

Abstract
Background
Probiotic bifidobacteria in combination with prebiotic carbohydrates have documented positive effects on human health regarding gastrointestinal disorders and improved immunity, however the selective routes of uptake remain unknown for most candidate prebiotics. The differential transcriptomes of Bifidobacterium animalis subsp. lactis Bl-04, induced by 11 potential prebiotic oligosaccharides were analyzed to identify the genetic loci involved in the uptake and catabolism of α- and β-linked hexoses, and β-xylosides.
Results
The overall transcriptome was modulated dependent on the type of glycoside (galactosides, glucosides or xylosides) utilized. Carbohydrate transporters of the major facilitator superfamily (induced by gentiobiose and β-galacto-oligosaccharides (GOS)) and ATP-binding cassette (ABC) transporters (upregulated by cellobiose, GOS, isomaltose, maltotriose, melibiose, panose, raffinose, stachyose, xylobiose and β-xylo-oligosaccharides) were differentially upregulated, together with glycoside hydrolases from families 1, 2, 13, 36, 42, 43 and 77. Sequence analysis of the identified solute-binding proteins that determine the specificity of ABC transporters revealed similarities in the breadth and selectivity of prebiotic utilization by bifidobacteria.
Conclusion
This study identified the differential gene expression for utilization of potential prebiotics highlighting the extensive capabilities of Bifidobacterium lactis Bl-04 to utilize oligosaccharides. Results provide insights into the ability of this probiotic microbe to utilize indigestible carbohydrates in the human gastrointestinal tract.
Keywords: Bifidobacterium lactis, Transcriptomics, ABC transporter, GPH transporter, Prebiotics, Glycoside hydrolase
Background
Health-promoting microbes, defined as probiotics [1], have gained increased interest for use in food and dietary supplement applications to improve health and well being. Clinical research has shown that bifidobacteria are an important genus for probiotic interventions through clinical studies [2,3]. Benefits reported using bifidobacteria include improvement of bowel functions [4], prevention of necrotizing enterocolitis in infants [5], treatment of Crohn’s disease [6] and modulation of immune functions in the elderly [7]. Understanding the mechanisms of action underlying the probiotic attribute of bifidobacteria on the molecular level has been restricted to functional extrapolation from genome sequencing [8]. Interestingly, from the 53 Bifidobacterium genomes that have been deposited publicly to date, comparative analysis has revealed the genetic diversity of bifidobacteria [9], leading to identification of genetic loci for colon adaptation by host mucin degradation in B. bifidum[10], foraging of dietary carbohydrates in e.g. B. longum[11] or the important feature of human milk utilization [12] enabling colonization of the infant GIT [11,13,14] .
Enhancement of beneficial microbes within the gastrointestinal tract (GIT) can be achieved by providing selectively utilizable carbohydrates [15], defined as prebiotics [16]. Prebiotics are dietary carbohydrates, resistant to the host digestive system and main commensal microbiota residing in the colon. To date, only a few carbohydrates have been documented as prebiotics, namely β-galactooligosaccharides (GOS), lactulose, fructo-oligosaccharides and inulin [17]. Several candidate prebiotics have been proposed, but there is a need for additional studies that document their selective utilization by beneficial microbes within the human GIT [18,19]. The diversity of prebiotics with respect to size, composition and glycosidic linkages require a multitude of transporters and hydrolytic enzymes, some of which are predicted to occur widely within bifidobacteria, based mostly on in silico analysis [20,21].
Bifidobacterium animalis subsp. lactis has been reported to exert positive effects as a probiotic microbe in clinical studies [22,23], or when supplemented as a synbiotic in combination with prebiotics [24]. The annotated genome sequence of B. lactis Bl-04 revealed putative prebiotic transport and catabolic pathways, suggesting the bacterium to be highly adapted to the GIT and capable of utilization of dietary-derived complex oligosaccharides [25]. In the present study, we used differential transcriptomics to identify genetic loci encoding uptake and hydrolytic pathways of potential prebiotics manifested by 11 structurally diverse galactosides, glucosides and xylosides within B. lactis Bl-04. This work validated and expanded the tentative in silico predictions of oligosaccharide transporters and specificities of glycoside hydrolases.
Furthermore the perspective of the study enables the combination of transcriptomics and genome mining to serve as a platform for future functional work within prebiotic utilization by bifidobacteria.
Results
Oligosaccharide induced global transcriptome profile of B. lactis BL-04
Global gene expression profiles were obtained for B. lactis Bl-04, exponentially growing on 11 potential prebiotic oligosaccharides and glucose (Table 1), representing α-galactosides (melibiose, raffinose and stachyose), β-galactosides (GOS), α-glucosides (isomaltose, maltotriose and panose), β-glucosides (cellobiose and gentiobiose) and β-xylosides (xylobiose and xylo-oligosaccharides (XOS)). Growth of B. lactis Bl-04 on various mono, di and oligosaccharides were previously published [26]. The gene expression levels were quantified by whole genome DNA microarrays showing an overall comparable gene expression profiles across all 12 carbohydrate treatments and with high technical reproducibility (Figure 1). Only a subset of genes were upregulated differentially in response to each oligosaccharide, although a slight deviation of the GOS and xylobiose samples was observed. The 10 % of the highest constitutively expressed genes for all carbohydrate treatments (163 genes) were assigned Clusters of Orthologous Groups (COG) categories [27] and emphasized main cellular functions of growth and energy turn-over (Figure 1). Notably, of the highly expressed single genes (listed by B. lactis Bl-04 locus tag numbers), several highlight molecular functions related to probiotic mechanisms in B. lactis putatively involved in fibronectin adhesion (Balac_1484–1485), host plasminogen interactions (Balac_1017 and Balac_1557), phage immunity [28] (Balac_1305), bile salt hydrolysis (Balac_0863) and peroxide reduction (Balac_0865). In addition, part of an oligosaccharide ATP-binding cassette (ABC) transporter encoding a solute-binding protein (SBP) (Balac_1565), and an ATP-binding protein associated with oligosaccharide uptake by ABC transporters (Balac_1610) were both found, linking catabolic adaptation to the primary physiological functions of B. lactis Bl-04. These findings correlate with previously proposed molecular functions related to probiotic mechanisms in B. lactis[29].
Table 1.
Carbohydrates, used for DNA microarray studies, listed with glycoside structure and type, supplier and purity
| Carbohydrate | Structure1 | Glycoside type | DP2 | Manufacturer or supplier | Purity (as given by Manufacturer or supplier) |
|---|---|---|---|---|---|
| Glucose |
Glcp |
glucoside |
1 |
Sigma |
> 99% |
| GOS |
[β-d-Galp-[1]–[4]]n-d-Glcp |
galactoside |
2–6 |
Dupont |
> 94% DP ≥ 2 |
| Melibiose |
α-d-Galp-[1]–[6]-d-Glcp |
galactoside |
2 |
Sigma |
> 98% |
| Raffinose |
α-d-Galp-[1]–[6]-d-Glcp-(α1,β2)-D-Fruf |
galactoside |
3 |
Sigma |
> 99% |
| Stachyose |
[α-d-Galp-[1]–[6]]2-d-Glcp-(α1,β2)-d-Fruf |
galactoside |
4 |
Sigma |
> 98% |
| Isomaltose |
α-d-Glcp-[1]–[6]-d-Glcp |
glucoside |
2 |
Sigma |
> 98% |
| Panose |
α-d-Glcp[1]–[6]-α-d-Glcp-[1]–[4]-d-Glcp |
glucoside |
3 |
Sigma |
> 98% |
| Maltotriose |
α-d-Glcp-[1]–[4]-α-d-Glcp-[1]–[4]-d-Glcp |
glucoside |
3 |
Dupont |
> 95% |
| Cellobiose |
β-d-Glcp-[1]–[4]-d-Glcp |
glucoside |
2 |
Fluka AG |
> 99% |
| Gentiobiose |
β-d-Glcp-[1]–[6]-d-Glcp |
glucoside |
2 |
Sigma |
> 98% |
| Xylobiose |
β-d-xylp-[1]–[4]-d-xylp |
xyloside |
2 |
Dupont |
> 95% |
| XOS | [β-d-xylp-[1]–[4]]m-d-xylp | xyloside | 2–7 | Shandong Longlive Bio-technology Co., Ltd, (China) | > 90%3 |
Figure 1.
