

Abstract
IRE1α, the most conserved transducer of the unfolded protein response, plays critical roles in many biological processes and cell fate decisions. Reporting in Science, Upton et al. (2012) broadened our understanding of IRE1a as a cell-death executioner, showing that upon ER stress, IRE1α degrades microRNAs to promote translation of caspase-2.
In eukaryotic cells, the endoplasmic reticulum (ER) is a highly specialized organelle responsible for the translation, folding, and modification of approximately one-third of the cell’s proteome. Upon accumulation of unfolded/misfolded proteins in the ER, cells activate the unfolded protein response (UPR) that, in metazoans, is initiated by three ER transmembrane protein sensor(s) in metazoans: inositol requiring enzyme 1 alpha (IRE1α), PKR-like ER kinase (PERK), and activated transcription factor 6 alpha (ATF6α). The UPR is essential for normal cellular and organismal physiology and contributes to the etiology of many diseases (Wang and Kaufman, 2012). Although initial UPR activation provides an adaptive response, severe or chronic UPR activation re-directs the adaptive response into a pro-apoptotic response, although the mechanisms are unknown. Among the ER stress sensors, IRE1α is conserved from yeast to humans. IRE1α has both protein kinase and endoribonuclease (RNase) activities that were originally characterized to initiate removal of a 26 base intron from X-box binding protein 1 (Xbp1) mRNA, thereby producing an active transcription factor that induces genes encoding adaptive functions to limit protein misfolding in the ER. However, IRE1α has a growing list of additional mRNA cleavage substrates identified through regulated IRE1-dependent degradation (RIDD) of mRNAs (Han et al., 2009; Hollien et al., 2009). In a recent report in Science, Upton et al. showed that IRE1α cleaves a new class of RNAs: microRNAs (miRs) that repress translation through binding to sequences in the 3’ end of mRNAs. IRE1α-mediated cleavage of miRs releases a translational block on Caspase 2 (Casp2) mRNA (Upton et al., 2012). Increased expression of Casp2 mRNA then contributes to apoptotic cell death through proteolytic cleavage of Bid, which causes cytochrome c release from mitochondria.
Upton et al. (2012) first demonstrated that treatment with brefeldin A, which causes protein accumulation in the ER, increases CASP2 protein expression in wild-type and Xbp1-/-, Perk-/-, and Atf6α-/- mouse embryo fibroblasts (MEFs), but not in Ire1α-/- MEFs. Although there was no change in the total level of Casp2 mRNA, polysome-associated Casp2 mRNA increased in the wild-type MEFs, but not in the Ire1α-/- MEFs. Importantly, sustained activation of IRE1α reduced levels of miRs −17, −34a, −96 and −125b, miRs that normally repress Casp2 mRNA translation. An in vitro nuclease assay demonstrated that IRE1α directly cleaves the miR-17 precursor at three sites distinct from those cleaved by DICER. Perhaps most convincingly, transfection of anti-miRs, which protect the miRNAs from degradation by IRE1α prevented Casp2 mRNA translational derepression, as shown by western blotting. In addition, anti-miR-17 expression was overcome by over-expression of IRE1α. The authors further showed that proteolytic cleavage of Bid occurs downstream of IRE1α-dependent Casp2 mRNA translational derepression.
The findings from Upton et al. (2012) show that IRE1a cleaves precursor miRs (pre-miR); an event that likely occurs in the nucleus or as the pre-miRs transit through the nuclear pore to the cytoplasm. Although IRE1α-mediated Xbp1 mRNA splicing occurs in the cytoplasm, IRE1α is localized to the inner nuclear envelope (Lee et al., 2002), consistent with a function in nuclear RNA processing. The studies of Upton et al. (2012) provide one example by which IRE1α activates apoptosis, but presumably there are others. Recently, PERK and IRE1α signaling were shown to induce pro-oxidant TXNIP. Whereas PERK signaling induces ATF5 to activate Txnip transcription, IRE1α nuclease cleaves miR-17 to stabilize Txnip mRNA (Lerner et al., 2012; Oslowski et al., 2012). As it is now evident that IRE1α regulates miR production, there may be a multitude of processes that are regulated through IRE1α that will be identified and characterized in the future.
