

Abstract
Pharmacophores are chemical scaffolds upon which changes in chemical moieties (R-groups) at specific sites are made to identify a combination of R-groups that increases the therapeutic potency of a small molecule inhibitor while minimizing adverse effects. We developed a pharmacophore based on a carbonyloxime (OXIM) scaffold for macrophage migration inhibitory factor (MIF), a protein involved in the pathology of sepsis, to validate that inhibition of a catalytic site could produce therapeutic benefits. We studied the crystal structures of MIF·OXIM-based inhibitors and found two opposite orientations for binding to the active site that were dependent on the chemical structures of an R-group. One orientation was completely unexpected based on previous studies with hydroxyphenylpyruvate and (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1). We further confirmed that the unexpected binding mode targets MIF in cellular studies by showing that one compound, OXIM-11, abolished the counter-regulatory activity of MIF on anti-inflammatory glucocorticoid action. OXIM-11 treatment of mice, initiated 24 h after the onset of cecal ligation and puncture-induced sepsis, significantly improved survival when compared with vehicle-treated controls, confirming that inhibition of the MIF catalytic site could produce therapeutic effects. The crystal structures of the MIF inhibitor complexes provide insight for further structure-based drug design efforts.
MIF4 is a pro-inflammatory cytokine critically involved in the pathogenesis of sepsis (1–4). Despite numerous advances in critical care medicine, sepsis remains a lethal inflammatory disorder and a substantial health care problem. Severe sepsis and septic shock constitute a progressive, systemic inflammatory response to infection, mediated by cytokines and other blood-borne factors (5–7).
Serum MIF concentrations positively correlate with the severity of Gram-negative and -positive sepsis in humans (3) and are significantly higher in non-survivors than in survivors from septic shock (1). Deletion of the mif gene in mice confers protection against lethal endotoxemia (8). MIF-deficient macrophages are hyporesponsive to bacterial stimuli, exhibiting a profound reduction in both TNF production and NF-κB activity (9). Treatment with neutralizing anti-MIF antibody (4, 10) or (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1) (11) protects mice from LPS-induced lethality, septic shock induced by Escherichia coli peritonitis, induced by cecal ligation and puncture (CLP). Counter-regulation of anti-inflammatory glucocorticoid activities is an important and unique feature of the MIF pro-inflammatory profile (12–14). Administration of recombinant MIF in mice completely blocks the protective effects of dexamethasone on endotoxin-induced lethality (12).
The three-dimensional x-ray crystallography of MIF shows that the molecule exists as a homotrimer with structural similarity to bacterial enzymes that catalyze various tautomerization reactions (15, 16). MIF does indeed catalyze the tautomerization of the non-physiological substrates D,L-dopachrome methyl esters into their corresponding indole derivatives (17). Crystallographic analysis of MIF complexed with p-hydroxyphenylpyruvic acid, another MIF “pseudo-substrate,” has revealed a catalytic site that lies in a hydrophobic cavity formed between two adjacent subunits of the homotrimer (18). Whether MIF functions as an enzyme in mammals is not known. However, numerous studies have now shown the importance of this site to the biological activities of MIF (11, 19–24). These studies also identify the active site of MIF as a pro-inflammatory site. Therefore, we reasoned that molecules that targeted this site could be useful to inhibit MIF actions in vitro and in vivo. Herein we define carbonyloxime (OXIM) as a new MIF small molecule binding scaffold and identify combinations of attached chemical R-groups with divergent MIF binding properties. Crystal structures of several such MIF·OXIM complexes reveal the surprising observation that this scaffold permits two opposite binding orientations depending on the chemical properties of the attached R-groups. We demonstrate that one derivative ((E)-4-Hydroxybenzaldehyde-O-cyclohexanecarbonyloxime (OXIM-11)) is a potent inhibitor of both MIF-mediated tautomerase activity (see Table 2) as well as several pro-inflammatory activities in vitro and in vivo. These findings and previous studies have identified the property of this binding site as a pro-inflammatory site. Significantly, late administration of OXIM-11 dramatically suppresses disease in an animal model of acute peritonitis. Taken together, these results underscore the therapeutic potential in intervening in the MIF pathway, the validity of a rational-based approach to the design of small molecule inhibitors, and the unanticipated and dramatic effects of the R-groups on the binding orientation to MIF.