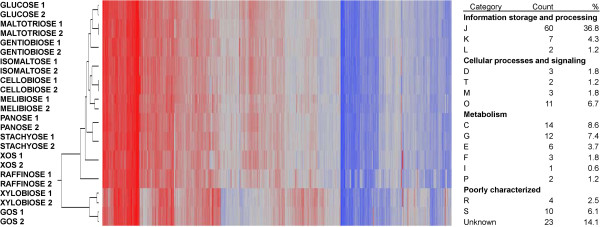
Two-way clustering of the global gene expression profiles and COG distribution of constitutively expressed genes. Gene expression intensities are represented by red coloring: up-regulation, blue coloring: down-regulation. Technical replicates for each carbohydrate are numbered and showed overall high reproducibility. The highest expressed decile (163 genes) of the global transcriptome across all carbohydrates conditions was assigned COG categories (assigned both as numbers and percentages), highlighting essential metabolic pathways of B. lactis Bl-04.
Functional grouping of global gene expression was observed based on the type of glycoside utilized (galactosides, glucosides or xylosides) from principal component analysis (Figure 2). A clear differentiation of the expressed global transcriptome was observed based on the type of glycoside utilized, indicating that prebiotics can affect the global transcriptome, and therefore physiological functions in B. lactis Bl-04. Furthermore, specific genetic loci were significantly differentially regulated by specific carbohydrates, which indicates their potential involvement in the uptake and catabolism of the respective glycosides (Figure 1).
Figure 2.

Principal component analysis of the global transcriptome for B. lactis Bl-04 cultivated with prebiotics showing differentiation of the global gene expression profiles depending on the glycoside type of carbohydrates utilized (pca2 = 22.2%). Galactosides in red (GOS, melibiose, raffinose, stachyose), glucosides in green (cellobiose, gentiobiose, glucose, maltotriose, isomaltose, panose) and xylosides in blue (XOS, xylobiose).
Differentially upregulated genes conferring potential prebiotic utilization
Analysis of the differential upregulation of specific genes mediating potential prebiotic utilization was conducted by one-way analysis of variance (ANOVA) and visualized by volcano plots (Figure 3) to identify statistically significant genes (cut off: p-value < 10-8.04) upregulated by each carbohydrate in the whole genome DNA microarray. An average of 56 genes were more than 2-fold differentially upregulated and above the statistical threshold for each pairwise comparison. Analysis revealed how subsets of genes involved with oligosaccharide metabolism were consistently differentially expressed throughout the ANOVA (Table 2, and Figure 4 for real time quantitative-PCR validation of selected genes). This led to the reconstruction of six putative gene clusters based on the differential upregulation of specific genes to specific oligosaccharide treatments. Gene clusters encoding a transporter and glycoside hydrolase(s) were linked to the uptake and degradation of substrates that varied in the degree of polymerization, glycosidic linkage or monosaccharide composition.
Figure 3.

Representative volcano plots of pairwise comparisons of the oligosaccharide-induced differential global transcriptome in B. lactis Bl-04. All genes are shown by solid grey circles, and putative carbohydrate-active protein encoding genes that were significantly up-regulated are highlighted with solid circles and color-coded by gene cluster and as listed in Table 2: cluster A (blue), cluster B (orange), cluster C (purple), cluster D (light blue), cluster E (green) and cluster F (red).
Table 2.
Statistically significant upregulated genes involved in carbohydrate uptake and catabolism
| ORF | Gene annotation | Inducing CHO type | Volcano plot (Figure3) | Highest inducing CHO | Gene cluster (Figure5) | Fold upregulated | -log10(P-value) |
|---|---|---|---|---|---|---|---|
| Balac_0053 |
β-galactosidase, GH42 |
Glucoside |
C |
gentiobiose |
A |
6.5 |
13.8 |
| Balac_0054 |
MFS permease |
Glucoside |
C |
gentiobiose |
A |
4.8 |
11.8 |
| Balac_0475 |
MFS permease |
Galactoside |
A |
GOS |
B |
21.8 |
19.9 |
| Balac_0476 |
β-galactosidase, GH2 |
Galactoside |
A |
GOS |
B |
36.9 |
17.9 |
| Balac_0484 |
β-galactosidase, GH42 |
Galactoside |
A |
GOS |
C |
11.4 |
17.2 |
| Balac_0485 |
ABC transporter, permease component |
Galactoside |
A |
GOS |
C |
8.4 |
16.2 |
| Balac_0486 |
ABC transporter, permease component |
Galactoside |
A |
GOS |
C |
5.7 |
12.9 |
| Balac_0511 |
Xylose isomerase |
xyloside |
A,C |
XOS |
D |
13.8 |
15.1 |
| Balac_0512 |
α-l-arabinofuranosidase, GH43 |
xyloside |
A,C |
XOS |
D |
6.8 |
13.6 |
| Balac_0513 |
Transcriptional regulator (lacI type) |
xyloside |
A,C |
Xylobiose |
D |
3.0 |
10.1 |
| Balac_0514 |
ABC transporter, oligosaccharide-binding protein |
xyloside |
A,C |
XOS |
D |
9.0 |
16.0 |
| Balac_0515 |
ABC transporter, permease component |
xyloside |
A,C |
XOS |
D |
16.8 |
16.5 |
| Balac_0516 |
ABC transporter, permease component |
xyloside |
A,C |
XOS |
D |
18.3 |
17.5 |
| Balac_0517 |
β-xylosidase, GH43 |
xyloside |
A,C |
XOS |
D |
17.9 |
16.1 |
| Balac_0518 |
Putative carbohydrate esterase |
xyloside |
A,C |
XOS |
D |
14.2 |
11.3 |
| Balac_0519 |
Esterase |
xyloside |
A,C |
XOS |
D |
6.9 |
15.1 |
| Balac_0520 |
α-l-arabinofuranosidase, GH43 |
xyloside |
A,C |
XOS |
D |
10.2 |
15.2 |
| Balac_0521 |
Xylulose kinase |
xyloside |
A,C |
Xylobiose |
D |
18.2 |
19.0 |
| Balac_1567 |
4-α-glucanotransferase |
glucoside |
B,D |
Maltriose |
E |
9.7 |
14.5 |
| Balac_1569 |
ABC transporter, permease component |
glucoside |
B,D |
Cellobiose |
E |
5.2 |
13.7 |
| Balac_1570 |
ABC transporter, permease component |
glucoside |
B,D |
Cellobiose |
E |
3.7 |
15.1 |
| Balac_1571 |
Transcriptional regulator (lacI type) |
glucoside |
B,D |
Cellobiose |
E |
3.7 |
13.1 |
| Balac_1572 |
ABC transporter, oligosaccharide-binding protein |
glucoside |
B,D |
Cellobiose |
E |
3.2 |
12.1 |
| Balac_1593 |
oligo-1,6-α-glucosidase, GH13 |
Galactoside, Glucoside |
B,D,E |
Isomaltose |
F |
4.5 |
14.0 |
| Balac_1597 |
ABC transporter, permease component |
Galactoside, Glucoside |
B,D,E |
Raffinose |
F |
14.1 |
13.9 |
| Balac_1598 |
ABC transporter, permease component |
Galactoside, Glucoside |
B,D,E |
Isomaltose |
F |
20.1 |
18.6 |
| Balac_1599 |
ABC transporter, oligosaccharide-binding protein |
Galactoside, Glucoside |
B,D,E |
Isomaltose |
F |
17.8 |
16.7 |
| Balac_1601 | α-galactosidase, GH36 | Galactoside, Glucoside | B,D,E | Raffinose | F | 8.1 | 16.4 |
The genes are listed by ascending locus tag numbers with the principle type of glycoside. Only the oligosaccharide that elicited the highest significance level (−log10(P-value)) is listed for genes that are upregulated by more than one oligosaccharide.