The IRE1α-dependent derepression of Casp2 mRNA translation through miR cleavage was shown to occur in MEFs, mouse insulinoma, and human kidney cell lines. If this IRE1α-dependent derepression of CASP2 occurs in additional cancerous and/or differentiated cell types that secrete high levels of protein, this pathway may be of greater physiological significance. In addition, chemical inhibitors of IRE1α RNase activity now exist (Mimura et al., 2012) that should be tested for the potential to divert apoptosis in response to ER stress. Finally, although IRE1α activation increases the expression of CASP2, there is another, yet unknown, signal that is required for its activation into a functional protease.
In summary, the authors have identified a pro-apoptotic pathway that emanates from IRE1α. This IRE1α-dependent pathway toward apoptosis adds to the other known IRE1α-mediated pathways including Xbp1 mRNA splicing, regulated IRE1-dependent decay (RIDD) of mRNAs, activation of the cJun N-terminal kinase (JNK), and nuclear factor kappa B (NFκB) pathways and inflammasome activation (Wang and Kaufman, 2012). Considering that the loss of IRE1α and/or XBP1 signaling is detrimental, especially for professional secretory cells (which includes pancreatic β cells, plasma cells, hepatocytes, gastric zymogenic cells, and Paneth cells in the small intestine), it appears IRE1α functions as a double-edged sword in the life versus death decision. The RIDD-dependent degradation of mRNAs by IRE1α is proposed to protect cells by reducing the protein-folding burden on the ER. However, RIDD can also perform the role of cell executioner by degrading mRNAs encoding pro-survival proteins during prolonged ER stress. In addition, the IRE1α-JNK pathway has been shown to cause apoptosis under some cellular stresses (Tabas and Ron, 2011). The findings of Upton et al. (2012) thus further our understanding of IRE1α as a regulatory hub of the cell fate decision. CASP2 is the most evolutionarily conserved of caspases identified to date. Although its role in the apoptotic cascade is still elusive, CASP2 regulates NFκB signaling and functions as a tumor suppressor (Bouchier-Hayes and Green, 2012). Given the critical role of IRE1 in NFκB activation, inflammation, and tumorigenesis, it is important to determine how caspase-2 and its downstream targets contribute to these cellular processes during ER stress.
Figure 1. IRE1α-mediated signaling of life and death.
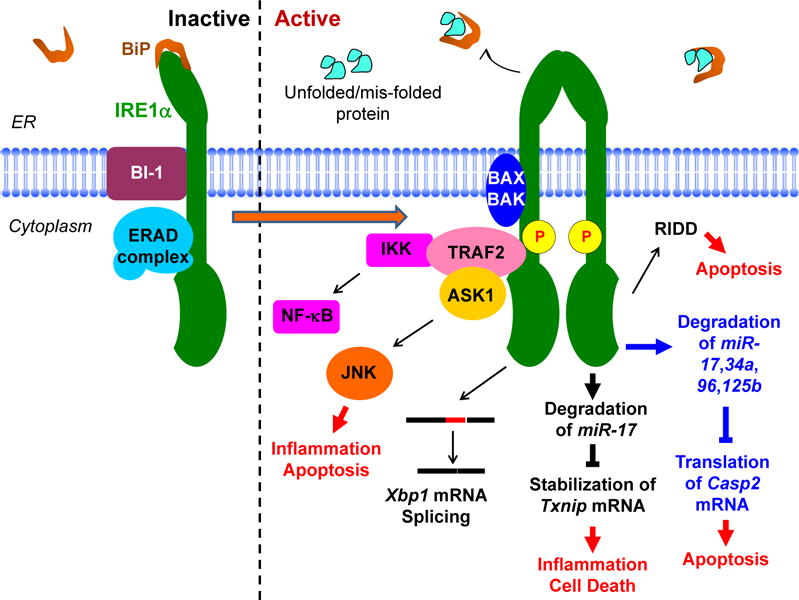
Dimerization of IRE1α induces its activation and initiates downstream signaling through the recruitment of TRAF2 and RNase activation to promote Xbp1 mRNA splicing, degradation of mRNAs, and degradation of miRNAs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bouchier-Hayes L, Green DR. Cell death and differentiation. 2012;19:51–57. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. Genes & development. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, et al. Blood. 2012;119:5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, et al. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al. Science. 2012 doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]