TABLE 2.
Crystallographic statistics
| MIF·OXIM-11 | MIF·fluoroOXIM-11 | MIF·OXIM-6 | |
|---|---|---|---|
| Data collection | |||
| Space group | P3121 | P212121 | P212121 |
| Unit cell |
a = b = 95.60 Å, c = 103.09 Å α = β = 90°, γ = 120° |
a = 67.61 Å, b = 67.79 Å, c = 87.25 Å, α = β = γ = 90° |
a = 67.80 Å, b = 67.77 Å, c = 87.86 Å, α = β = γ = 90° |
| Resolution (high resolution shell) | 1.85 Å (1.92-1.85 Å) | 1.75 Å (1.81-1.75 Å) | 1.80 Å (1.86-1.8 Å) |
| Unique reflections | 46,116 (4,450) | 40,995 (4,021) | 37,780 (3,746) |
| Completeness | 98.2% (96.0%) | 100.0% (100.0%) | 99.0% (100.0%) |
| Avg. I/Avg. σ | 24.5 (3.61) | 27.7 (2.83) | 25.6 (2.7) |
| Rmerge | 0.061 (0.350) | 0.060 (0.441) | 0.056 (0.417) |
|
| |||
| Refinement statistics | |||
| R (working) | 19.1% | 19.8% | 21.3% |
| R-free | 21.8% | 20.4% | 22.5% |
| Average B factors (Å2) | |||
| Overall | 22.76 | 23.84 | 26.75 |
| Protein (number of residues) | 20.93 (114 × 3 subunits) | 22.02 (114 × 3) | 25.39 (114 × 3) |
| Water | 32.23 (305) | 35.12 (260) | 36.95 (210) |
| Sulfate | 49.61 (8) | 48.26 (5) | 60.53 (5) |
| Isopropanol | NA (0) | 28.87 (3) | 41.65 (1) |
| Glycerol | 29.15 (2) | 38.30 (6) | 41.35 (3) |
| Inhibitors | 43.02 (2) | 41.35 (2) | 39.80 (2) |
NA, not applicable.
MATERIALS AND METHODS
Synthesis of OXIM-11
OXIM-11 was synthesized in two steps as follows (25). A mixture of 4-hydroxybenzaldehyde oxime (1.75 g, 12.8 mmol) and cyclohexanecarboxylic acid chloride (1.8 ml, 13.4 mmol) in 70 ml of dry dichloromethane was cooled to 0 °C. To this suspension, pyridine (1.03 ml, 12.8 mmol) was added dropwise, which resulted in a pale yellow solution. The solution was stirred at 0 °C for 10 min and then was allowed to warm to room temperature for 18 h. The mixture was diluted with CH2Cl2 and water, and the layers were separated. The aqueous portion was washed with CH2Cl2, and the combined organic fraction was washed with saturated NaCl and dried over MgSO4. Filtration and evaporation in vacuo afforded a white solid, which was purified by flash chromatography (40% EtOAc/hexanes). Crystallization from EtOAc/hexanes afforded 1.2 g (38%) of the desired white solid product: 1H NMR (270 MHz, acetone-d6) δ 9.04 (br, 1H), 8.42 (s, 1H), 7.65 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 2.46 (m, 1H), 1.2–2.0 (m, 10H); 13C NMR (67.5 MHz, acetone-d6) δ 172.23, 160.56, 155.92, 130.06, 122.23, 115.88, 41.67, 25.19.
Dopachrome Tautomerase Assay
MIF tautomerase activity was measured as described previously (21). Briefly, l-dopachrome methyl ester was prepared at 2.4 mm through oxidation of l-3,4-dihydroxyphenylalanine methyl ester with sodium periodate as described previously. Activity was determined at room temperature by adding dopachrome methyl ester to MIF (50 nm in 50 mm potassium phosphate buffer, pH 6, 0.5 mm EDTA) and measuring the decrease in absorbance for 20 s at 475 nm.
