). Interestingly, both clones contain long CDR-H3 loops with a distribution of aromatic (Trp/Tyr/Phe) and flexible (Gly/Ser) residues. In addition, FabUNCL contains several Pro residues in the CDR-H3 segment (Figure 2A). The length of the CDR-H3 regions suggest that this segment constitutes a major element of the structural paratope for both Fabs.
Binding and Neutralization Profiles
FabCL and FabUNCL proteins were expressed periplasmically in Escherichia coli and purified. The binding profiles toward GPCL, GPUNCL, and BSA were assessed by ELISA (Figures 2B and 2C). FabCL demonstrated preferential binding to GPCL, with a half-maximal binding titer (EC50) of 1.7 nM for GPCL, but also exhibited cross-reactivity with GPUNCL (EC50 of 13 nM). No binding was observed to BSA. FabUNCL was specific for GPUNCL, with an EC50 of 75 nM and only low levels of binding to GPCL. FabUNCL had no appreciable binding to BSA.
We used a vesicular stomatitis virus particle bearing EBOV GP in place of the native envelope glycoprotein (VSV-GP pseudotype) to determine whether these two antibodies had neutralization activity for GP-mediated viral entry. As with the purified ectodomain, the VSV-GP construct lacks the mucin-like domain (“VSV-GPUNCL”). Furthermore, the viral genome encodes the enhanced green fluorescent protein (eGFP), allowing quantification of infection by fluorescence microscopy. VSV-GP particles can be treated with thermolysin in situ to mimic the cleaved form (“VSV-GPCL”)[14,16], allowing antibody neutralization potential to be tested against viruses displaying both forms of GP. For these experiments, the IgG1 versions of both FabCL and FabUNCL (IgGCL and IgGUNCL) were produced in HEK293T cells and purified. IgGCL had enhanced stability relative to FabCL and was therefore used for the neutralization studies. The FabUNCL and IgGUNCL were equally well behaved; therefore, FabUNCL was used due to higher yield. Vero cells were used as the infection host.
As shown in Figure 3A, IgGCL exhibited dose-responsive neutralization of VSV-GPCL with an IC50 of 87 nM. At the highest IgGCL concentrations tested (670 nM), the maximum neutralization observed was ~ 75%. Previous studies by Shedlock et al. identified a monkey antibody, JP3K11, that bound to and neutralized Cat L-cleaved GP, suggesting that epitopes on GPCL could be targeted by antibodies[44]. Our results with IgGCL confirm that epitopes on GPCL can be targeted for virus neutralization. However, unlike JP3K11, which did not discriminate between viruses containing uncleaved and cleaved GP, IgGCL had no appreciable neutralization activity for VSV-GPUNCL at the highest antibody concentration tested (Figure 3C). These results indicate that the neutralization epitope for IgGCL is not exposed prior to endosomal uptake. Furthermore, the specific neutralization activity of IgGCL toward the cleaved form of the glycoprotein matches the preferential binding of FabCL for GPCL observed by ELISA.
Figure 3.

Neutralization profiles of FabUNCL and FabCL for infection of Vero cells. A) IgGCL vs VSV-GPCL. IgGCL inhibited VSV-GPCL in a dose dependent manner, with an IC50 of 87 nM. B) FabUNCL vs VSV-GPUNCL. FabUNCL inhibited VSV-GPUNCL with an IC50 of 1 μM. C) Neutralization profiles of IgGCL and FabUNCL against VSV-GPUNCL and VSV-GPCL at high antibody concentrations. Neutralization profiles of the two antibodies are completely orthogonal, and match the recognition specificities determined by ELISA.
FabUNCL showed specific neutralization of VSV-GPUNCL with an IC50 of 1 μM, and up to ~ 80% neutralization at the highest Fab concentration (3.2 μM) (Figure 3B). No neutralization of the VSV-GPCL particle was observed, even at the highest FabUNCL concentration tested. Therefore, IgGCL and FabUNCL have orthogonal neutralization profiles for the cleaved and prefusion forms of GP, respectively.
