

Abstract
Background
Glutaminyl cyclase (QC) forms the pyroglutamyl residue at the amino terminus of numerous secretory peptides and proteins. We previously proposed the mammalian QC has some features in common with zinc aminopeptidases. We now have generated a structural model for human QC based on the aminopeptidase fold (pdb code 1AMP) and mutated the apparent active site residues to assess their role in QC catalysis.
Results
The structural model proposed here for human QC, deposited in the protein databank as 1MOI, is supported by a variety of fold prediction programs, by the circular dichroism spectrum, and by the presence of the disulfide. Mutagenesis of the six active site residues present in both 1AMP and QC reveal essential roles for the two histidines (140 and 330, QC numbering) and the two glutamates (201 and 202), while the two aspartates (159 and 248) appear to play no catalytic role. ICP-MS analysis shows less than stoichiometric zinc (0.3:1) in the purified enzyme.
Conclusions
We conclude that human pituitary glutaminyl cyclase and bacterial zinc aminopeptidase share a common fold and active site residues. In contrast to the aminopeptidase, however, QC does not appear to require zinc for enzymatic activity.
Background
The synthesis of biologically active peptide hormones and neurotransmitters begins with precursor proteins undergoing maturation via different processing enzymes. Prohormone convertase catalyzes cleavage within precursor proteins at paired basic amino acid residues [1] while carboxypeptidase removes these basic residues [2]. Several peptide hormones and neurotransmitters, such as thyrotropin-releasing hormone, are further modified at their termini. These termini modifications appear to be necessary for recognition of a receptor-binding site or protection of the peptide from exopeptidases. A frequent terminus modification is the oxidative cleavage by peptidyl α-amidating monooxygenase [3] of a carboxyl terminal glycine to form a terminal amide. The amino terminus can be acylated or, in the case of an N-terminal glutaminyl residue, can be cyclized to the pyroglutamyl residue with the accompanying release of the amide nitrogen as ammonia. This last reaction is catalyzed by glutaminyl cyclase (EC 2.3.2.5).
In 1965 Messer and Ottesen identified and purified the first known form of glutaminyl cyclase (QC) from papaya latex [4]. The laboratories of J.S. Kizer and J. Spiess concurrently published the initial identification of QC in porcine and bovine pituitaries, respectively [5,6] and, as expected, QC was found to be localized within secretory vesicles [7]. Subsequent cloning of the bovine and human QC cDNA revealed very similar proteins of 361 amino acids, including a nearly thirty amino acid endoplasmic reticulum signal sequence [8,9]. QC homologs can be discerned in the genomes of a wide variety of organisms including yeast [10], with the human QC gene residing on chromosome 2 (NCBI LocusID: 25797).
Human pituitary QC has been expressed in bacteria, although this has often resulted in protein with little activity, stability, or solubility, or active protein with a non-cleavable GST fusion [9-12]. Thus, functional and structural characterization of QC has been limited. Site directed mutagenesis of the two conserved cysteines revealed neither was essential to enzyme function [12]. Recent mutational analysis of four highly conserved histidine residues indicated that two residues participated in catalysis and two in substrate binding [10]. Taken together with the apparent lack of nucleophilic essential amino acids [5,12], this suggests catalysis by mammalian QC proceeds via general acid/base promotion of the direct cyclization of the terminal glutamine as we have proposed for the papaya enzyme [13,14].
Glutaminyl cyclase has also been purified from papaya latex, but animal and plant forms of QC have no discernable primary sequence identity [15]. We hypothesized previously [10] that, while animal and plant forms of QC may be utilize similar catalytic mechanisms, they have different tertiary structures and probably represent two independent families of enzymes. Taken together with previous work on papaya QC, this study demonstrates that the mammalian and plant QC forms are indeed structurally unrelated, while providing evidence of a structural relationship between mammalian QC and the bacterial zinc aminopeptidase family.
Results
Features of the model of human QC
We predict that human QC and the bacterial zinc exopeptidases share a similar fold (Figure 1). This consists of a central 8-stranded β-sheet, which is of a mixed parallel/anti-parallel nature. The sheet is flanked by six α-helices on one side and three on the other. The proposed active site lies at the C-terminal end of the central two β-strands, and is mostly comprised of residues from loop regions.
Figure 1.