Purification of MIF and Crystallization of the MIF·OXIM Complexes
Recombinant MIF was overexpressed and purified in a similar manner to that previously described (21). Inhibitors were dissolved in Me2SO (hexadeuterated Me2SO in the case of the fluorinated OXIM) and co-crystallized with MIF using the hanging drop vapor diffusion technique. Protein inhibitor solutions contained 1.2 mm MIF:1.6 mm OXIM-11 and 0.39 mm MIF:3.2 mm inhibitor for the other complexes. Two microliters of MIF inhibitor complex were mixed with two microliters of reservoir consisting of 50% saturated ammonium sulfate, 0.1 m tris(hydroxymethyl)aminomethane (pH 7.5), 4% isopropyl alcohol. Crystals of the MIF·OXIM-11 complex formed in space group P3121. The other complexes crystallized in space group P212121. Diffraction data were collected at −180 °C. Prior to freezing, crystals were briefly soaked in a cryo-protective solution containing 25% glycerol, 50% saturated ammonium sulfate, 0.1 m tris(hydroxymethyl)aminomethane (pH 7.5), and 3 or 4% isopropyl alcohol. HKL2000 was used to integrate and scale the diffraction data to 1.85, 1.75, and 1.8 Å for complexes of OXIM-11, the fluorinated OXIM-11, and OXIM-6, respectively. Unit cell parameters are presented in Table 2.
Crystallographic Refinement
The structure of MIF bound to ISO-1 (Protein Data Bank entry 1LJT) (21), with the ISO-1 and water molecules removed, was used as a starting model for the OXIM-11 complex refinement. Protein coordinates from the OXIM-11 complex were used as a starting model for refinement of the other complexes. Rotation and translation searches using AMoRe (27) were performed to properly position the model in the unit cell prior to refinement. This proved to be necessary in a number of cases, probably due to differences in unit cell parameters and imperfect non-crystallographic symmetry. Refinement was performed using CNS (28), and manual manipulation of the model was done using O (29) and COOT (30). Five percent of reflections in the OXIM-11 data set, and 2% in the others, were set aside for cross-validation. Non-crystallographic restraints were employed during the refinement, which were eventually dropped for the OXIM-11 and OXIM-6 complexes according to R-free values. In one of the three active sites of each complex in space group P212121, there is no inhibitor present due to the close proximity of the active site to neighboring copies of MIF in the crystal. Nonetheless, there was electron density visible in this active site, which could be modeled very well with a glycerol molecule. Statistics for all three structures are presented in Table 2. Coordinates have been deposited to the RCSB Protein Data Bank. Entry codes are 2OOH, 2OOW, and 2OOZ for the OXIM-11, fluorinated OXIM, and OXIM-6 complexes, respectively.
Glucocorticoid Override Assay
Human mononuclear cells were isolated from whole blood by Ficoll density gradient centrifugation, and monocytes were purified by adherence. Non-adherent cells were removed by washing each well twice at 24 h with RPMI containing 10% heat-inactivated fetal bovine serum. Cells were trypsinized, counted, and plated in 24-well plates at 1 × 106 cells/well incubated for 1 h with dexamethasone (10−9 m), dexamethasone plus MIF (100 ng/ml recombinant MIF), or dexamethasone plus MIF and OXIM-11 (0.1–10 μm) before the addition of 0.5 μg/ml LPS (E. coli 0111:B4, Sigma). After 16 h, cell culture supernatants were collected for determination of TNF-α concentrations by enzyme-linked immunosorbent assay (R & D Systems).
Measurement of NF-κB Activation by Electrophoretic Mobility Shift Assay
RAW 267.4 macrophages were treated with various concentrations of OXIM-11 (1–100μm) 30 min prior to LPS (endotoxin, E. coli 0111:B4, Sigma) addition. Macrophages were collected 2 h after activation with LPS and washed 1× in phosphate-buffered saline, pH 7.4. Nuclear extract was isolated using NE-PER nuclear and cytoplasmic extraction reagents according to the manufacturer’s instructions (Pierce Biotechnology). For detection of NF-κB binding, nuclear extract from cells (~5μg of protein) was incubated with 0.2 ng of 32P-labeled double-stranded oligonucleotide sequence in a 10-μl reaction volume containing 5× gel shift binding buffer (20% glycerol, 5 mm MgCl2, 2.5 mm EDTA, 2.5 mm dithiothreitol, 250 mm NaCl, 50 mm Tris-HCl (pH 7.5), and 0.25 mg/ml poly(dI-dC)-poly(dI-dC)) for 30 min at room temperature. The samples were resolved on a 4% polyacrylamide gel and visualized directly by autoradiography after drying the gel. The NF-κB consensus sequence (Promega) was labeled with 10 units of T4 polynucleotide kinase (Promega) per 25 ng of oligonucleotide, 1× kinase buffer, and 5 μl of [γ-32P]ATP (Amersham Biosciences, catalog number PB10168, 10 mCi/ml) for 30 min at 37 °C. The binding bands were quantified by scanning densitometry of a bio-image analysis system. The results for each treatment were expressed as relative intensity when compared with the control (saline-treated) intensity.