Figure 4.
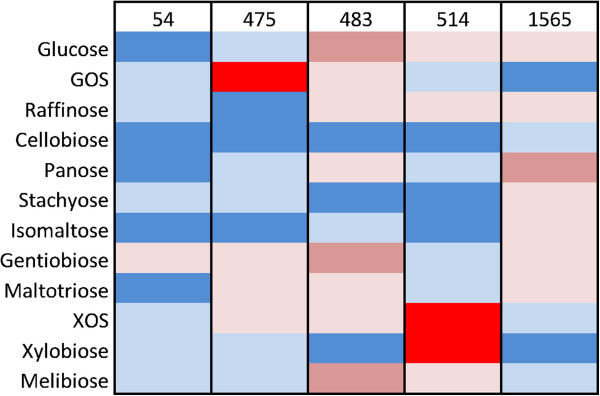
Heat map representation of RT-qPCR validation of microarray gene expression values. The mRNA transcript level were quantified for each of the five genes in each of the 12 carbohydrate conditions and color-coded as the relative fold upregulation of each gene: Blue [1–2], light blue [5–9], red [9–17] and strong red [>16].
Moreover, the relative induction of gene clusters involved in carbohydrate uptake and catabolism (Figure 5) strongly supports the identification of the differential specificities of upregulated proteins involved with utilization of the various oligosaccharides.
Figure 5.
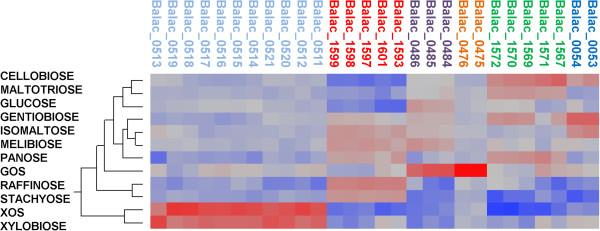
Two-way clustering of the expression profile for genes identified to be differentially upregulated by ANOVA. The coloring of each ORF corresponds to the gene cluster of Figure 2 and Table 2.
Gene cluster analysis and functional assignment
Analysis of differentially up-regulated loci enabled the identification of six gene clusters conferring the uptake and hydrolysis of the oligosaccharides used in the study (Figure 6A–F). Common to all the identified loci is that they encoded a transport system, a transcriptional regulator and one or more glycoside hydrolases (GHs) as predicted from the glycoside hydrolase family annotation in the CAZy database [30] (Figure 3). Four ATP-binding cassette (ABC) systems and two major facilitator superfamily (MFS) transporters, including one putative glycoside-pentoside-hexuronide (GPH) system were identified [31], supporting the differential expression of gene clusters being induced by multiple oligosaccharides (Figure 3).
Figure 6.

Organization of differentially expressed gene clusters encoding proteins predicted to be involved with prebiotic utilization. Genes are listed with locus tag numbers and gene functions are colored as: glycoside hydrolases in red, ABC transporter SBPs- and permeases (perm) in green, MFS transporters in blue, transcriptional regulators (reg) in light grey and hypothetical proteins (hypo), carbohydrate esterases (ester1 and ester2), xylose isomerase (xyl.iso) and xylulose kinase (xyl.kin) all in dark gray. A short putative nonfunctional ORF is highlighted by a black triangle.
Gene cluster A, differentially up-regulated by gentiobiose, encoded an MFS transporter (Balac_0054), with only 25% amino acid sequence identity to a sucrose permease from Arabidopsis thaliana (uniprot: Q9FG00), and an intracellular (as predicted by SignalP 4.0 [32]) putative β-galactosidase of GH42 (Balac_0053), adjacent to a GH30 subfamily 1 (GH30_1) putative β-glucosidase (Balac_0052) [33] not identified from the ANOVA. Interestingly, GH42 enzymes are only reported to be active on β-galactoside linkages [34], whereas GH30_1 enzymes harbor several specificities including endo-β-[1,4]-glucosidases rendering the GH30_1 a likely candidate for gentiobiose hydrolysis. Interestingly, co-occurrence of the GH42 and the GH30_1 genes is also observed in other bifidobacteria e.g. B. adolescentis ATCC 15703 and B. dentium Bd1. The rationale for the co-occurrence of these two GH genes is currently unclear, but the data identifies a novel route for gentiobiose uptake via the MFS transporter and hydrolysis by the putative GH30_1 family β-[1,4]-glucosidase.
The GOS substrate upregulated the expression of two loci: an MFS transporter (Balac_0475) homologous to the lactose transporter from B. longum NCC2705 [35], and a typical GH2 β-galactosidase (Balac_0476, cluster B) and cluster C (Balac_0483–0486) encoding the heterodimeric permease and the solute binding protein of an ABC transport system along with a GH42 putative β-galactosidase. The upregulation of two loci with a similar architecture was also reported in B. breve[36].
Xylobiose and XOS induced locus D encoding an ABC transporter, a putative GH43 β-xylosidase (Balac_0517) and two putative GH43 arabinofuranosidases (Balac_0512 and Balac_0520) identified based on homology to characterized bifidobacterial enzymes [37,38]. This suggests that the gene cluster mediates the transport and hydrolysis of both undecorated and arabinosyl-decorated XOS. The gene cluster also encodes a xylose isomerase and a xylulose kinase necessary to convert xylose to 2-xylulose-5 phosphate for entry into the bifid-shunt pathway [39]. The removal of acetyl sidechains that typically occur at the C2 or C3 of mainchain xyloxyl residues and feruloyl esters at the C5 or C2 of arabinosyl decorations in arabinoxylan is a prerequisite for the utilization of decorated arabinoxylo-oligosaccharides [40]. Notably, two putative carbohydrate esterases (Balac_0518 and Balac_0519) were upregulated highlighting the ability of the B. animlais subsp. lactis to remove acetyl or feruloyl sidechains from imported arabinoxylan fragments.
The organization and type of genes in cluster E showed resemblance to maltose operons from B. longum NCC2705 [35]. In the current study, however, the gene cluster was also upregulated by the trisaccharides panose, maltotriose and remarkably the β-linked disaccharide cellobiose. The gene landscape of this maltooligosaccharide gene cluster differed in the types of glycoside hydrolases encoded in the comparison to counterparts reported in other Gram positive bacteria [41]–[43] suggesting divergence in α-glucan metabolism.