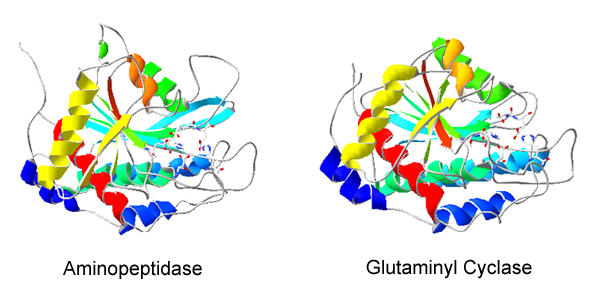
Ribbon diagram of hQC theoretical model. Ribbon diagram of secondary and tertiary structures of Vibrio aminopeptidase (PDB 1AMP) and human glutaminyl cyclase (PDB 1MOI). Structures are oriented so that active sites are on the right of the molecule. Illustration was generated with DeepView 3.7 using solid open GL rendering and colored by secondary structure succession.
The sequence identity between human QC and members of the aminopeptidase family is low: between QC and Vibrio proteolyticus aminopeptidase (pdb code 1AMP, [16]), for example, it is 16% over the entire length of the alignment. Nevertheless the fold recognition program "Fugue" assigns QC to the "bacterial exopeptidases" family with a Z-score of 26.5. Z-scores with values above 6 correspond to "certain" assignments (>99% probability). Fugue is amongst the most sensitive fold recognition programs currently available [17]. It considers not just the amino acid type, but also its environment, up-weighting those amino-acid residues likely to be conserved through evolution. Conservation of these important features leads Fugue to give the confident assignment in this case.
The sequence alignment (Figure 2) and model have several convincing features. The Psipred prediction of secondary structure corresponds extremely well to that of the aminopeptidase family (Figure 2). This family consists of aminopeptidases from Vibrio proteolyticus (pdb code 1AMP) and Streptomyces griseus (pdb code 1XJO, [18], and the catalytic domain of carboxypeptidase G2 from Pseudomonas (pdb code 1CG2, [19]). Fugue does not use predicted secondary structure to determine either the most likely family assignment for a given sequence nor the alignment, and so the correlation gives increased confidence in the fold assignment and alignment. The N-linked glycosylation sites (Asn 49 and Asn 296) are solvent exposed in the model. The cysteine residues are positioned such that they are able to form a disulfide bond. This bond is in a different position in the QC model as compared to that in template structures 1AMP and 1XJO (the disulfide is absent in 1CG2). In the QC model the disulfide is between α-helix 4 and the third β-strand of the sheet, and as such is completely buried in the core and unlikely to play any direct role in catalysis. The reported effect on activity produced by reducing the disulfide [11] is therefore likely to be structural.
Figure 2.

Sequence alignment of hQC and structural templates. Alignment of amino acid sequence of Vibrio aminopeptidase (PDB code 1AMP), Streptomyces aminopeptidase (PDB code 1XJO) Pseudomonas carboxypeptidase G2 (PDB code 1CG2) and human pituitiary glutaminyl cyclase (bottom line). Numbers along the top indicate the alignment position. Numbers in parentheses on the left hand side indicate the sequence numbering for the first amino acid in on that line for each sequence. Bars indicate secondary structure (dark bars for helices, light bars for strands) for those sequences with experimentally determined structures. Bars underneath the alignment (labeled "Psipred") indicate predicted secondary structure for the human QC sequence.
We predict from the model, therefore, that human QC has a mixed α-helix/ β-strand structure that, consistent with the previous prediction, Asn 49 is the likely glycosylation site, and that the two cysteine residues (139 and 164) should be in the disulfide-bonded rather than free thiol form. It should be noted that these features were not used as constraints when aligning the sequences. Their experimental validation can, therefore, serve as a confirmation of the model.
Interestingly, the active site residues (His 140, Asp 159, Glu 201, Glu 202, Asp 248 and His 330, QC numbering) in 1AMP are conserved in the QC model (Figure 3), and probably adopt a similar conformation. Five of these residues form a metal binding site for two zinc ions in 1AMP, and it would appear that a metal binding site could also be present in human QC. However, there is conflicting evidence for the presence of zinc in human QC (see discussion).
Figure 3.
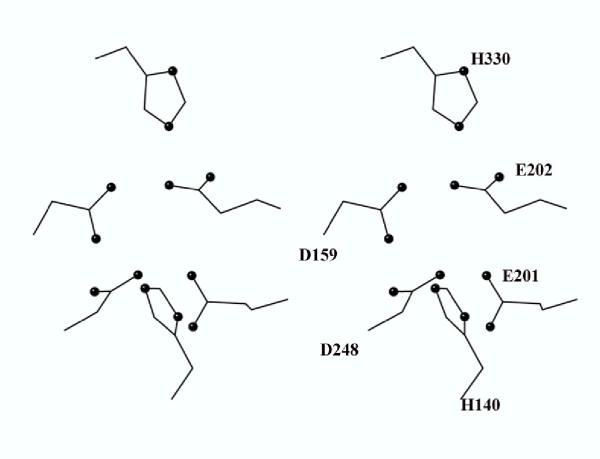
Predicted active site of human glutaminyl cyclase. Figure 5 represents the five conserved human QC residues corresponding to the zinc ligands of the bacterial aminopeptidase plus the conserved glutamate corresponding to the general base of the aminopeptidase. QC numbering from the top (clockwise): His 330, Glu 202, Glu 201, His 140, Asp 248, Asp 159. See Table 1 for comparisons with 1AMP.