Cecal Ligation and Puncture Induced Polymicrobial Sepsis
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Feinstein Institute. In anesthetized mice (ketamine 100 mg/kg and xylazine 8 mg/kg administered intramuscularly), the cecum was ligated and given a single puncture. Abdominal access was gained via a midline incision. The cecum was isolated and ligated with a 6-0 silk ligature below the ileocecal valve, and the cecum was punctured once with a 22-gauge needle, stool (~1 mm) was extruded from the hole, and the cecum was placed back into the abdominal cavity. The abdomen was closed with two layers of 6-0 Ethilon sutures. Antibiotics were administered immediately after CLP (Premaxin 0.5 mg/kg, subcutaneously, in a total volume of 0.5 ml/mouse) and a single dose of resuscitative fluid (normal saline solution administered subcutaneously (20 ml/kg of body weight)) immediately after CLP surgery (11). Control (aqueous 20% dimethyl sulfoxide as a vehicle) or OXIM-11 (3.5 mg/kg; intraperitoneal) treatment was started 24 h after the induction of sepsis and repeated twice daily for 3 days. Animal survival was monitored for 2 weeks.
Statistical Analysis
Data are expressed as mean ± S.D. Multiple groups were compared using analysis of variance and Dunn’s post hoc analysis.
RESULTS
The Design of OXIM Compounds
We reasoned that a compound mimicking the indole intermediate of MIF tautomerase activity could bind to and thereby block the pro-inflammatory site. Thus, Schiff base adducts were synthesized by coupling amino acid methyl ester with p-hydroxybenzaldehyde to furnish an amino acid Schiff base-type compound (26). Based on the effectiveness of the Schiff base inhibition of the tautomerase activity (26), we modified the structure to produce a parent pharmacophore (the oxime scaffold, OXIM) as shown in Fig. 1, which was then sequentially synthesized using 12 different R-groups. Table 1 shows the IC50 for the synthesized molecules. OXIM-11 inhibited the dopachrome tautomerase activity in a dose-dependent manner with an IC50 of 1.3 μm.
FIGURE 1. Design of MIF inhibitor.
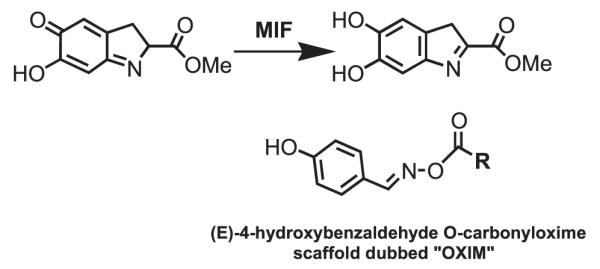
Above, MIF tautomerizes dopachrome methyl esters. Below, the structure of (E)-4-hydroxybenzaldehyde O-carbonyloxime scaffold as a novel and potent inhibitor of MIF.
TABLE 1.