An ABC transporter was identified in cluster F and was induced by the raffinose family oligosaccharides (RFO) melibiose, raffinose and stachyose representing α-1,6 linked galactosides, along with the α-1,6 linked glucosides isomaltose and panose. The GH36 subfamily 1 (GH36_1, Balac_1601) α-galactosidase [44] confers the hydrolysis of the α-1,6 linked galactosides, while the GH13 oligo-α-1,6-glucosidase (Balac_1593) catalyzes the hydrolysis of α-1,6 linked glucosides [45]. B. lactis Bl-04 encoded a total of three GH36, yet the remaining two (Balac_1537 and Balac_1596) were not differentially expressed. Further analysis of the global transcriptome (Figure 1) showed low basal expression of Balac_1537 in all conditions suggesting that the gene product plays a continuous metabolic role in cell function, while Balac_1596, assigned to GH36_2 (homologous to plant raffinose synthases [44]) was not expressed under these conditions.
In summary, all proposed pathways deduced from the identified gene clusters are shown in Figure 7, where potential prebiotic oligosaccharides are internalized and hydrolyzed into products that can be metabolized by the bifid shunt pathway [39]. Consistently, the majority of the bifid shunt genes were found to be highly expressed in all conditions as marked in Figure 7. Notably, a single putative phosphoketolase gene is encoded in B. lactis Bl-04, suggesting that this gene product could phosphorolyse both fructose-6P and xylulose-5P as the initial step of the bifid shunt, as previously described within B. lactis[46].
Figure 7.

Proposed pathways for oligosaccharide uptake and catabolism into monosaccharides for entry into the bifid shunt. Transporters are colored as in Figure 6 and all genes are given by their locus tag. The schematic pathways for glucose (entering as glucose-1P), galactose, fructose (entering as fructose-6P) and xylose are shown with the main steps of the bifid shunt. All constitutive highly expressed genes (Figure 1) are denoted with an asterisk (*)
Differentiation of transporter functionalities by transmembrane topology and sequence diversity
To differentiate the functionality of ABC and MFS transporters, the putative α-helical topology of the membrane spanning domains of all predicted oligosaccharide transporters in B. lactis Bl-04 was mapped (Table 3). Notably, the gentiobiose-specific MFS transporter (Balac_0054) possesses 11 predicted transmembrane helices, indicating structural-functional divergence from previously identified homologous MFS permeases displaying mainly 12 transmembrane helix topology [47]. Furthermore, one permease protein (Balac_1570) constituting part of the maltotriose upregulated ABC transporter was found to be N-terminally truncated and lacking two helices implicated in heterodimer formation in the permease domain of the maltose ABC transporter from Escherichia coli[48]. Comparison to an additional putative B. lactis Bl-04 maltose transporter (Balac_1563–1565) and the experimentally verified maltose ABC transporters from Lactobacillus casei[42] and Streptococcus pneumoniae[49] showed that all harbored the additional two α-helical domain, suggesting the divergence of the maltotriose ABC transporter (Balac_1569, 1570 and 1572) from known maltose ABC transporters.
Table 3.
Prediction of α-helical topology within oligosaccharide transporters identified in B. lactis Bl-04
| ORF | Predicted substrates | Class | Predicted TMH1 | Sequence length (aa) |
|---|---|---|---|---|
| 0054 |
Gentiobiose |
MFS |
11 |
384 |
| 0139 |
Sucrose (putative) |
MFS |
12 |
537 |
| 0475 |
GOS |
GPH homolog of MFS |
12 |
505 |
| 1240 |
FOS (putative) |
MFS |
12 |
441 |
| 1588 |
arabinofuranosides (putative) |
GPH homolog of MFS |
12 |
481 |
| 0485 |
GOS |
ABC |
6 |
326 |
| 0486 |
GOS |
ABC |
6 |
322 |
| 0515 |
XOS |
ABC |
6 |
352 |
| 0516 |
XOS |
ABC |
6 |
289 |
| 1563 |
Maltose (putative) |
ABC |
6 |
322 |
| 1564 |
Maltose (putative) |
ABC |
8 |
457 |
| 1569 |
Maltotriose |
ABC |
6 |
278 |
| 1570 |
Maltotriose |
ABC |
6 |
284 |
| 1597 |
RFO + IMO |
ABC |
6 |
301 |
| 1598 | RFO + IMO | ABC | 6 | 330 |
1Transmembrane α-helices (TMH) predicted using the Phobius tool [71].
Four of the five in silico annotated ABC transporters [25] were found to be differentially upregulated, while the remaining putative maltose ABC transporter discussed above (Balac_1563–1565) was found to be constitutively expressed to a comparable level. The transcriptomics data enabled the identification of novel specificities and multiple ligand recognition by the SBPs, recognized as specificity determinants for ABC transporters [50]. This is in agreement with the binding plasticity proposed for ABC-mediated transport [50]. To elaborate on these findings, the phylogenetics of the SBPs were compared to known protein orthologs (Supplemental Table) identified from bifidobacteria and pathogenic GIT-associated bacteria (Figure 8), hence displaying the functional and taxonomical distribution of oligosaccharide SBPs.
Figure 8.

Functional comparison of the identified oligosaccharide SBPs of ABC transporters in B. lactis Bl-04. The phylogenetic tree was rooted by a characterized fructose-binding protein [69] as a functional and structural outlier group of oligosaccharide-binding proteins [50]. Sub-clusters were defined by bootstrap values in percentages and the characterized SBP(s) identified within each sub-cluster, where the number of sequences are listed in brackets. The tree was colored by substrate specificity (maltose-binding proteins (green), cellodextrins (orange), GOS and lacto-N-biose (blue), XOS (light blue) and raffinose family oligosaccharides (red)) and sub-clusters were denoted by numbers as given in Table 4. The raffinose family oligosaccharide cluster was seemingly sub-clustering into two parts but with the current data could not be supported by boot-strap analysis.
Discussion
Bifidobacteria have been shown to exert a positive influence on the human gut [51] and may selectively utilize oligosaccharides of plant and milk-derived prebiotics [52]. Despite significant advances in bacterial genomics, understanding of carbohydrate uptake and catabolism mechanisms remains elusive, mainly because of poor overall annotation of oligosaccharide transporters where recent advances in uptake of human derived glycans [53] combined with the present study will enable improved functional overview of the Bifidobacterium genus with respect to carbohydrate utilization as an important factor for competitive GIT colonization and pathogen inhibition.
The global transcriptome of B. lactis Bl-04
The catabolic adaptation potential of B. lactis Bl-04 became apparent from the global comparison of oligosaccharide induced gene expression by principal component analysis (Figure 2). The altered global gene expression by the type of glycoside metabolized (galactoside, glucoside or xyloside) was not influenced by the differentially expressed gene clusters involved in the uptake and catabolism of oligosaccharides. It is likely that global gene expression, induced by carbohydrate source, involves modulation of the metabolic equilibrium within the bacterium. This was observed in B. longum for glycoside-induced changes in exopolysaccharide production [54] and the inhibition of enteropathogens by acidification when metabolizing fructose rather than glucose [55]. These findings underscore the effects associated with the catabolism of glycoside type on the overall behavior and potentially probiotic functionality of bifidobacteria in the GIT. Because of the importance for selective utilization of oligosaccharides, we hypothesize a vital role of ABC transporters for prebiotic uptake. Interestingly, a sole oligosaccharide ABC transporter-specific ATP-binding protein was identified in the genome and found to be constitutively highly expressed, consistent with a single ATP-binding protein energizing multiple oligosaccharide ABC transporters as previously described [56]. Likewise, various genes encoding proteins linked to proposed probiotic mechanisms of action, such as adhesion (Balac_1484–1485), phage immunity (Balac_1305) and bile salt hydrolysis (Balac_0863) were found to be highly expressed, supporting the clinically proven probiotic nature of B. lactis Bl-04 and reflecting the adaptation to the conditions of the GIT.