Competition ELISAs demonstrate that FabCL and FabUNCL bind novel epitopes
We next explored if the two synthetic antibodies bound to known structural epitopes on the prefusion or proteolytically-cleaved forms of GP. Although the neutralization profiles of the two Fabs were specific, the moderate cross-reactivity of FabCL and FabUNCL for the different forms of GP (Figure 2B) suggested that there might be some overlap in their structural epitopes. We therefore performed competition ELISAs to dissect recognition profiles. FabCL was biotinylated (bFabCL) and binding to immobilized GPCL (detected with a strepavidin/horseradish peroxidase conjugate) was quantified in the presence of four non-biotinylated competitors: FabCL (positive control), FabUNCL, KZ52 IgG (an antibody isolated from an EBOV human survivor library[45]), and the soluble isolated form of the 248-residue luminal domain C of NPC-1 (Loop C). Recently, it has been established that Loop C binds specifically and directly to GPCL[19]. Furthermore, Loop C is able to inhibit the infection of VSV-GPCL[19]. These results indicated that Loop C acts as an essential determinant for the recognition of NPC1 by cleaved GP within endosomes. We included Loop C in the competition ELISA to see if Loop C would inhibit bFabCL binding since one potential mechanism for inhibition of VSV-GPCL entry would be direct competition for NPC1. We found that binding of FabCL was not reduced in the presence of Loop C, even at concentrations of ~ 1 μM. Therefore, the FabCL binding epitope is distinct from that of Loop C. Additionally, neither FabUNCL nor KZ52 IgG reduced the binding of bFabCL indicating that FabCL binds a structural epitope distinct from FabUNCL and KZ52. As expected, FabCL (non-biotinylated) reduced the binding of bFabCL in a concentration-dependent manner with an IC50 of 90 nM (Figure 4A).
Figure 4.

Competition ELISAs. A) Biotinylated FabCL (bFabCL) versus four unbiotinylated competitors: FabCL, FabUNCL, KZ52 and the luminal domain C of the endosomal cholesterol transporter NPC-1 (Loop C)[19]. FabCL competed with bFabCL; the data were fit to a four-parameter logistic equation, yielding an IC50 of 90 nM. None of the other proteins reduced the binding of bFabCL and therefore the data were not fit to an equation in these cases. B) Biotinylated FabUNCL (bFabUNCL) versus three unbiotinylated competitors: FabUNCL, FabCL, and KZ52 IgG. Neither FabCL nor KZ52 IgG had an effect on bFabUNCL binding. FabUNCL competed with bFabUNCL with an IC50 of 200 nM.
Similar experiments were performed with biotinylated FabUNCL (bFabUNCL) against GPUNCL, although Loop C was not included in this competition because Loop C does not bind GPUNCL[19]. GPUNCL is heavily glycosylated in the mucin-like domain and glycan cap, though all constructs investigated here were with GP lacking the mucin-like domain. Recently, a “hotspot” for neutralization was described; the structural epitopes for KZ52 and 16F6, a murine antibody isolated from vaccinated mice, were found to bind similar regions at the base of the chalice in the prefusion forms of GP from the Zaire and Sudan species, respectively[36,37]. We therefore sought to determine if FabUNCL bound Zaire GPUNCL at a similar location as KZ52. Again, as expected, FabUNCL (non-biotinylated) was able to compete with bFabUNCL with an IC50 of 200 nM. However, neither KZ52 IgG nor FabCL were able to reduce binding of bFabUNCL, indicating that neither KZ52 nor FabCL have overlapping structural epitopes with FabUNCL.