Analysis of papaya QC
In contrast to human QC, the papaya enzyme produces hits with lower confidence levels. It is predicted by Fugue [17] as having similarity to the cytochrome cd1-nitrite reductase family, specifically to the β-sheet domain, which is an eight-bladed β-propeller structure (pdb codes 1AOQ and 1NIR, Z-score = 8.1) and to bacterial amine dehydrogenase (pdb code 1JJU, chain B, Z-score = 7.4), which is a seven-bladed β-propeller. Again in contrast to human QC, papaya QC is predicted by Psipred to have an entirely β-sheet structure, which is consistent with the CD spectrum [20]. Given the relatively low score, the ambiguity regarding the number of blades of the propeller and the difficulty of building accurate models of proteins with repetitive secondary structure [21], we have not built a model of papaya QC. However, it is clear that the papaya QC is structurally distinct from the human QC having an all-beta structure, possibly a β-propeller.
Secondary structure and metal analyses
Secondary structural characteristics of DES hQC and the mutant forms (data not shown) were examined by circular dichroism (Figure 4). Samples were prepared as described in the methods section and then analyzed at Tulane University with an OLIS CD spectrophotometer. Eight scans from 260 nm to 196 nm were obtained on a 6 μM sample. The resulting spectrum indicated that DES hQC contains 32% α-helix, and 17% β-sheet, a secondary structure composition that correlates well with the Vibrio proteolyticus aminopeptidase, which contains 35% α-helix, and 17% β-sheet.
Figure 4.
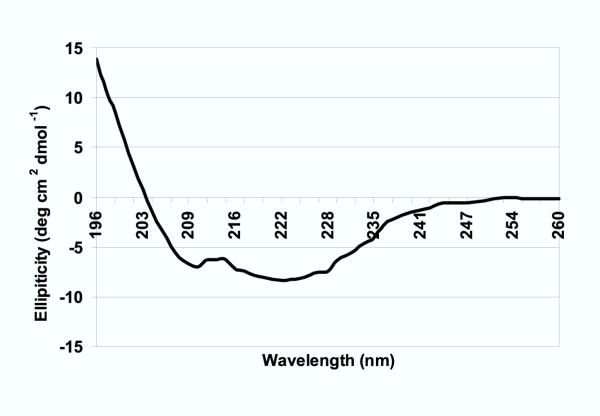
Circular dichroism spectrum of DES hQC. Circular dichroism of 6 μM DES hQC in 5 mM sodium phosphate buffer, pH 6.0 from 196 to 260 nm.
Metal content was determined by ICP-MS as described in Methods. An average of 2.0 nmol/ml Zn were obtained for every 6.2 nmol/ml QC, giving a molar stoichiometry of 0.32:1 Zn:QC.
Active site identification
Four QC acidic amino acid residues were selected for site-directed mutagenesis to alanine based on their conservation between species [10] as well as their presence in the bacterial aminopeptidase active site (see figure 3 and model analysis above). Mutations in either glutamate 201 or 202 resulted in completely inactive QC forms. These mutants interact as well as unmutated enzyme with the affinity column, suggesting that the binding site was not disrupted; furthermore, fluorescence scans and CD spectroscopy revealed no apparent changes in structure. Kinetic studies on aspartate 159 and 248 mutants revealed no significant differences in the Km from unmutated DES hQC (1.4 ± 0.2 (unmutated), 1.5 ± 0.3 (D159), and 1.4 ± 0.4 (D248) mM, respectively) and only a slight decrease in the Vmax of D159 (3.7 ± 0.4 (unmutated), 1.5 ± 0.2 (D159), and 3.3 ± 0.5 (D248) μmol/min/mg, respectively).