Analysis of the differentially expressed genes of B. lactis Bl-04 involved in prebiotic utilization revealed upregulation of explicit gene clusters, as was also observed from previous studies of oligosaccharide utilization in probiotic bacteria [35,57]. The uptake of oligosaccharides was facilitated by ABC and MFS types of oligosaccharide transporters, by the lack of phosphoenolpyruvate-dependent phosphotransferase systems in the B. lactis Bl-04 genome [25], all associated with glycoside hydrolases.
Oligosaccharide ABC transporters
The phylogenetic analysis of homologs of identified ABC transporters allowed the assignment of the SBPs identified in the current study into functional clusters harboring experimentally identified counterparts. Within each functional cluster, protein orthologs segregated based on taxonomic distance. Evolutionary adaptation was evident from this analysis as the milk disaccharide lacto-N-biose specificity defined by cluster LacN is almost exclusively found within bifidobacteria, while XOS SBP orthologs were dominated by soil bacteria and few GIT associated bacteria with the majority originating from Actinobacteria. This suggests that XOS utilization by bifidobacteria shares a metabolic niche within the GIT with xylan utilizing commensal bacteria [58].
The phylogenetic analysis indicates convergent evolution of a subset of maltose ABC transporter, based on the upregulated maltotriose ABC transporter (Balac_1569, 1570 and 1572). The diversity of canonical maltose SBP orthologs was illustrated by their segregation into four sub-clusters (clusters Mal1–Mal4), where a taxonomical sub-clustering was observed. A distant sub-cluster (Mal5) was defined by the maltotriose upregulated binding protein (Balac_1572). Notably, the corresponding permease domain of this ABC transporter was distinguished from identified maltose specific counterparts by the lack of two N-terminal α-helices (Balac_1570, Table 3), supporting the proposed convergent nature of this type of maltose transporter, which seems to share topological features with the raffinose and XOS type binding proteins (Figure 8).
Novel specificities of glycoside hydrolases for GIT adaptation
Identification of single gene being differentially upregulated by specific oligosaccharides revealed novel enzyme substrate specificities as compared to the initial in silico annotation of the hydrolytic capabilities of B. lactic Bl-04 [25]. Interestingly, the observation of a GH42 β-galactosidase being induced by the β-1,6-glucoside gentiobiose was intriguing as only β-galactosidases have been reported in this family. Thus, the GH30_1 putative β-glucosidase is the more likely candidate for gentiobiose hydrolysis. Nonetheless, the transcriptomics data suggests that gentiobiose is specifically transported by the MFS permease, thus defining a novel specificity for this MFS transporter. It remains to be investigated if additional substrates are taken up by the MFS permease including possible substrates for the upregulated GH42, which exhibits modest sequence identity (≈30%) to characterized GH42 enzymes.
No putative glycoside hydrolase was differentially up-regulated on cellobiose. However, transcriptional mining of B. lactis Bl-04 identified a constitutively expressed GH1 β-glucosidase (Balac_0151). The β-glucosidase displayed 51% amino acid identity to the GH1 β-glucosidase from B. brevis UCC2003 shown to be active on cellobiose and cellodextrin (β-1,4-glucooligosaccharides) initially transported by an ABC transporter [59], supporting the suggested function of Balac_0151. Furthermore, the only transporter differentially up-regulated on cellobiose was the above ABC transporter (Balac_1572) which was also up-regulated by maltotriose (Figure 5). This indicates a potentially dual specificity of the transporter likely to have evolved from multiple sugar metabolism-types of oligosaccharide ABC transporters (Figure 8).
The uptake and catabolism of XOS within bifidobacteria was recently proposed [37,60]. Comparative genomic of genes involved with XOS utilization within bifidobacteria (Figure 9) reflected a core gene structure of the XOS ABC transporter with a GH43 β-1,4-xylosidase (Balac_0517), while the occurrence of arabino-furanosidases, xylanases of GH8 and GH120 and carbohydrate esterases suggested more species and strain specific adaptation to utilize specific types of XOS e.g. arabinosyl decorated fragments. The multiplicity of GH43 arabino-furanosidases reflects the complexity of arabinosyl decorations that occurs naturally in arabinoxylan and its degradation products. Two putative oligosaccharide esterases, distantly related to previously identified xylan acetyl esterases [61] and conserved among bifidobacteria, were upregulated by XOS and xylobiose in B. lactis Bl-04. This implicates these putative esterases in de-esterification of xylan fragments transported into bifidobacteria. Taken together, this suggests an exquisite metabolic versatility in the uptake and utilization of xlyan degradation fragments that occur naturally with a diversity of arabinosyl and esterified side chain decorations.
Figure 9.

Genomic content and organization of XOS utilization gene clusters identified within bifidobacteria. All strains were ordered top down by highest sequence similarity of the XOS-binding proteins to the XOS-binding protein of B. lactis Bl-04 (balac_0514). Gene functions are colored as: Glycoside hydrolases (red), XOS ABC transporters (green), xylose ABC transporters (light green), transcriptional regulators (light grey), and putative XOS esterases, xylose isomerases (xyl.iso), xylulose kinases (xyl.kin), alcohol dehydrogenases (Reductase) and a putative secreted amidase (lysM) (all dark gray). All GH43 enzymes were annotated and differentiated by protein similarity to previously characterized xylosidases (xyln) or arabinofuranosidases (arab) together with the GH8 and GH120 enzymes [37,38]. An insertion in B. longum JDM301 is shown by below the gene cluster with an arrow indicating the position of the insert.
The ABC transporter mediated uptake of GOS coupled with co-induction of a GH42 showed homology to a B. longum NCC2705 gene cluster upregulated by lactose [35]. Interestingly this gene cluster diverges from those identified for human milk oligosaccharide uptake [62] both by the similarity of the associated SBP (Figure 8, LacN versus GOS) and the GH encoded in the gene clusters (GH42 versus GH112). Therefore, B. lactis Bl-04 has evolved a broad oligosaccharide utilization profile for potential prebiotics and dietary fibers.
Conclusion
In conclusion, the overall global gene expression of B. lactis Bl-04 was dependent of the type of glycoside utilized (galactosides, glucosides or xylosides) potentially linking the prebiotic catabolism of the bacteria to the overall behavior in the GIT. From the transcriptional analyses, we identified the genetic loci encoding MFS and ABC transporters concurrently with glycoside hydrolases for utilization of potential prebiotic oligosaccharides of α- and β-linkages and varying glycoside composition. This highlights the metabolic versatility of B. lactis Bl-04 and offers a means of enhancing probiotic effects by dietary supplementation with novel prebiotics. Furthermore, this study provides molecular level support for utilization of potential prebiotics, some of which are already known to be bifidogenic, and paves the way for expanding synbiotic formulations targeting specific groups of probiotic bacteria.
Methods
Culture preparation
B. animalis subsp. lactis Bl-04 (ATCC SD5219) was originally isolated from a human fecal sample [25]. Cultures prepared for transcriptional analysis were propagated in 0.22 μm filtered LABSEM media [63] pretreated by the Hungate method for oxygen removal [64]. The media was supplemented with 1% (w/v) of the 12 tested carbohydrates (Table 1) and each culture was transferred for five passages, under anaerobic conditions, on each carbohydrate before being harvested in the early logarithmic growth phase (OD600 = 0.3–0.5) by centrifugation at 4°C (3,000 g for 15 min) and flash freezing of the cell pellet for storage.
RNA isolation and microarray hybridization
Cells were mechanically disrupted by beadbeating and total RNA was isolated using Trizol-chloroform extraction (Invitrogen, Carlsbad, CA). Genomic DNA was removed with Turbo DNAse (Ambion, Austin, TX), followed by RNA purification using a RNeasy Mini Kit (Qiagen Inc., Valencia, CA) [65].