pH-Dependent Recognition Properties of FabUNCL and FabCL
Following initial cell surface-binding events, EBOV is taken up into the endosome via macropinocytosis, where GP proteolytic cleavage and GP-mediated viral membrane fusion occurs [46,47]. It is believed that antibodies targeting prefusion GP (e.g., KZ52 and 16F6) function by stabilizing the prefusion conformation and preventing transformations (conformational or other) that are required for membrane fusion[37]. It therefore follows that such neutralizing antibodies must retain binding activity under the acidic conditions of the endosome for a long enough period of time to sequester GP. Presumably, neutralizing antibodies that target other viruses that enter via the endosome and prevent membrane fusion must also have the capacity to function under endosomal conditions. For example, it has been reported that an antibody that targets the envelope protein of West Nile Virus (E16) is colocalized to the endosomal compartments along with the virus during entry[48]. Therefore, the likely mechanism of E16 action is to inhibit acid-dependent fusion-associated conformational changes. Endosomal pH is thought to be required for GP-mediated entry for several reasons. First, most endosomal cysteine proteases have optimal activity at pH ~5 and their activity is critical for generation of the proteolytic intermediate[11]. Second, pH-dependent conformational transitions result in activation of the GP2 fusion loop[13]. Third, folding of the GP2 six-helix bundle that provides the driving force for membrane fusion is more stable under acidic conditions[49,50].
Treatment of VSV-GPUNCL with thermolysin primes GP for fusion by mimicking endosomal proteolytic cleavage; therefore VSV-GPCL can bypass the requirement for cathepsin cleavage within endosomes[14]. However, the entry of VSV-GPCL as well as VSV-GPUNCL likely occurs in the endosome and not at the cell surface[16–19,46]. We therefore explored the recognition profiles of FabCL and FabUNCL at pH 4.5 (the pH of the late endosome/lysosome) to gain insight into the stability of the Fab-GP complexes under conditions that mimic those found in the endocytic pathway (Figure 5). It was recently reported that the binding of KZ52 to Zaire EBOV GPCL was not affected at endosomal pH, suggesting that KZ52 remains bound to GP in the endosome[16]. These data thus support an endosomal mode of action for KZ52[16]. We found that FabCL retained affinity for GPCL at pH 4.5 (EC50 = 10 nM). Interestingly, under these conditions, the specificity of FabCL for GPCL over GPUNCL was markedly attenuated relative to pH 7.5 (at pH 4.5, EC50 for GPCL was less than 2-fold lower than GPUNCL). One possible explanation for this result is that GPUNCL undergoes a subtle conformational change upon exposure to low pH, which can be detected by an increase in FabCL binding. However, we cannot rule out the possibility that pH-induced structural effects on the FabCL CDR segments are responsible for this pH-dependent recognition profile. pH-dependent conformational changes have been reported in full-length IgGs and Fab fragments, but these generally occur in conditions below pH 3.5[51,52]. Therefore, we believe it is less likely that Fab conformational changes are responsible for the change in recognition profile observed here (pH 4.5). In contrast to FabCL, FabUNCL retained its selectivity for GPUNCL over GPCL at pH 4.5, but had lower overall affinity for GPUNCL (EC50 = 340 nM). Again, these results raise the possibility that there are some conformational differences in GPUNCL at neutral versus endosomal pH that are detected by the binding of FabUNCL. Neither FabCL nor FabUNCL had any appreciable affinity for BSA under these conditions.
Figure 5.
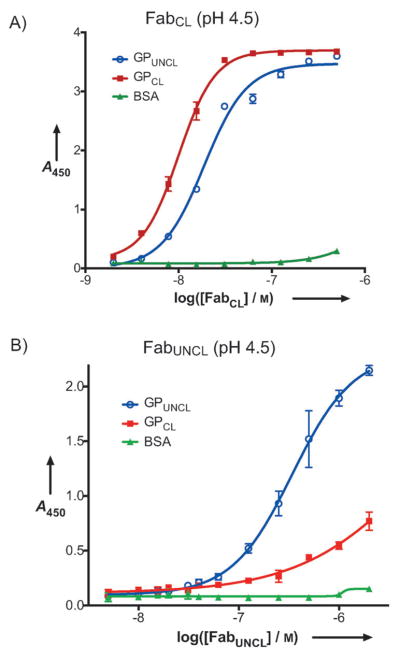
Fab Binding to GPUNCL and GPCL at pH 4.5. BSA was included as a control. The half-maximal binding titers were: A) 10 nM for FabCL against GPCL and 18 nM against GPUNCL; B) 340 nM for FabUNCL against GPUNCL.