Discussion
As a peptide-processing enzyme in the mammalian secretory pathway, QC is susceptible to a variety of posttranslational modifications. Recently we showed that at least one of the two predicted N-linked glycosylation sites is indeed glycosylated in the insect-expressed protein and one, if not both, sites in the native tissue QC contain glycans as well [22]. Mutagenesis of both sites showed that the presence or absence of glycans had no apparent effect on QC enzymatic activity. The glycans, then, either have a cellular role such as localization or they are simply fortuitous. Also, consistent with the oxidizing environment of the secretory pathway is the finding that the two cysteine residues in QC are disulfide-bonded [22]. The presence of a disulfide bond was also recently reported by Schilling [11] in human QC expressed in yeast. They postulated a role for this disulfide bond in enzyme activity since addition of high concentrations of reducing agents result in loss of activity. Our previous mutagenesis of these two cysteine residues [12] indicate that they are not directly involved in catalysis, making it likely that they play a role in stabilizing an active form of enzyme tertiary structure. Indeed, the model predicts the disulfide bond is immediately behind the enzyme active site and crosslinks two pieces of secondary structure directly connected to loops containing active site residues.
Tertiary structure examination of QC began with the development of a predicted model (pdb code 1MOI) based on a bacterial zinc aminopeptidase fold as described in the methods section and predicted previously [10]. DES hQC secondary structure composition determined using circular dichroism shows DES hQC to contain 32% α-helix, 17% β-sheet, and 52% random coil, a secondary structure composition that corresponds well both to the CD analysis of the bacterial aminopeptidase which is 35% α-helix, 17% β-sheet, and 48% coil [23] and the secondary structure composition of the model (33% α-helix, 24% β-sheet, and 43% coil). Not only does the proportion of each secondary structure type correspond well between the QC model and the bacterial aminopeptidase family, but also the predicted linear arrangement and position of each secondary structural element from Psipred corresponds well with the aminopeptidases (Figure 2). In addition, the model predicts that the conserved cysteine residues are in position to form a disulfide bond, a prediction experimentally confirmed in our previous study [22]. Further, both N-linked glycosylation sites are located on the exterior of the protein model.
In contrast with human QC, papaya QC is an all β-sheet protein. This has been shown by Oberg [20], based on CD spectra. The computational methods used here are in agreement with this result, in that β-structure is predicted by Psipred and the weak similarity to a β-propeller structure is also suggestive of a β-structure. In addition, the lack of sequence similarity between the two enzymes [15], and the dramatic differences in susceptibility to inhibition by both competitive inhibitors and diethylpyrocarbonate [10,24] suggest that the two enzymes are unlikely to be divergently evolved.
Convergently evolved functions and/or mechanisms are not without precedent. The most widely known example is the catalytic triad, which was first found in protease chymotrypsin [25], and subsequently in the structurally unrelated but functionally similar subtilisin [26]. It has since been observed in lipases [27] and recently in acetylhydrolase [28]. All of these enzymes share related functions and similar mechanisms, but have quite different folds.
Holbrook and Ingram [29] have suggested a set of tests for distinguishing divergent and convergent evolution in enzymes. In descending order of importance they are: sequence similarity at the DNA level, sequence similarity at the amino-acid level, similarity of three-dimensional structure, similarity of enzyme-substrate interactions, similar catalytic mechanism and segments of the chain essential for catalysis are in the same sequence. The catalytic triad containing enzymes fail every test except for similar mechanism and so are generally accepted as examples of convergent evolution. Similar function with different fold and lack of sequence similarity is currently used as evidence for convergence (for example [30]). Russell [31] has looked for similar spatial arrangements of catalytic residues in proteins that share no fold similarity, and therefore are unlikely to be divergently evolved. In a search of the protein structure database he identified 22 pairs of proteins that have similar function but different folds, and are therefore likely to be convergently evolved. Similarly, we find that human and papaya QCs are unrelated at the DNA and amino-acid levels, are likely to have different structures, and yet have similar functions. In the absence of more structural studies we are unable to tell whether the least important of Holbrook and Ingram's criteria are met, but given the known structural differences this seems highly unlikely.
Six amino acid residues are predicted to be at the active site of QC based on their location in the zinc aminopeptidase fold (Figure 3, Table 1). These are picked up quickly in a PSI-BLAST [32] search with the human QC protein sequence. Two of these, QC histidines 140 and 330, were previously shown to be essential catalytic residues [10]. The other four residues, glutamates 201 and 202 and aspartates 159 and 248, were examined in this study. Mutagenesis of the two glutamates showed both to be essential for QC enzymatic activity. The aminopeptidase counterpart of QC Glu 201 acts as a general base to promote water attack on the peptide bond in aminopeptidase catalysis. A glutamate residue plays the same role in other well-studied zinc peptide hydrolases such as carboxypeptidase A and thermolysin. It is tempting to speculate that QC Glu 201 may perform a similar function in the transamidation reaction of glutaminyl cyclization by promoting attack of the alpha amine on the gamma carboxamide.
Table 1.