Reverse transcription of total RNA, fragmentation and 3’ biotin labeling of cDNA was done using 10 μg of total RNA in duplicates for each of the 12 conditions and microarray hybridizations were performed using the Affymetrix GeneChip® system (Affymetrix, Santa Clara, CA). Total RNA was reverse transcribed using random primers and SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California) and cDNA was purified using MinElute PCR Purification kit (QIAGEN, Inc., Valencia, CA) with a final elution volume of 12 μl. Subsequently, cDNA fragmentation into 50–100 bp was performed using DNase I (GE Healthcare, Waukesha, WI) and cDNA fragments were biotin-labeled using GeneChip DNA labeling reagent (Affymetrix) and terminal deoxynucleotidyl transferase (Promega, Madison, WI).
Labeled cDNA fragments were hybridized at Utah State University using Affymetrix custom-made chips. All extracted data was imported into SAS JMP Genomics (SAS Institute Inc, Cary, NC) before being quantile normalized and modeled using a one-way ANOVA for identification of differentially upregulated genes using a threshold value of α = 0.005 and Bonferroni correction.
The full genome transcriptome (98.1% of the total ORF) for each of the 12 growth conditions was used for the one-way ANOVA analysis. The pairwise analysis was done by comparing each single condition to the other 11 for a total of 66 pairwise conditions. To each of these 66 comparisons, the ANOVA identified a number of genes that were significantly upregulated, for further in silico validation by real-time quantative PCR of selected genes. The identified upregulated genes from the ANOVA were then annotated for potential involvement in carbohydrate utilization.
Real-time quantitative PCR (RT-qPCR) validation of microarray
RT-qPCR was performed on five selected genes (Table 3) found to be differentially upregulated. The DNAse-treated total RNA, identical to the RNA used in microarray sample preparation, was used as template for each of the above 12 growth conditions, measured in triplicates. Experiments were conducted with a QRT-PCR thermal cycler (I-cycler; Bio-Rad, Hercules, CA) in combination with the iScript One-Step RT-PCR Kit with SYBR Green (Biorad).
Construction of phylogenetic tree of carbohydrate SBPs
The sequence dataset was compiled from oligosaccharide-binding proteins all identified from previous works or from the current study (Table 4). Sequence homologs for each protein entry were identified by BLAST [66] and restricted to either 100 hits or an e-value of 10-3 against the non-redundant database. All redundant sequences were removed and the remaining sequences together with a monosaccharide (fructose) binding proteins were aligned with ClustalX [67] using the Blosum series substitution matrix and a gap opening penalty of 2, compared to the standard penalty of 10. The resulting phylogenetic tree file was visualized using Dendroscope [68]. Bootstrap values were calculated by ClustalX using standard conditions (1000 iterations).
Table 4.
Identified clusters of oligosaccharide-binding proteins from Figure8
| Cluster | Sub-cluster | Substrate specificity | Identified Organism | Reference |
|---|---|---|---|---|
| Malto-oligosaccharides |
1 |
α-(1,4)-gluco-oligosaccharides |
Listeria monocytogenes |
[72] |
|
Streptococcus pneumoniae |
[73] |
|||
| |
2 |
β-Cyclodextrin and maltose |
Bacillus subtilis |
[74] |
| |
3 |
Maltose |
L. casei BL23 |
[42] |
| |
4 |
Putative maltose |
B. animalis subsp lactis Bl-04 |
This study |
| |
5 |
Maltose |
B. longum NCC2705 |
[35] |
| Maltotriose |
B. lactis Bl-04 |
This study |
||
| β-glucosides |
- |
β-(1,4)-gluco-oligosaccharides |
B. breve UCC2003 |
[59] |
| β-galactosides |
A |
Lactose and |
B. longum NCC2705 |
[35] and |
| β-galacto-oligosaccharides |
B. lactis Bl-04 |
This study |
||
| |
B |
Lacto-N-biose |
B. bifidum |
[75] |
| |
B. longum |
[76] |
||
| XOS |
- |
β-(1,4)-xylo-oligosaccharides |
B. lactis Bl-04 |
This study |
| RFO |
A |
Raffinose and isomaltose |
Streptococcus mutans |
[43] |
| |
B |
Raffinose |
B. longum NCC2705 |
[35] |
| Raffinose and Isomaltose1 |
B. lactis Bl-04 |
This study |
||
| Root | - | Fructose | B. longum NCC2705 | [69] |
Clusters are shown by numbers and if possible sub-clusters are listed with letters. The experimentally identified oligosaccharide-binding proteins used to generate the tree are listed in the corresponding cluster and sub-cluster if possible.
1Including melibiose, panose and stachyose.
Table 5.
Primer pairs used for RT-qPCR
| ORF | Primer 5' – 3' | Product size (bp) |
|---|---|---|
| 0054 |
CACACTCGCTCGAGATTC |
140 |
| AGGCCAATCATGCATACG | ||
| 0475 |
GCTGACGATGGGAATGAC |
160 |
| GCTCGACGTGTTCTACTC | ||
| 0483 |
CGTCGGAGTTCTTGATGG |
142 |
| CAGGCAGCCTATGACTTC | ||
| 0514 |
GGCTGACCTTGGATTCTT |
145 |
| CTTCTCGCCCATGTAGTTG | ||
| 1565 |
GAACGCCGTAGATCTTGC |
148 |
| ATGTTCGCCAATGACCAG |
Microarray submission
All raw data have been deposited in the GEO database (Accession number: GSE41906) and complies with the MIAME guidelines.
Abbreviations
(ANOVA): Analysis of variance; (ABC): ATP-binding cassette; (GOS): β-galacto-oligosaccharides; (COG): Clusters of Orthologous Groups; (GH): Glycoside hydrolase; (MFS): Major Facilitator Superfamily; (RFO): Raffinose family oligosaccharides; (XOS): Xylo-oligosaccharides
Authors’ contributions
Designed research: RB, MAH, SL, BS, TRKl; Performed research: JMA, YJG; Contributed new reagents (SL) and analytic tools: YJG, TRK; Analyzed data: JMA, RB, YJG, TRK; Wrote the paper: JMA, RB, MAH, SL, YJG, BS, TRK. All authors read and approved the final manuscript.
Contributor Information
Joakim M Andersen, Email: joa@bio.dtu.dk.
Rodolphe Barrangou, Email: rodolphe.barrangou@danisco.com.
Maher Abou Hachem, Email: maha@bio.dtu.dk.
Sampo J Lahtinen, Email: sampo.lahtinen@danisco.com.
Yong Jun Goh, Email: yjgoh@ncsu.edu.
Birte Svensson, Email: bis@bio.dtu.dk.
Todd R Klaenhammer, Email: trk@ncsu.edu.
Acknowledgements
Sarah O’Flaherty is greatly acknowledged for her assistance in RT-qPCR design. This research was funded by DuPont Nutrition and Health, North Carolina Dairy Foundation and the FøSu grant from the Danish Strategic Research Council to the project “Gene discovery and molecular interactions in prebiotics/probiotics systems. Focus on carbohydrate prebiotics”. J.M.A. is funded by a joint Ph.D. stipend from DuPont, the FøSu grant, and the Technical University of Denmark.