Conclusion
We describe the isolation and characterization of two synthetic antibodies (FabCL/IgGCL and FabUNCL) that have distinct recognition and neutralization properties for mimics of the prefusion and proteolytically cleaved forms of EBOV GP. Several antibodies have been described against the prefusion form of GP, but the two that have been characterized structurally (KZ52 and 16F6) bind to a localized position at the base of the GP trimer[36,37]. We found that FabUNCL likely binds a novel epitope on GPUNCL since it does not compete for binding with KZ52 IgG. Additionally, FabCL/IgGCL binds a novel epitope on GPCL. While some cross-reactivity for GPCL and GPUNCL was observed for FabCL binding, IgGCL had a very distinct specificity for the neutralization of VSV-GPCL. Interestingly, although the ability of FabCL to bind GPCL was retained at low pH, its ability to distinguish GPCL from GPUNCL was diminished relative to pH 7.5. Therefore, the likely mechanism of specificity for neutralization of GPCL involves preferential binding at pH 7.5 prior to VSV-GPCL uptake. These results confirm that epitopes on GPCL are available for neutralization, although such epitopes may be exposed only within the endosome. Additionally, the differences in binding of FabCL and FabUNCL to GPUNCL at low pH raises the possibility that GPUNCL undergoes a subtle conformational change upon exposure to low pH, perhaps facilitating proteolytic cleavage.
A growing body of work has demonstrated the utility of synthetic antibody and fragment repertoires for identification of antibodies for therapeutic or research purposes[38, 40–42,53]. Here, we have expanded the scope to include targeting suspected structural intermediates in viral entry into host cells. In the specific case of EBOV, these results provide access to novel reagents for the dissection of the viral entry and membrane fusion pathway. Furthermore, this work suggests that future immunotherapeutic development could center around novel epitopes on GPUNCL and GPCL.
Experimental Section
Antibody Selection on Phage
Biopanning, direct phage ELISAs, and single-point competitive phage ELISAs were performed as described previously[39]. Briefly, in sorting for GPCL specific Fabs, phage pools representing a phage-displayed synthetic antibody library (Library F) were cycled through five rounds of binding selection with GPCL immobilized on 96-well Maxisorp immunoplates (Fisher Scientific, Nepean, ON, Canada) as the capture target. Stringency was increased in the panning process by 1) pre-incubation of the phage pool in immunoplates coated with GPUNCL (5 μg/mL) for 1 hour at room temperature before the phage pool was transferred to GPCL-immobilized immunoplates for rounds 2–5 and 2) decreasing antigen concentration used for coating the immunoplates from 5 μg/mL for rounds 2 and 3 to 2 μg/mL for rounds 4 and 5. The same strategy was also applied for the selection against GPUNCL, except that only four rounds of selection were performed. After selection, 96 clones each from rounds 4 and 5 for the GPCL selection and 96 clones from round 4 of the GPUNCL selection were grown overnight in 96-well deep well plates with 2YT broth supplemented with carbenicillin and M13K07. The culture supernatants were directly used in phage ELISAs to identify binding clones specifically targeting GPCL over GPUNCL or vice versa. Clones exhibiting phage ELISA signals of at least 10-fold higher than control (BSA) and at least 2-fold higher than the other form of GP were subjected to DNA sequence analysis.