IC50 of (E)-4-hydroxybenzaldehyde O-carbonyloxime derivatives for MIF d-dopachrome activity
| Entry | Compounds Oxime Derivatives |
IC50 μM |
|---|---|---|
| 1 |
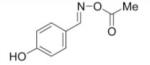
|
147 |
| 2 |
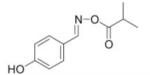
|
25 |
| 3 |
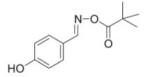
|
113 |
| 4 |
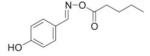
|
14.5 |
| 5 |
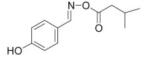
|
7 |
| 6 |
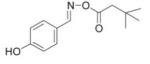
|
7 |
| 7 |
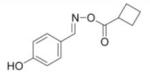
|
11 |
| 8 |
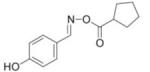
|
4 |
| 9 |
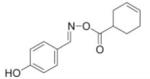
|
3.4 |
| 10 |
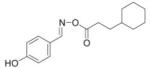
|
3.0 |
| 11 |

|
1.3 |
| 12 |
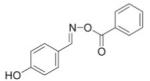
|
7 |
X-ray Crystallography of MIF·OXIM Compounds
To confirm that OXIM-11 binds to the pro-inflammatory site and also to analyze the interactions that it makes with the active site, we co-crystallized MIF with OXIM-11. The complex was refined to 1.85 Å (crystallographic statistics are presented in Table 2). Although refinement of the protein proceeded well, fitting of OXIM-11 in an orientation similar to that of ISO-1 (21) or p-hydroxyphenylpyruvic acid (18) was difficult. The hydroxyphenyl groups of both ISO-1 and p-hydroxyphenylpyruvic acid bind deep in the active site with the hydroxyl group making hydrogen bonds with Asn-97. Interestingly, the original fit of OXIM-11 to the density did not result in any significant interactions with the active site amino acid residues of MIF that could explain its potency relative to other OXIM derivatives, ISO-1, or p-hydroxyphenylpyruvic acid. Reversal of the orientation of OXIM-11 with respect to the active site such that the cyclohexyl ring was deep in the cleft and the phenyl moiety pointed outside the active site led to a better fit of the Fo – Fc density (Fig. 2a). This orientation is confirmed by the density for the carbonyl group that is visible proximal to the cyclohexyl ring. To confirm that the p-hydroxyphenyl group of OXIM-11 was not deep in the active site, an OXIM-11 derivative fluorinated ortho to the hydroxyl group was synthesized, x-ray data were collected to 1.75 Å resolution, and the structure was refined to an Rfree of 20.4%. The electron density deep in the active site (data not shown) was no different from that shown in Fig. 2a, suggesting that the phenyl ring was outside the active site. To determine whether the R-groups of OXIM-11 led to the unexpected binding conformation, we also co-crystallized MIF with OXIM-6, which has R-groups of a hydroxyphenyl and neopentyl group (Table 2). The structure of this complex was solved to 1.80 Å and refined to an Rfree of 22.5%. It is clear in this complex that the hydroxyphenyl group occupies the active site (Fig. 2b). The neopentyl R-group, which extends out of the active site, is not clearly visible in the electron density and was omitted from the model. The hydrogen-bonding interactions of MIF with OXIM-11 and OXIM-6 are shown in Fig. 2c.
FIGURE 2. Structure of the MIF/OXIM-scaffold complexes.

a and b, stereo views of OXIM-11 (a) and OXIM-6 (b), shown in σA weighted Fo – Fc electron density maps, calculated omitting all inhibitor molecules. The maps are contoured at 3.5σ. The figures were prepared using Molscript (29) and Bobscript (26). c, diagrams displaying hydrogen-bond interactions between MIF and OXIM-11 (left) and between MIF and OXIM-6 (right). The neopentyl R-group, which extends out of the active site, is not clearly visible in the electron density and was omitted from the model. For clarity, it is shown in panels b and c in a possible orientation. Panel c was made using ChemDraw.
OXIM-11 Preserves the Anti-inflammatory Activities of Glucocorticoids
The potent binding of OXIM-11 to the active site of MIF led us to study its effects on physiological pathways in which MIF is involved. We therefore examined the effect of OXIM-11 on the counter-regulatory action of MIF on glucocorticoid suppression of TNF release. We pretreated MIF with various concentrations of OXIM-11 (0.1–10 μm) prior to dexamethasone and MIF treatment of endotoxin-stimulated human macrophages. In a dose-dependent manner, OXIM-11 abolished the MIF counter-regulatory action on this potent, anti-inflammatory glucocorticoid, as shown in a TNF secretion assay (Fig. 3a). We further studied the mechanisms underlying the inhibitory effects of OXIM-11 on TNF release. The nuclear transcription factor NF-κB occupies a central role in the regulation of TNF and of other pro-inflammatory cytokines. MIF has been shown to play a pivotal role in mediating endotoxin-activated signaling pathways, including NF-κB activation (9). To determine the effect of OXIM-11 on the NF-κB signaling pathway, we analyzed nuclear extracts from endotoxin-stimulated macrophages by electrophoretic mobility shift assay. As shown in Fig. 3b, OXIM-11 dose-dependently inhibited NF-κB activation, an effect consistent with the previously reported MIF antisense-mediated suppression of NF-κB (9).