Active site residue comparison. Comparison of positionally identical active site residues in Vibrio proteolyticus aminopeptidase and human pituitary glutaminyl cyclase. A designation of "none" means the residue plays no discernable role in the enzymatic reaction.
| Aminopeptidase Residue | Function in Aminopeptidase | QC residue | Function in QC |
| Asp 117 | Zn bridge | Asp 159 | None |
| His 256 | Coordinates Zn #1 | His 330 | Catalytic |
| Glu 152 | Coordinates Zn #1 | Glu 202 | Catalytic |
| His 97 | Coordinates Zn #2 | His 140 | Catalytic |
| Asp 179 | Coordinates Zn #2 | Asp 248 | None |
| Glu 151 | Catalytic | Glu 201 | Catalytic |
| His 230 | None | His 307 | Substrate binding |
| Met 242 | None | His 319 | Substrate binding |
Does human QC contain active site zinc? Our ICP-MS analysis indicated a stoichiometry of 0.3 mol Zn/mol QC, effectively ruling out the presence of two zinc atoms and suggesting the residual zinc presence is adventitious, although admittedly it is possible some zinc was lost during enzyme purification. QC is refractory to metal chelators such as EDTA, nitrolotriacetic acid [5,10], peptide thiols and boronic acids [24], Glu-Trp, and gamma-glutamate hydroxamate (unpublished results). In addition, the enzyme refolds into an active form after denaturation with urea [6,9]. Finally, an absence of metal is consistent with the observation that mutation of the two aspartates, which are zinc ligands in the aminopeptidase, had no effect on QC enzymatic activity. Taken together, the above evidence argues strongly against a requirement for zinc at the QC active site.
Schilling et al [24] have recently proposed that QC is a metalloenzyme based on time dependent inhibition by o-phenanthroline and dipicolinic acid, as well as the activation of the inactive QC "apoenzyme" by added zinc. In contrast, we have observed competitive inhibition (Ki 20 μM) with o-phenanthroline. As Schilling et al. point out, both o-phenanthroline and dipicolinic acid are planar heterocyclic compounds. Both can therefore potentially act as reversible inhibitors just as the imidizole derivatives they report [5]. It is also curious that cobalt did not reactivate QC in the Schilling study, since it commonly reactivates zinc peptidases. It remains to be seen, therefore, whether the reported reactivation by zinc is due to replacement of an active site metal, binding of zinc to an alternative site such as the polyhistidine expression tag, or an experimental artifact.
The bacterial zinc aminopeptidase fold is found in proteins such as carboxypeptidase G2 [19] as well as the human tranferrin receptor [3], which exhibits no known enzymatic activity. A VAST search of the protein databank using the Vibrio proteolyticus aminopeptidase (1AMP) structure reveals a number of similar tertiary structures including, intriguingly, a pyroglutamate aminopeptidase (1A2Z) from Thermococcus litoralis. The VAST superimposition of 1A2Z with 1AMP shows good structural alignment with the central beta sheet and connecting helices, with an alpha carbon RMSD of 3.3 angstroms. Pyroglutamate aminopeptidase is a cysteine peptidase, with the catalytic triad only slightly offset from the zinc aminopeptidase active site residues described in this paper. It is clear, then, that the bacterial zinc aminopeptidase fold is available for a variety of functions other than zinc-catalyzed peptide bond hydrolysis. In the case of QC, it appears that four of the six critical active site residues of the aminopeptidase are also essential for QC catalysis, possibly representing the distant recruitment of catalytic residues during evolution among hydrolases/amidotranferases that operate on peptide termini.
Conclusions
A theoretical model of human pituitary glutaminyl cyclase has been generated based on the tertiary structure of the bacterial zinc aminopeptidase family. Six key active site residues in the aminopeptidase structure are present in the QC model, with four of these six residues essential for QC enzymatic activity. Despite the structural similarity with the zinc aminopeptidase family, our site directed mutagenesis, inhibitor studies, and metal analysis argue strongly against QC being a metalloenzyme.
The structural model proposed here will provide the basis for interpreting further mutagenic and spectroscopic studies of this mammalian peptide processing enzyme, as well as facilitate structural comparisons with the plant glutaminyl cyclase family.
Methods
Materials
Peptide substrates were obtained from Bachem. All restriction enzymes and their corresponding buffers were obtained from Promega. Oligonucleotide primers were made by Integrated DNA technologies, Inc. Automated sequencing was performed at UF DNA Core Lab. Mutagenesis reagents were obtained from Amersham Pharmacia Biotech. Human pituitary glutaminyl cyclase cDNA was expressed in Drosophila cell culture and purified from serum-free media by affinity chromatography as described previously [22].