Fab Protein Expression
The phage display vectors were converted to Fab protein expression vectors by insertion of a stop codon and hexahistidine tag upstream of the P3 gene fusion. Fab proteins were expressed periplasmically in E. coli BL21(DE3) (Invitrogen, Carlsbad, CA) by growth in low-phosphate media at 30°C for 18 – 22 hours. The cells were harvested by centrifugation and lysed using the “Bug Buster” reagent according to the manufacturer’s instructions (Novagen, Madison, WI). The lysate was subjected to centrifugation and the supernatent was applied to a Nickel column (Ni-NTA resin, Qiagen, Valencia, CA). The column was washed twice, first with 20 mM imidazole and then with 50 mM imidazole. The protein was eluted with 500 mM imidazole. The eluent was dialyzed into phosphate buffered saline (PBS), pH 7.4 and applied to a protein-A affinity column (beads from Pierce Thermo Scientific, Rockford, IL) to remove unpaired heavy or light chains. The beads were washed with PBS, pH 8.0 (2–4 column volumes), and the Fab proteins were eluted with 100 mM glycine, pH 2.0. The eluted Fab proteins were neutralized immediately to ~pH 7.4 using 1 M Tris, pH 8.0. Fractions containing the Fab proteins were dialyzed overnight into PBS, pH 7.4 and used directly in binding or neutralization assays, or flash-frozen and stored at −80°C for later use. The Fab protein concentration was determined by measuring the absorbance at 280 nm.
IgG Protein Expression
DNA for the variable domains were subcloned into pMAZ-IgL and pMAZ-IgH vectors for expression of full-length IgG1 molecules[55]. Vectors for the heavy chain and light chain were transfected into HEK293F cells using the Freestyle Max Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cell cultures were incubated at 37°C for 5–7 days post-transfection. After incubation, the cultures were centrifuged and the supernatant was applied to a protein-A affinity column (Pierce, Thermo Scientific, Rockford, IL). The eluent was dialyzed into PBS, pH 7.4 overnight and the IgG protein was concentrated and used directly in neutralization assays.
Binding ELISAs
The target proteins were directly immobilized on 96-well Maxisorp plates (Corning Incorporated, Corning, NY) (GPUNCL = 0.4 μg/well; GPCL = 0.25 μg/well) by incubating in NaHCO3, pH 8.0 at room temperature for 1 hour. Phosphate-buffered saline (PBS, pH 7.4) containing 3% BSA was used to block the wells after target immobilization (incubation for 45 minutes at room temperature). The Fabs were diluted into PBS containing 0.05% (v/v) Tween 20 (“PBS-T”) buffer, pH 7.4, applied to the wells, and incubated at room temperature for 1 hour. The plates were washed with PBS-T and incubated for 45 minutes with horseradish peroxidase/anti-HIS antibody conjugate (1:3000 dilution in PBS-T). The wells were washed with PBS-T, developed with 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate, and quenched with 1.0 M H2SO4. The absorbance at 450 nm was determined. The data was fit to a standard four-parameter logistic equation using Graphpad Prism (GraphPad Software, La Jolla, CA). The half-maximal binding (EC50) values were obtained from the inflection point of the curve.
Similar experiments were conducted at pH 4.5; in these experiments, the pH of the PBS-T buffer used for dilution of the Fabs was pH 4.5. The rest of the ELISA was conducted as described above.