FIGURE 3. Effect of OXIM-11 on MIF regulation of glucocorticoid activity and on NF-κB activation from LPS-treated macrophages.

a, OXIM-11 decreased the level of TNF-α production in combination with dexamethasone (Dex) from MIF stimulation of LPS-treated human macrophages. Monocyte-derived macrophages from human peripheral blood were preincubated with dexamethasone (10−9 m) or dexamethasone plus MIF (100 ng/ml) and various concentrations of OXIM-11 before the addition of 0.5 μg/ml LPS. The data shown are mean ± S.D. of triplicate wells in experiments that were repeated twice. b, monocyte-derived macrophages from human peripheral blood were treated with various concentrations of OXIM-11 (1–100 μm) 30 min prior to LPS (10 ng/ml) addition. Nuclear extract was isolated using NE-PER nuclear and cytoplasmic extraction reagents. For detection of NF-κB binding, nuclear extract from cells (~5 μg of protein) was incubated with 0.2 ng of 32P-labeled double-stranded oligonucleotide sequence, and the samples were resolved on a 4% polyacrylamide gel and visualized directly by autoradiography after drying the gel. The results for each treatment were expressed as relative intensity when compared with the control (saline-treated) intensity.
Effect of OXIM-11 on CLP-induced Sepsis
We also examined the time course of MIF release in mice with CLP-induced peritonitis, a widely used model of sepsis. As shown in Fig. 4a, serum MIF levels increased to 70% of maximal value within 24 h after CLP and peaked at 36 h. This finding identifies MIF as a late mediator in the inflammatory response and defines a new therapeutic window for the treatment of experimental sepsis. Based on these data, we reasoned that the late administration of OXIM-11 could confer protection against mortality in sepsis. Intraperitoneal administration of OXIM-11 (3.5 mg/kg) initiated 24 h after operation and continued twice daily for 3 days significantly improved the survival rate to 70% when compared with 10% survival in the control (vehicle-treated) group of mice (Fig. 4b). These results are similar to those recently published on the MIF inhibitor (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester, with the exception that OXIM-11 is a more potent inhibitor (11). The similar results by two unrelated compounds support the hypothesis that the MIF catalytic site is a pro-inflammatory site and an important drug target for sepsis, septic shock, and other inflammatory diseases.
FIGURE 4. Therapeutic targeting of MIF.

a, kinetics of MIF appearance in the serum after CLP surgery. Blood was collected after 1, 12, 24, 36, and 48 h after CLP, and the serum was analyzed by Western blot to determine the circulating MIF levels. (n = 5 mice/time point). b, OXIM-11 is protective after 24 h of late treatment in a CLP model. Mice were injected intraperitoneally with OXIM-11 (3.5 mg/kg) (n = 13, **, p < 0.01) or vehicle 24 h after CLP (n = 13). An additional two injections were given on day 2 and 3.
DISCUSSION
The interactions of OXIM-11 with MIF are shown in Fig. 2. There are hydrogen-bond interactions between 1) the imino group of Pro-1 and ester oxygen of OXIM-11, 2) the hydroxyl group of Tyr-95 and the OXIM-11 carbonyl oxygen, and 3) Lys-32 with the imine functional group of OXIM-11. A different conformation seen in another active site is due to crystal contacts perturbing the orientation of OXIM-11. Nonetheless, the cyclohexyl group is also found buried in the cleft in this active site. The active site displayed in Fig. 2a is solvent-exposed and therefore is representative of OXIM-11 binding to MIF in solution. The new orientation raised the issue of the contribution of each chemical moiety in binding to the active site. If OXIM-11 were to bind in the conventional orientation, with the hydroxyphenyl ring buried in the binding pocket, fewer polar groups would be in favorable positions for hydrogen bonding (Fig. 2c). We therefore suggest that the preference for the cyclohexyl moiety of OXIM-11 to occupy the active site is at least partially driven by the optimization of hydrogen bonds of carbonyl oxime linker. The carbonyl oxime derivative can be considered a new pharmacophore that contains two R-groups at each end that can be optimized for MIF inhibition.