Comparative modeling
Secondary structure predictions were calculated using Psipred [33]. Homologous proteins of known structure were identified, and initial sequence alignments were produced using the Fugue program [17]. Clear homologues were found: aminopeptidase from Vibrio proteolyticus (pdb code 1AMP) and Streptomyces griseus (pdb code 1XJO), and carboxypeptidase G2 from Pseudomonas sp. (pdb code 1CG2). These all belong to the "bacterial aminopeptidase" family in the structural alignment database Homstrad [34]. The initial alignment was adjusted manually (Figure 2) and verified using Harmony3 (J. Shi, unpublished), which matches observed substitution frequency in the alignment with predicted substitution frequency from standard and environment specific substitution tables. Models were built with Modeller 6.1 [35,36]. Since Cys 139 and Cys 164 were within bonding distance in the initial models, in subsequent models they were built in the disulfide bonded rather then free thiol form. Five loops (residues 177–186, 203–215, 227–237, 273–269, 298–304) contained insertions in the QC model relative to the template structures. These loops were rebuilt with Rapper [37,38]. As these loops had no equivalent in the parent structures, they should be considered the least reliable parts of the model. Similarly, no homologous residues were available for the N-terminal 35 residues and these were therefore omitted. The final model had only a single Ramachandran outlier (Phe 146), according to the Rampage program [39]. It has been submitted to the Protein Data Bank and assigned pdb code 1MOI.
Site-directed mutagenesis
A mutation was introduced into the DES hQC expression vector using Unique Site Elimination Mutagenesis kit (Amersham Pharmacia Biotech). This method uses two primers to incorporate mutations into the same strand of DNA by in vitro DNA synthesis with T4 DNA polymerase [40]. Primers were designed to replace the glutamic acids at postions 201 or 202, and aspartic acids at positions 159 and 248 with alanine while introducing a silent mutation in the expression vector that deletes a restriction site. Mutant constructs were confirmed by automated sequencing at UF DNA Core Lab (Gainsville, FL).
Activity assay
A spectrophotometric assay was used to detect QC activity as reported by Bateman [41] with the modification that 5 mM Gln-NH2 was routinely used as substrate and 10 μM bestatin was included. For kinetic analysis, substrate concentrations from 0.3 mM to 4 mM were used.
Protein samples (1–5 μg) were run in 10 % NuPAGE as described in the manufacturers instructions except the reducing agent, 50 μM DTT, was omitted in non-reduced samples.
Metal analysis
Purified protein samples were analyzed for metal content with ICP-MS by the University of Georgia Chemical Analysis Laboratory. Two independent purifications were used, with the buffers treated with Chelex 100 to remove residual zinc. Blanks included buffer alone as well as elution buffer from a complete purification protocol using media from nontransfected S2 cells which had been treated in a manner identical to the QC-expressing S2 cells. Protein was quantified using the predicted molar extinction coefficient (55 310 M-1cm-1 at 280 nm) and by SDS-PAGE followed by staining with Coomassie blue, then densitometry with BSA standards. Both methods agreed closely.
Secondary structure analysis
Purified protein samples were dialyzed against 5 mM potassium phosphate buffer, pH 6.0 and then concentrated to 6 μM using Microcon Centricon-10 (Amicon). Data was collected using an OLIS circular dichroism (CD) spectrophotometer. Fifty data points were collected for each scan from 196–260 nm with a total of eight scans per sample. Baseline corrected data were smoothed with a Gavitsky-Golay filter. The Y-scale was measured in ellipiticity, θ, and set from -40 to 60. Molar ellipiticity, [θ], was calculated for each data point using the following formula: [θ] = (100 * θ) / (C * n * l) where C = concentration in mM, n = number of residues, and l = path length of cell in cm. The molar ellipiticity values were imported into the K2d program [42] which uses a database comparison to predict the % α-helix, % β-sheet, and % random coil.
List of abbreviations
QC, Glutaminyl Cyclase
ICP-MS, Inductively Coupled Plasma Mass Spectrometry
DES, Drosophila Expression System
PDB, Protein Data Bank
DTT, Dithiothreitol
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis
BSA, bovine serum albumin
Authors' contributions
REB carried out the expression, purification, mutagenesis, and spectral studies. SCL performed the modeling studies. SAM performed the kinetic experiments. RCB designed and oversaw the study. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
Funding from the NSF EPSCOR Program, the state of Mississippi, and the University of Southern Mississippi is gratefully acknowledged. SCL is a Wellcome Trust fellow of mathematical biology. We thank Iris Lindberg and Jae Hwang at the LSU Medical Center for advice on the S2 cell cultures. We also thank Pernilla Wittung-Stafshede and Irina Pozdnyakova at Tulane for assistance with the circular dichroism experiments.
Contributor Information
Rachell E Booth, Email: rbooth@swt.edu.
Simon C Lovell, Email: simon.lovell@man.ac.uk.