Virus Neutralization Assay
We used a vesicular stomatitis virus that is pseudotyped to display EBOV GP on its surface in place of its native glycoprotein, G (VSV-GP). The viral genome encodes the enhanced green fluorescent protein (eGFP); therefore, infection can be scored by counting fluorescent cells after infection. VSV-GP was made in 293T cells as has been described previously[14,56]. Briefly, the virus-containing supernatants were harvested and concentrated by pelleting through a 10% sucrose cushion. Virus stocks were titered by infecting Vero (African Green Monkey Kidney) cells with serial dilutions and counting GFP-positive cells by fluorescence microscopy. Typical titers for VSV-GP were 109 infectious units (IU)/mL. Aliquots were stored at −80°C in 10% sucrose. VSV-GP was used to infect Vero cells at approximate multiplicities of infection (MOIs) of 0.1 to 1.0 in Dulbecco’s modified Eagle medium (DMEM) containing 5% fetal bovine serum, such that 20–200 cells were infected per well. VSV-GP was either used directly in neutralization experiments (VSV-GPUNCL) or pre-treated with thermolysin in situ, resulting in pseudotyped virus displaying GPCL (VSV-GPCL). To generate VSV-GPCL, VSV-GP was treated with thermolysin (200 μg/mL, Sigma) in NT buffer (10 mM Tris Cl, 135 mM NaCl) at pH 7.5 for 1 hour at 37°C. The reactions were neutralized with phosphoramidon (1 mM, Peptide International, Louisville, KY) and incubated on ice for at least 15 minutes before use. Vero cell monolayers (~ 7.5e4 cells/well in a 48 well plate) were exposed to pseudotyped virus that had been pre-incubated with dilutions of FabUNCL or IgGCL. Infection was scored by manually counting GFP-positive cells under a fluorescence microscope at 14 – 18 hours post-infection.
Competition ELISAs
FabUNCL and FabCL proteins were biotinylated (bFabUNCL and bFabCL, respectively) using a NHS-PEG4-BIOTIN labeling kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. GPUNCL or GPCL was immobilized on 96-well Maxisorp plates (Corning Incorporated, Corning, NY) at concentrations of 0.4 μg/well or 0.25 μg/well, respectively. For the FabUNCL competition ELISAs, bFabUNCL was diluted to 6 μg/mL in PBS-T buffer ± varying amounts of three unbiotinylated competitors: FabUNCL, FabCL, and KZ52 IgG. For the FabCL competition ELISAs, bFabCL was diluted to 6 μg/mL in PBS-T buffer ± varying amounts of four unbiotinylated competitors: FabCL, FabUNCL, KZ52 IgG, and the luminal domain C of the endosomal cholesterol transporter NPC-1 (Loop C)[17]. The mixtures of bFabUNCL or bFabCL ± competitors were applied to the wells and incubated at room temperature for 1 hour. The plates were washed with PBS-T and then horseradish peroxidase/strepavidin conjugate (1:1000 dilution in PBS-T buffer) was added and incubated for 45 minutes. The plates were washed with PBS-T, developed with TMB substrate, and quenched with 1.0 M H2SO4. Absorbance at 450 nm was measured. For the dose-responsive curves, the data was fit to a standard four-parameter logistic equation using Graphpad Prism (GraphPad Software, La Jolla, CA). The half-maximal binding (EC50) values were obtained from the inflection point of the curve.
Acknowledgments
We thank Professor Dennis Burton for supplying the KZ52 IgG used in this work and Dr. Rodolfo Chaparro and the Albert Einstein Protein Production Facility for assistance with production of IgGCL and IgGUNCL. We also thank members of the Lai Lab for critical reading of this manuscript. This work was funded by the Albert Einstein College of Medicine, National Institutes of Health (NIH) Grants R01-AI088027 and R01-Al101436 (K.C.), R01-A1067927 and R01-Al081982 (E.O.S.) and R01-AI090249 (J.R.L), and the Canadian Institutes for Health Research Grant MOP-93725 (S.S.S.). E.O.S. acknowledges an Investigators in the Pathogenesis of Infectious Disease Award from The Burroughs Wellcome Fund and The Skaggs Institute for Chemical Biology. J.R.L. acknowledges a Young Investigator Award from the Arnold and Mabel Beckman Foundation. J.F.K. was supported in part by NIH Medical Scientist Training Grant T32-GM007288.
Contributor Information
Prof. Dr. Kartik Chandran, Email: kartik.chandran@einstein.yu.edu.