References
- Reid G. et al. New scientific paradigms for probiotics and prebiotics. J Clin Gastroenterol. 2003;37:105–118. doi: 10.1097/00004836-200308000-00004. [DOI] [PubMed] [Google Scholar]
- Turroni F, van Sinderen D, Ventura M. Genomics and ecological overview of the genus Bifidobacterium. Int J Food Microbiol. 2011;149:37–44. doi: 10.1016/j.ijfoodmicro.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Szajewska H, Guandalini S, Morelli L, Van Goudoever JB, Walker A. Effect of Bifidobacterium animalis subsp. lactis supplementation in preterm infants: A systematic review of randomized controlled trials. J Pediatr Gastroenterol Nutr. 2010;51:203–209. doi: 10.1097/MPG.0b013e3181dc0d93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller PA. et al. Dose–response effect of Bifidobacterium lactis HN019 on whole gut transit time and functional gastrointestinal symptoms in adults. Scand J Gastroenterol. 2011;46:1057–1064. doi: 10.3109/00365521.2011.584895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguli K, Walker WA. Probiotics in the prevention of necrotizing enterocolitis. J Clin Gastroenterol. 2011;45(Suppl):S133–138. doi: 10.1097/MCG.0b013e318228b799. [DOI] [PubMed] [Google Scholar]
- Steed H. et al. Clinical trial: The microbiological and immunological effects of synbiotic consumption - a randomized double-blind placebo-controlled study in active crohn’s disease. Aliment Pharmacol Ther. 2010;32:872–883. doi: 10.1111/j.1365-2036.2010.04417.x. [DOI] [PubMed] [Google Scholar]
- Ouwehand AC. et al. Bifidobacterium microbiota and parameters of immune function in elderly subjects. FEMS Immunol Med Microbiol. 2008;53:18–25. doi: 10.1111/j.1574-695X.2008.00392.x. [DOI] [PubMed] [Google Scholar]
- Ventura M. et al. Genome-scale analyses of health-promoting bacteria: Probiogenomics. Nat Rev Microbiol. 2009;7:61–71. doi: 10.1038/nrmicro2047. [DOI] [PubMed] [Google Scholar]
- Bottacini F. et al. Comparative genomics of the genus Bifidobacterium. Microbiology. 2010;156:3243–3254. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- Turroni F. et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci USA. 2010;107:19514–19519. doi: 10.1073/pnas.1011100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell M. et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci USA. 2002;99:14422–14427. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González R, Klaassens ES, Malinen E. de Vos W,M. & Vaughan EE. Differential transcriptional response of Bifidobacterium longum to human milk, formula milk, and galactooligosaccharide. Appl Environ Microbiol. 2008;74:4686–4694. doi: 10.1128/AEM.00122-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F. et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One. 2012;7:e36957. doi: 10.1371/journal.pone.0036957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens ES. et al. Mixed-species genomic microarray analysis of fecal samples reveals differential transcriptional responses of bifidobacteria in breast- and formula-fed infants. Appl Environ Microbiol. 2009;75:2668–2676. doi: 10.1128/AEM.02492-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LM, Martinez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One. 2011;6:e25200. doi: 10.1371/journal.pone.0025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberfroid M. Prebiotics: The concept revisited. J Nutr. 2007;137:830S–837S. doi: 10.1093/jn/137.3.830S. [DOI] [PubMed] [Google Scholar]
- Roberfroid M. et al. Prebiotic effects: Metabolic and health benefits. Br J Nutr. 2010;104(Suppl 2):S1–S63. doi: 10.1017/S0007114510003363. [DOI] [PubMed] [Google Scholar]
- Gibson GR, Probert HM, Loo JV, Rastall RA, Roberfroid M. Dietary modulation of the human colonic microbiota: Updating the concept of prebiotics. Nutr Res Rev. 2004;17:259–275. doi: 10.1079/NRR200479. [DOI] [PubMed] [Google Scholar]
- Makelainen H, Hasselwander O, Rautonen N, Ouwehand AC. Panose, a new prebiotic candidate. Lett Appl Microbiol. 2009;49:666–672. doi: 10.1111/j.1472-765X.2009.02698.x. [DOI] [PubMed] [Google Scholar]
- van den Broek LA, Hinz SW, Beldman G, Vincken JP, Voragen AG. Bifidobacterium carbohydrases-their role in breakdown and synthesis of (potential) prebiotics. Mol Nutr Food Res. 2008;52:146–163. doi: 10.1002/mnfr.200700121. [DOI] [PubMed] [Google Scholar]
- Pokusaeva K, Fitzgerald GF, van Sinderen D. Carbohydrate metabolism in bifidobacteria. Genes & Nutrition. 2011;6:285–306. doi: 10.1007/s12263-010-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paineau D. et al. Effects of seven potential probiotic strains on specific immune responses in healthy adults: A double-blind, randomized, controlled trial. FEMS Immunol Med Microbiol. 2008;53:107–113. doi: 10.1111/j.1574-695X.2008.00413.x. [DOI] [PubMed] [Google Scholar]
- Ouwehand AC. et al. Specific probiotics alleviate allergic rhinitis during the birch pollen season. World J Gastroenterol. 2009;15:3261–3268. doi: 10.3748/wjg.15.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch S, Woodmansey EJ, Paterson JC, McMurdo ME, Macfarlane GT. Microbiological effects of consuming a synbiotic containing Bifidobacterium bifidum, Bifidobacterium lactis, and oligofructose in elderly persons, determined by real-time polymerase chain reaction and counting of viable bacteria. Clin Infect Dis. 2005;40:28–37. doi: 10.1086/426027. [DOI] [PubMed] [Google Scholar]
- Barrangou R. et al. Comparison of the complete genome sequences of Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04. J Bacteriol. 2009;191:4144–4151. doi: 10.1128/JB.00155-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zanten GC. et al. The effect of selected synbiotics on microbial composition and short-chain fatty acid production in a model system of the human colon. PLoS One. 2012;7(10):e47212. doi: 10.1371/journal.pone.0047212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL. et al. The COG database: An updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Gilad O. et al. Insights into physiological traits of Bifidobacterium animalis subsp. lactis BB-12 through membrane proteome analysis. J Proteomics. 2012;75:1190–200. doi: 10.1016/j.jprot.2011.10.031. [DOI] [PubMed] [Google Scholar]
- Cantarel BL. et al. The carbohydrate-active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH Jr, Yen MR, Noto K, Tamang DG, Elkan C. The transporter classification database: Recent advances. Nucleic Acids Res. 2009;37:D274–278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- St John FJ, Gonzalez JM, Pozharski E. Consolidation of glycosyl hydrolase family 30: A dual domain 4/7 hydrolase family consisting of two structurally distinct groups. FEBS Lett. 2010;584:4435–4441. doi: 10.1016/j.febslet.2010.09.051. [DOI] [PubMed] [Google Scholar]
- Shipkowski S, Brenchley JE. Bioinformatic, genetic, and biochemical evidence that some glycoside hydrolase family 42 beta-galactosidases are arabinogalactan type I oligomer hydrolases. Appl Environ Microbiol. 2006;72:7730–7738. doi: 10.1128/AEM.01306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parche S. et al. Sugar transport systems of Bifidobacterium longum NCC2705. J Mol Microbiol Biotechnol. 2007;12:9–19. doi: 10.1159/000096455. [DOI] [PubMed] [Google Scholar]
- O’Connell Motherway M, Kinsella M, Fitzgerald GF, van Sinderen D. Transcriptional and functional characterization of genetic elements involved in galacto-oligosaccharide utilization by Bifidobacterium breve UCC2003. Microb Biotechnol. 2013;6(1):67–79. doi: 10.1111/1751-7915.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagaert S. et al. Substrate specificity of three recombinant α-l-arabinofuranosidases from Bifidobacterium adolescentis and their divergent action on arabinoxylan and arabinoxylan oligosaccharides. Biochem Biophys Res Commun. 2010;402:644–650. doi: 10.1016/j.bbrc.2010.10.075. [DOI] [PubMed] [Google Scholar]