Stephanie A Misquitta, Email: stephaniemisquitta@yahoo.com.
Robert C Bateman, Jr, Email: robert.bateman@usm.edu.
References
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/S1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Reznik SE, Fricker LD. Carboxypeptidases from A to z: implications in embryonic development and Wnt binding. Cell Mol Life Sci. 2001;58:1790–1804. doi: 10.1007/PL00000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge ST, Mains RE, Eipper BA, Amzel LM. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell Mol Life Sci. 2000;57:1236–1259. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer M, Ottesen M. Isolation and properties of glutamine cyclotransferase of dried papaya latex. C R Trav Lab Carlsberg. 1965;35:1–24. [PubMed] [Google Scholar]
- Busby WH, Jr, Quackenbush GE, Humm J, Youngblood WW, Kizer JS. An enzyme that converts glutaminyl-peptides into pyroglutamyl-peptides. J Biol Chem. 1987;262:8532–8536. [PubMed] [Google Scholar]
- Fischer WH, Spiess J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc Natl Acad Sci U S A. 1987;84:3628–3632. doi: 10.1073/pnas.84.11.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockers TM, Kreutz MR, Pohl T. Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J Neuroendocrinol. 1995;7:445–453. doi: 10.1111/j.1365-2826.1995.tb00780.x. [DOI] [PubMed] [Google Scholar]
- Pohl T, Zimmer M, Mugele K, Spiess J. Primary structure and functional expression of a glutaminyl cyclase. Proc Natl Acad Sci U S A. 1991;88:10059–10063. doi: 10.1073/pnas.88.22.10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Chuang CZ, Bateman RC., Jr Molecular cloning, sequence analysis and expression of human pituitary glutaminyl cyclase. J Mol Endocrinol. 1994;13:77–86. doi: 10.1677/jme.0.0130077. [DOI] [PubMed] [Google Scholar]
- Bateman RC, Jr, Temple JS, Misquitta SA, Booth RE. Evidence for essential histidines in human pituitary glutaminyl cyclase. Biochemistry. 2001;40:11246–11250. doi: 10.1021/bi011177o. [DOI] [PubMed] [Google Scholar]
- Schilling S, Hoffmann T, Rosche F, Manhart S, Wasternack C, Demuth HU. Heterologous expression and characterization of human glutaminyl cyclase: evidence for a disulfide bond with importance for catalytic activity. Biochemistry. 2002;41:10849–10857. doi: 10.1021/bi0260381. [DOI] [PubMed] [Google Scholar]
- Temple JS, Song I, Burns KH, Bateman RC., Jr Absence of an Essential Thiol in Human Glutaminyl Cyclase: Implications for Mechanism. Korean J Biol Sci. 1998;2:243–248. [Google Scholar]
- Gololobov MY, Wang W, Bateman RC., Jr Substrate and inhibitor specificity of glutamine cyclotransferase (QC) Biol Chem Hoppe Seyler. 1996;377:395–398. [PubMed] [Google Scholar]
- Gololobov MY, Song I, Wang W, Bateman RC., Jr Steady-state kinetics of glutamine cyclotransferase. Arch Biochem Biophys. 1994;309:300–307. doi: 10.1006/abbi.1994.1117. [DOI] [PubMed] [Google Scholar]
- Dahl SW, Slaughter C, Lauritzen C, Bateman RC, Jr, Connerton I, Pedersen J. Carica papaya glutamine cyclotransferase belongs to a novel plant enzyme subfamily: cloning and characterization of the recombinant enzyme. Protein Expr Purif. 2000;20:27–36. doi: 10.1006/prep.2000.1273. [DOI] [PubMed] [Google Scholar]
- Chevrier B, Schalk C, D'Orchymont H, Rondeau JM, Moras D, Tarnus C. Crystal structure of Aeromonas proteolytica aminopeptidase: a prototypical member of the co-catalytic zinc enzyme family. Structure. 1994;2:283–291. doi: 10.1016/s0969-2126(00)00030-7. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Greenblatt HM, Almog O, Maras B, Spungin-Bialik A, Barra D, Blumberg S, Shoham G. Streptomyces griseus aminopeptidase: X-ray crystallographic structure at 1.75 A resolution. J Mol Biol. 1997;265:620–636. doi: 10.1006/jmbi.1996.0729. [DOI] [PubMed] [Google Scholar]
- Rowsell S, Pauptit RA, Tucker AD, Melton RG, Blow DM, Brick P. Crystal structure of carboxypeptidase G2, a bacterial enzyme with applications in cancer therapy. Structure. 1997;5:337–347. doi: 10.1016/s0969-2126(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Oberg KA, Ruysschaert JM, Azarkan M, Smolders N, Zerhouni S, Wintjens R, Amrani A, Looze Y. Papaya glutamine cyclase, a plant enzyme highly resistant to proteolysis, adopts an all-beta conformation. Eur J Biochem. 1998;258:214–222. doi: 10.1046/j.1432-1327.1998.2580214.x. [DOI] [PubMed] [Google Scholar]