Prof. Dr. Sachdev S. Sidhu, Email: sachdev.sidhu@utoronto.ca.
Prof. Dr. Jonathan R. Lai, Email: jon.lai@einstein.yu.edu.
References
- 1.Feldmann H, Geisbert TW. Lancet. 2011;377:849– 862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. Arch Virol. 2010;155:2083– 2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan N, Yang ZY, Nabel GJ. J Virol. 2003;77:9733–9737. doi: 10.1128/JVI.77.18.9733-9737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartman AL, Towner JS, Nichol ST. Clin Lab Med. 2010;30:161– 177. doi: 10.1016/j.cll.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Wamala JF, Lukwago L, Malimbo M, Nguku P, Yoti Z, Musenero M, Amone J, Mbabazi W, Nanyunja M, Zaramba S, Opio A, Lutwama JJ, Talisuna AO, Okware SI. Emerg Infect Dis. 2010;16:1087–1092. doi: 10.3201/eid1607.091525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.www.cdc.gov/ncidod/dvrd/spb/outbreaks/
- 7.Harrison SC. Nat Struct Mol Biol. 2008;15:690– 698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White JM, Delos SE, Brecher M, Schornberg K. Crit Rev Biochem Mol Biol. 2008;43:189– 219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckert DM, Kim PS. Annu Rev Biochem. 2001;70:777– 810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 10.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Nature. 2008;454:177– 182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JE, Saphire EO. Future Virol. 2009;4:621– 635. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. J Virol. 2012;86:364– 372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandan K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Science. 2005;308:1643– 1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. J Virol. 2006;80:4174– 4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misasi J, Chandran K, Yang JY, Considine B, Sullivan NJ, Filone CM, Hensley LE, Cunningham J. J Virol. 2012;86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bale S, Liu T, Li S, Wang Y, Abelson D, Fusco M, Woods VL, Jr, Saphire EO. PLoS Negl Trop Dis. 2011;5:e1395. doi: 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Nature. 2011;477:340– 343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Nature. 2011;477:344– 348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. J Virol. 2010;84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burton DR, Pyarti J, Koduri R, Sharp SJ, Thornton GB, Parren PWHI, Sawyer LSW, Hendry RM, Dunlop N, Nara PL, Lamacchia M, Garratty E, Stiehm ER, Bryson YJ, Cao Y, Moore JP, Ho DD, Barbas CF., 3rd Science. 1994;266:1024– 1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 22.Pantophlet R, Wilson IA, Burton DR. Protein Eng Des Sel. 2004;17:749– 758. doi: 10.1093/protein/gzh085. [DOI] [PubMed] [Google Scholar]
- 23.Pantophlet R, Saphire EO, Poignard P, Parren PW, Wilson IA, Burton DR. J Virol. 2003;77:642– 658. doi: 10.1128/JVI.77.1.642-658.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mor A, Segal E, Mester B, Arshava B, Rosen O, Ding FX, Russo J, Dafni A, Schvartzman F, Scherf T, Naider F, Anglister J. Biochemistry. 2009;48:3288– 3303. doi: 10.1021/bi802308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekiert DC, Bhabha G, Elsliger MA, Friesen RHE, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Science. 2009;324:246– 251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ekiert DC, Friesen RHE, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJWM, Brandenburg B, Vogels R, Brakenhoff JPJ, Kompier R, Koldijk MH, Cornelissen MH, Poon LAHM, Peiris LLMM, Koudstaal W, Wilson IA, Goudsmit IAJ. Science. 2011;333:843– 850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JPM, Skehel JJ, Lanzavecchia A. Science. 2011;333:850– 856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 28.Burton DR, Barbas CF, Persson MA, Koenig S, Chanock RM, Lerner RA. Proc Natl Acad Sci USA. 1991;88:10134– 10137. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashyap AK, Steel J, Oner AF, Dillon MA, Swale RE, Wall KM, Perry KJ, Faynboym A, IIhan M, Horowitz M, Horowitz L, Palese P, Bhatt RR, Lerner RA. Proc Natl Acad Sci USA. 2008;105:5986– 5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O’Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. Science. 2010;329:856– 861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. AIDS Res Hum Retroviruses. 2001;17:1757– 1765. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- 32.Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, Burt FJ, Leman PA, Khan AS, Rowe AK, Mukunu R, Sanchez A, Peters CJ. J Infect Dis. 1999;179(Suppl 1):S177–187. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 33.Dolnik O, Volchkova V, Garten W, Carbonnelle C, Becker S, Kahnt J, Ströher U, Klenk HD, Volchkov V. EMBO J. 2004;23:2175–2184. doi: 10.1038/sj.emboj.7600219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francica JR, Varela-Rohena A, Medvec A, Plesa G, Riley JL, Bates P. PLoS Pathog. 2010;6:e1001098. doi: 10.1371/journal.ppat.1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, Peters CJ, Parren PW, Burton DR. J Virol. 1999;73:6024–6030. doi: 10.1128/jvi.73.7.6024-6030.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dias JM, Kuehne AI, Abelson DM, Bale S, Wong AC, Halfmann P, Muhammad MA, Fusco ML, Zak SE, Kang E, Kawaoka Y, Chandran K, Dye JM, Saphire EO. Nat Struct Mol Biol. 2011;18:1424– 1427. doi: 10.1038/nsmb.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JE, Saphire EO. Curr Opin Struct Biol. 2009;19:408– 417. doi: 10.1016/j.sbi.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koide S, Sidhu SS. ACS Chem Biol. 2009;4:325–334. doi: 10.1021/cb800314v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. J Mol Biol. 2007;373:924– 940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Gao J, Sidhu SS, Wells JA. Proc Natl Acad Sci USA. 2009;106:3071– 3076. doi: 10.1073/pnas.0812952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Proc Natl Acad Sci USA. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koldobskaya Y, Duguid EM, Schechner DM, Suslov NB, Ye J, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. Nat Struct Mol Biol. 2011;18:100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, White JM. J Virol. 2009;83:2883– 2891. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shedlock DJ, Bailey MA, Popernack PM, Cunningham JM, Burton DR, Sullivan NJ. Virology. 2010;401:228–235. doi: 10.1016/j.virol.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, Peters CJ, Parren PWHI, Burton DR. J Virol. 1999;73:6024– 6030. doi: 10.1128/jvi.73.7.6024-6030.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. PLoS Pathogens. 2010;6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. Virology. 2011;419:72– 83. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson BS, Moesker B, Smit JM, Wilschut J, Diamond MS, Fremont DH. PLoS Pathogens. 2009;5:e1000453. doi: 10.1371/journal.ppat.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison JS, Higgins CD, Chandran K, Lai JR. Protein Sci. 2011;20:1587– 1596. doi: 10.1002/pro.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrison JS, Koellhoffer JF, Chandran K, Lai JR. Biochemistry. 2012;51:2515–2525. doi: 10.1021/bi3000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welfle K, Misselwitz R, Hausdorf G, Hohne W, Welfle H. Biochem Biophys Acta. 1999;1431:120– 131. doi: 10.1016/s0167-4838(99)00046-1. [DOI] [PubMed] [Google Scholar]
- 52.Buchner J, Rudolph R, Lilie H. J Mol Biol. 2002;318:829– 836. doi: 10.1016/S0022-2836(02)00171-7. [DOI] [PubMed] [Google Scholar]
- 53.Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S. Nat Struct Mol Biol. 2010;17:519– 527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Mol Cell. 1998;2:605– 616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 55.Mazor Y, Barnea I, Keydar I, Benhar I. J Immunol Methods. 2007;321:41– 59. doi: 10.1016/j.jim.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 56.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. Proc Natl Acad Sci USA. 1997;94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]