- Lagaert S. et al. Recombinant expression and characterization of a reducing-end xylose-releasing exo-oligoxylanase from Bifidobacterium adolescentis. Appl Environ Microbiol. 2007;73:5374–5377. doi: 10.1128/AEM.00722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. et al. Proteomics analysis of Bifidobacterium longum NCC2705 growing on glucose, fructose, mannose, xylose, ribose, and galactose. Proteomics. 2011;11:2628–2638. doi: 10.1002/pmic.201100035. [DOI] [PubMed] [Google Scholar]
- Taylor EJ. et al. Structure and activity of two metal ion-dependent acetylxylan esterases involved in plant cell wall degradation reveals a close similarity to peptidoglycan deacetylases. J Biol Chem. 2006;281:10968–10975. doi: 10.1074/jbc.M513066200. [DOI] [PubMed] [Google Scholar]
- Nakai H. et al. The maltodextrin transport system and metabolism in Lactobacillus acidophilus NCFM and production of novel alpha-glucosides through reverse phosphorolysis by maltose phosphorylase. FEBS J. 2009;276:7353–7365. doi: 10.1111/j.1742-4658.2009.07445.x. [DOI] [PubMed] [Google Scholar]
- Monedero V, Yebra MJ, Poncet S, Deutscher J. Maltose transport in Lactobacillus casei and its regulation by inducer exclusion. Res Microbiol. 2008;159:94–102. doi: 10.1016/j.resmic.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Ajdic D, Pham VT. Global transcriptional analysis of Streptococcus mutans sugar transporters using microarrays. J Bacteriol. 2007;189:5049–5059. doi: 10.1128/JB.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredslund F. et al. Crystal structure of alpha-galactosidase from Lactobacillus acidophilus NCFM: Insight into tetramer formation and substrate binding. J Mol Biol. 2011;412:466–480. doi: 10.1016/j.jmb.2011.07.057. [DOI] [PubMed] [Google Scholar]
- Pokusaeva K, O’Connell-Motherway M, Zomer A, Fitzgerald GF, van Sinderen D. Characterization of two novel alpha-glucosidases from Bifidobacterium breve UCC2003. Appl Environ Microbiol. 2009;75:1135–1143. doi: 10.1128/AEM.02391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meile L, Rohr LM, Geissmann TA, Herensperger M, Teuber M. Characterization of the D-xylulose 5-phosphate/D-fructose 6-phosphate phosphoketolase gene (xfp) from Bifidobacterium lactis. J Bacteriol. 2001;183:2929–2936. doi: 10.1128/JB.183.9.2929-2936.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008;62:289–305. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Fa Q, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Espinosa M, Puyet A. The maltose / maltodextrin regulon of Streptococcus pneumoniae. Biochemistry (N Y ) 1997;272:30860–30865. doi: 10.1074/jbc.272.49.30860. [DOI] [PubMed] [Google Scholar]
- Berntsson RP, Smits SHJ, Schmitt L, Slotboom D, Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Cani PD, Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Ther. 2011;130:202–212. doi: 10.1016/j.pharmthera.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Macfarlane GT, Steed H, Macfarlane S. Bacterial metabolism and health-related effects of galacto-oligosaccharides and other prebiotics. J Appl Microbiol. 2008;104:305–344. doi: 10.1111/j.1365-2672.2007.03520.x. [DOI] [PubMed] [Google Scholar]
- Turroni F. et al. Analysis of predicted carbohydrate transport systems encoded by Bifidobacterium bifidum PRL2010. Appl Environ Microbiol. 2012;78:5002–5012. doi: 10.1128/AEM.00629-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audy J, Labrie S, Roy D, Lapointe G. Sugar source modulates exopolysaccharide biosynthesis in Bifidobacterium longum subsp. longum CRC 002. Microbiology. 2010;156:653–664. doi: 10.1099/mic.0.033720-0. [DOI] [PubMed] [Google Scholar]
- Fukuda S. et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- Marion C, Aten AE, Sa W, King SJ. Identification of an ATPase, MsmK, which energizes multiple carbohydrate ABC transporters in Streptococcus pneumoniae. Infect Immun. 2011;79:4193–4200. doi: 10.1128/IAI.05290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R. et al. Global analysis of carbohydrate utilization by Lactobacillus acidophilus using cDNA microarrays. Proc Natl Acad Sci USA. 2006;103:3816–3821. doi: 10.1073/pnas.0511287103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC. et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokusaeva K, Connell-motherway M, Zomer A, Fitzgerald GF, Sinderen DV. Cellodextrin utilization by Bifidobacterium breve UCC2003. Appl Environ Microbiol. 2011;77:1681–1690. doi: 10.1128/AEM.01786-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad O. et al. Combined transcriptome and proteome analysis of Bifidobacterium animalis subsp. lactis BB-12 grown on xylo-oligosaccharides and a model of their utilization. Appl Environ Microbiol. 2010;76:7285–7291. doi: 10.1128/AEM.00738-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabel M. et al. Biochemical characterization and relative expression levels of multiple carbohydrate esterases of the xylanolytic rumen bacterium Prevotella ruminicola 23 grown on an ester-enriched substrate. Appl Environ Microbiol. 2011;77:5671–5681. doi: 10.1128/AEM.05321-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushinobu S. Unique sugar metabolic pathways of bifidobacteria. Biosci Biotechnol Biochem. 2010;74:2374–2384. doi: 10.1271/bbb.100494. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Altermann E, Hutkins R, Cano R, Klaenhammer TR. Functional and comparative genomic analyses of an operon involved in fructooligosaccharide utilization by Lactobacillus acidophilus. Proc Natl Acad Sci USA. 2003;100:8957–8962. doi: 10.1073/pnas.1332765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels L, Zeikus JG. Improved culture flask for obligate anaerobes. Appl Microbiol. 1975;29:710–711. doi: 10.1128/am.29.5.710-711.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh YJ. et al. Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol. 2009;75:3093–3105. doi: 10.1128/AEM.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Larkin MA. et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Huson DH. et al. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinforma. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X. et al. Fructose uptake in Bifidobacterium longum NCC2705 is mediated by an ATP-binding cassette transporter. J Biol Chem. 2012;287:357–367. doi: 10.1074/jbc.M111.266213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JM. et al. Transcriptional and functional analysis of galactooligosaccharide uptake by lacS in Lactobacillus acidophilus. Proc Natl Acad Sci USA. 2011;108:17785–17790. doi: 10.1073/pnas.1114152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction-the phobius web server. Nucleic Acids Res. 2007;35:W429–32. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal S. et al. Maltose and maltodextrin utilization by Listeria monocytogenes depend on an inducible ABC transporter which is repressed by glucose. PLoS One. 2010;5:e10349. doi: 10.1371/journal.pone.0010349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott DW. et al. The molecular basis of glycogen breakdown and transport in Streptococcus pneumoniae. Mol Microbiol. 2010;77:183–199. doi: 10.1111/j.1365-2958.2010.07199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanert S. et al. Maltose and maltodextrin utilization by Bacillus subtilis. J Bacteriol. 2006;188:3911–3922. doi: 10.1128/JB.00213-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido D, Kim JH, German JB, Raybould HE, Mills DA. Oligosaccharide binding proteins from Bifidobacterium longum subsp. infantis reveal a preference for host glycans. PLoS One. 2011;6:e17315. doi: 10.1371/journal.pone.0017315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R. et al. Structural and thermodynamic analyses of solute-binding protein from Bifidobacterium longum specific for core 1 disaccharide and lacto-N-biose I. J Biol Chem. 2008;283:13165–13173. doi: 10.1074/jbc.M709777200. [DOI] [PubMed] [Google Scholar]