- Williams MG, Shirai H, Shi J, Nagendra HG, Mueller J, Mizuguchi K, Miguel RN, Lovell SC, Innis CA, Deane CM, Chen L, Campillo N, Burke DF, Blundell TL, de Bakker PI. Sequence-structure homology recognition by iterative alignment refinement and comparative modeling. Proteins. 2001;Suppl 5:92–97. doi: 10.1002/prot.1169. [DOI] [PubMed] [Google Scholar]
- Booth RE, Misquitta SA, Bateman RC., Jr Human pituitary glutaminyl cyclase: expression in insect cells and dye affinity purification. Protein Expr Purif. 2003;32:141–146. doi: 10.1016/S1046-5928(03)00226-2. [DOI] [PubMed] [Google Scholar]
- Ustynyuk L, Bennett B, Edwards T, Holz RC. Inhibition of the aminopeptidase from Aeromonas proteolytica by aliphatic alcohols. Characterization of the hydrophobic substrate recognition site. Biochemistry. 1999;38:11433–11439. doi: 10.1021/bi991090r. [DOI] [PubMed] [Google Scholar]
- Schilling S, Niestroj AJ, Rahfeld JU, Hoffmann T, Wermann M, Zunkel K, Wasternack C, Demuth HU. Identification of human glutaminyl cyclase as a metalloenzyme: Potent inhibition by imidazole derivatives and heterocyclic chelators. J Biol Chem. 2003. [DOI] [PubMed]
- Blow DM. The study of α-chymotrypsin by x-ray diffraction. The third Ciba medal lecture. Biochemical Journal. 1969;112:261–268. doi: 10.1042/bj1120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alden RA, Birktoft JJ, Kraut J, Robertus JD, Wright CS. Atomic coordinates for subtilisin BPN' (or Novo) Biochem Biophys Res Commun. 1971;45:337–344. doi: 10.1016/0006-291x(71)90823-0. [DOI] [PubMed] [Google Scholar]
- Brady L, Brzozowski AM, Derewenda ZS, Dodson E, Dodson G, Tolley S, Turkenburg JP, Christiansen L, Huge-Jensen B, Norskov L. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature. 1990;343:767–770. doi: 10.1038/343767a0. [DOI] [PubMed] [Google Scholar]
- Ho YS, Swenson L, Derewenda U, Serre L, Wei Y, Dauter Z, Hattori M, Adachi T, Aoki J, Arai H, Inoue K, Derewenda ZS. Brain acetylhydrolase that inactivates platelet-activating factor is a G-protein-like trimer. Nature. 1997;385:89–93. doi: 10.1038/385089a0. [DOI] [PubMed] [Google Scholar]
- Holbrook JJ, Ingram VA. Ionic properties of an essential histidine residue in pig heart lactate dehydrogenase. Biochem J. 1973;131:729–738. doi: 10.1042/bj1310729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuth B, Niefind K, Schomburg D. Crystal structure of creatininase from Pseudomonas putida: a novel fold and a case of convergent evolution. J Mol Biol. 2003;332:287–301. doi: 10.1016/S0022-2836(03)00860-X. [DOI] [PubMed] [Google Scholar]
- Russell RB. Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J Mol Biol. 1998;279:1211–1227. doi: 10.1006/jmbi.1998.1844. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Mizuguchi K, Deane CM, Blundell TL, Overington JP. HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci. 1998;7:2469–2471. doi: 10.1002/pro.5560071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, DePristo MA, Burke DF, Blundell TL. Ab initio construction of polypeptide fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Born solvation model. Proteins. 2003;51:21–40. doi: 10.1002/prot.10235. [DOI] [PubMed] [Google Scholar]
- DePristo MA, de Bakker PI, Lovell SC, Blundell TL. Ab initio construction of polypeptide fragments: efficient generation of accurate, representative ensembles. Proteins. 2003;51:41–55. doi: 10.1002/prot.10285. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Ray FA, Nickoloff JA. Site-specific mutagenesis of almost any plasmid using a PCR-based version of unique site elimination. Biotechniques. 1992;13:342–348. [PubMed] [Google Scholar]
- Bateman RC., Jr A spectrophotometric assay for glutaminyl-peptide cyclizing enzymes. J Neurosci Methods. 1989;30:23–28. doi: 10.1016/0165-0270(89)90070-8. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]