Our results demonstrate that although not every OXIM R-group readily binds in the MIF active site, such as that of OXIM-6, aromatic groups are not the only groups that are able to bind in the mainly hydrophobic MIF binding cleft. For example, cyclohexyl rings are also able to bind. We propose that in such a case, in which there are chemical groups that can bind in the pocket at each end of a molecule, separated by a hydrophilic scaffold, the orientation of binding is dictated not only by the relative affinities of the hydrophobic “head groups” but also by the orientation providing more optimum hydrogen bonding of the protein with the hydrophilic scaffold, in this case, the OXIM template. This is the only known example where a pharmacophore has the versatility to bind to its target site by a 180° rotation dependent on two R-groups.
In summary, OXIM-11 potently binds to MIF and thereby reduces MIF-induced cell activation and inflammation. The mechanism underlying the anti-inflammatory effect of OXIM-11 may be related, at least in part, to the ability of the compound to inhibit MIF-mediated counter-regulation of the anti-inflammatory glucocorticoid activity and to decrease NF-κB activation. Importantly and consistent with the profile of MIF appearance in the serum, OXIM-11 treatment initiated 24 h after CLP markedly improved survival in established polymicrobial sepsis. Our data with ISO-1, an MIF inhibitor, and now with OXIM-11, support the hypothesis that inhibition of the MIF pro-inflammatory site can significantly improve survival in sepsis in a clinically relevant time frame. Structural studies of the MIF·OXIM complex indicate that the carbonyl oxime linker interacts with the site in a way that selects between two orientations of binding that are 180° apart. The factors responsible for this are the accommodation of different hydrophobic groups in the active site and the hydrogen bonds made by the linker. This is generally instructive in regards to rational drug design. Modifying inhibitors by attaching or replacing side chains can cause a reversal of orientation based upon 1) the ability of the “core” of the inhibitor to bind to the protein in more than one orientation and 2) the ability of the new side chain to bind in a pocket normally occupied by another moiety. This can be used to guide optimization of lead compounds based on structural studies.
The carbonyl oxime now provides a new pharmacophore scaffold to design more potent inhibitors of the MIF pro-inflammatory activity and raises the possibility that the use of OXIM-11 or a derivative may provide a key breakthrough in the treatment of sepsis. Future studies will reveal whether co-treatments with OXIM-11 (or a derivative) and other inflammatory regimes such as low dose glucocorticoids would additionally improve survival.
Acknowledgments
We gratefully acknowledge and thank Randi and Mark Jacobson for generous philanthropic support of the studies conducted at The Feinstein Institute for Medical Research.
Footnotes
This research was supported by National Institutes of Health Grants HL081655 (to E. J. M. and Y. A.-A.) and AI065029 (to E. L.) and by National Institutes of Health Training Grant T32 CA09085 (to G. V. C.).
The atomic coordinates and structure factors (code 2OOH, 2OOW, and 2OOZ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: MIF, macrophage migration inhibitory factor; OXIM, carbonyloxime; ISO-1, (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester; CLP, cecal ligation and puncture; TNF, tumor necrosis factor; LPS, lipopolysaccharide.
REFERENCES
- 1.Beishuizen A, Thijs LG, Haanen C, Vermes I. J. Clin. Endocrinol. Metab. 2001;86:2811–2816. doi: 10.1210/jcem.86.6.7570. [DOI] [PubMed] [Google Scholar]
- 2.Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J. Microbes Infect. 2002;4:449–460. doi: 10.1016/s1286-4579(02)01560-5. [DOI] [PubMed] [Google Scholar]
- 3.Calandra T. J. Chemother. 2001;13(Spec. No. 1):173–180. doi: 10.1179/joc.2001.13.Supplement-2.173. [DOI] [PubMed] [Google Scholar]
- 4.Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP. Nat. Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 5.Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. Ann. Intern. Med. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 6.Bone RC. Ann. Intern. Med. 1991;115:457–469. doi: 10.7326/0003-4819-115-6-457. [DOI] [PubMed] [Google Scholar]
- 7.Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Ann. Intern. Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 8.Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. J. Exp. Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roger T, David J, Glauser MP, Calandra T. Nature. 2001;414:920–924. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- 10.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 11.Al-Abed Y, Dabideen D, Aljabari B, Valster A, Messmer D, Ochani M, Tanovic M, Ochani K, Bacher M, Nicoletti F, Metz C, Pavlov VA, Miller EJ, Tracey KJ. J. Biol. Chem. 2005;280:36541–36544. doi: 10.1074/jbc.C500243200. [DOI] [PubMed] [Google Scholar]
- 12.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 13.Calandra T, Bucala R. Crit. Rev. Immunol. 1997;17:77–88. doi: 10.1615/critrevimmunol.v17.i1.30. [DOI] [PubMed] [Google Scholar]
- 14.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R. Am. J. Pathol. 1997;150:235–246. [PMC free article] [PubMed] [Google Scholar]
- 15.Sun HW, Bernhagen J, Bucala R, Lolis E. Proc. Natl. Acad. Sci. U. S. A. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Nat. Struct. Biol. 1996;3:259–266. doi: 10.1038/nsb0396-259. [DOI] [PubMed] [Google Scholar]
- 17.Rosengren E, Bucala R, Aman P, Jacobsson L, Odh G, Metz CN, Rorsman H. Mol. Med. 1996;2:143–149. [PMC free article] [PubMed] [Google Scholar]
- 18.Lubetsky JB, Swope M, Dealwis C, Blake P, Lolis E. Biochemistry. 1999;38:7346–7354. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 19.Meyer-Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. J. Immunol. 2006;177:8730–8739. doi: 10.4049/jimmunol.177.12.8730. [DOI] [PubMed] [Google Scholar]
- 20.Nicoletti F, Creange A, Orlikowski D, Bolgert F, Mangano K, Metz C, Di Marco R, Al Abed Y. J. Neuroimmunol. 2005;168:168–174. doi: 10.1016/j.jneuroim.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, Lolis E, Al-Abed Y. J. Biol. Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 22.Cvetkovic I, Al-Abed Y, Miljkovic D, Maksimovic-Ivanic D, Roth J, Bacher M, Lan HY, Nicoletti F, Stosic-Grujicic S. Endocrinology. 2005;146:2942–2951. doi: 10.1210/en.2004-1393. [DOI] [PubMed] [Google Scholar]
- 23.Sakuragi T, Lin X, Metz CN, Ojamaa K, Kohn N, Al-Abed Y, Miller EJ. Surg. Infect. 2007;8:29–40. doi: 10.1089/sur.2006.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dabideen DR, Cheng KF, Aljabari B, Miller EJ, Pavlov VA, Al-Abed Y. J. Med. Chem. 2007;50:1993–1997. doi: 10.1021/jm061477+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue CB, Wityak J, Sielecki TM, Pinto DJ, Batt DG, Cain GA, Sworin M, Rockwell AL, Roderick JJ, Wang S, Orwat MJ, Frietze WE, Bostrom LL, Liu J, Higley CA, Rankin FW, Tobin AE, Emmett G, Lalka GK, Sze JY, Di Meo SV, Mousa SA, Thoolen MJ, Racanelli AL, Olson RE, Hausner EA, Reilly TM, DeGrado WF, Wexler RR. J. Med. Chem. 1997;40:2064–2084. doi: 10.1021/jm960799i. [DOI] [PubMed] [Google Scholar]
- 26.Dios A, Mitchell RA, Aljabari B, Lubetsky J, O’Connor K, Liao H, Senter PD, Manogue KR, Lolis E, Metz C, Bucala R, Callaway DJ, Al-Abed Y. J. Med. Chem. 2002;45:2410–2416. doi: 10.1021/jm010534q. [DOI] [PubMed] [Google Scholar]
- 27.Navaza J. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. Sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]