

Abstract
Diet influences health as a source of nutrients and toxins, and by shaping the composition of resident microbial populations. Previous studies have begun to map out associations between diet and the bacteria and viruses of the human gut microbiome. Here we investigate associations of diet with fungal and archaeal populations, taking advantage of samples from 98 well-characterized individuals. Diet was quantified using inventories scoring both long-term and recent diet, and archaea and fungi were characterized by deep sequencing of marker genes in DNA purified from stool. For fungi, we found 66 genera, with generally mutually exclusive presence of either the phyla Ascomycota or Basiodiomycota. For archaea, Methanobrevibacter was the most prevalent genus, present in 30% of samples. Several other archaeal genera were detected in lower abundance and frequency. Myriad associations were detected for fungi and archaea with diet, with each other, and with bacterial lineages. Methanobrevibacter and Candida were positively associated with diets high in carbohydrates, but negatively with diets high in amino acids, protein, and fatty acids. A previous study emphasized that bacterial population structure was associated primarily with long-term diet, but high Candida abundance was most strongly associated with the recent consumption of carbohydrates. Methobrevibacter abundance was associated with both long term and recent consumption of carbohydrates. These results confirm earlier targeted studies and provide a host of new associations to consider in modeling the effects of diet on the gut microbiome and human health.
Introduction
Humans live in association with immense populations of bacteria, viruses, fungi and archaea [1]–[8]. Many groups have now contributed surveys using deep sequencing to characterize these populations, revealing that the human microbiome differs radically at different body sites and among individuals [9]–[12]. Differences in body sites are associated with availability of nutrients, water, oxygen, and other site-specific features. The origin of differences between individuals is less clear, however, potentially reflecting distinct colonization early in life and different environmental exposures such as antibiotic use [13]–[15]. Another environmental exposure, ubiquitous but incompletely understood, is diet.
Recently, we reported correlations of long-term dietary patterns in 98 individuals and the bacterial lineages present in the gut microbiota [10]. Two genera, Prevotella and Bacteroides, were shown to have reciprocal patterns of abundance, paralleling several reports from others [16]–[18]. Abundant Prevotella correlated with consumption of carbohydrates, while abundant Bacteroides correlated with consumption of choline, fats, and amino acids. A short term controlled feeding study showed changes in the gut microbiota associated with the dietary interventions, but not a change in the overall structure of the bacterial community analyzed, supporting a role for long-term diet in determining the structure of the gut microbiome [10]. Another study recently reported that the diversity of the gut microbiota was linked with long-term diet, where a more diverse diet was correlated with an increased gut bacterial diversity [19].
Bacteria are abundant members of the gut microbiome, but not the only residents. Bacteriophage particles within the intestinal tract are present in potentially greater numbers than bacterial cells [20], [21]. Recently changes in bacteriophage communities in gut have been correlated with dietary interventions [5].
Archaea are also present in human gut, the most frequently occurring of which is Methanobrevibacter smithii [22]–[25], a methane producer from byproducts of bacterial fermentation [23]. Reported colonization rates by methanogenic archaea range from 25% to 95% of humans [26], [27]. In microbial ecosystems such as the human gut, when H2 accumulates due to bacterial catabolism, archaeal growth is stimulated associated with incorporation of H2 into methane [23]. Support for such syntrophy in the mammalian gut has been shown in a gnotobiotic mouse model, where co-colonization by M. smithii and Bacteroides thetaiotaomicron promoted increased growth of both species compared to mono-colonization [28].
Yeasts have been detected in human stool samples at least since 1917 [29], and by the mid 20th century their presence in the human intestine had been proposed to have a saprotrophic role [30]. Gut fungi may also be involved in pathogenic processes. Anti-Saccharomyces antibodies are detected in inflammatory bowel disease cohorts and are used as a predictor of disease progression [31], [32]. Recent work using a murine model has suggested that normally mutualistic or commensal fungi species may exacerbate intestinal inflammation in mice with sensitized genotypes [33]. In mice, over 14 fungal genera have been reported to be present within the mucus layer lining the intestinal epithelium [34]. Available data is likely incomplete, because of reliance mostly on culture-based methods. Recent reports using next generation sequencing also suggest diverse fungal communities in humans [35]–[37].
Based on the above, we hypothesized that the gut archaea and fungi are influenced by both diet and the other microorganisms present. Here we investigated these ideas in a cohort of 96 healthy individuals who were previously characterized for their bacteria/diet relationships [10]. Fungi were characterized by sequencing the Internal Transcribed Spacer region 1 (ITS1) of the rRNA locus and the archaea by sequencing a segment of the 16S rRNA gene. Short-term diet was characterized using a Recall questionnaire, and long-term diet characterized using a Food Frequency questionnaire. Analysis showed notable correlations of the three Domains of life with each other, and with dietary components–thus these data begin to specify potential multi-domain trophic interactions in the human gut microbiota.
Results
Samples for Analysis of the Relationship between Human Diet and Gut Microbial Populations
A total of 98 samples were collected from healthy volunteers, and sequences from 96 of these samples were used in this analysis after quality filtering. The archaeal and fungal components of the microbiota were assessed using the 16S rRNA gene and the ITS1 rRNA gene tags, respectively [38]–[40]. The bacterial population of these samples was characterized previously by 454 pyrosequencing of V1V2 segments of the 16S rRNA gene [10]. Volunteers were screened to be free of chronic gastrointestinal disease, cardiac disease, diabetes mellitus or immunodeficiency diseases, to have a normal bowel frequency (minimum once every 2 days, maximum 3 times per day), and body mass index (BMI) between 18.5 and 35.
The Archaea of the Gut Microbiome
A total of 99,131 archaeal sequence reads were obtained, resulting in the detection of 5 genera (Figure 1). A total of 44 of the 96 samples analyzed were positive for at least one species. Methanobrevibacter sp was detected in 30 samples, and Nitrososphaera sp was detected in 16 samples (Figure 1A). The two genera were usually mutually exclusive, coexisting in only 6 samples.
Figure 1. The archaeal and fungal components of the human gut microbiome.

The heatmaps show the relative proportions of microbial lineages detected by pyrosequencing. The lineages are marked on the right, with Phylum (abbreviated), Class, and Genus. Archaeal genera are shown in (A), representative bacterial genera in (B), and fungal genera in (C). The top two rows show the DNA yield from PCR amplification reactions, which serves as a rough indicator of abundance. Proportions were calculated within each amplicon (archaeal 16S, bacterial 16S, or fungal ITS) for each sequencing study separately. The abbreviations for phyla were as follows (Eur: Euryarchaeota; Tha: Thaumarchaeota; Act: Actinobacteria; Bac: Bacteroidetes; Fir: Firmicutes; Asc: Ascomycota; Bas; Basidiomycota). Other Ascomycota and Other Basidiomycota are composed of genera which were detected in only one sample (see Table S7 and Figure S2 for a complete list of detected genera and their prevalence).
The detection of Nitrososphaera was surprising and so was investigated further. Comparison of amplification efficiency suggested that the Methanobrevibacter when present was relatively abundant, while the Nitrososphaera was less abundant (Figure 1, top). Nitrosophaera was not detected after sequencing control amplifications with archaeal primers using products of blank DNA purifications as template (no positives out of eight tested). To validate the detection of Nitrososphaera, we used a nested PCR assay to detect the AmoA gene, which encodes the ammonia mono-oxyenase enzyme, and is distinctive for Nitrososphaera. We found an association between an AmoA positive PCR and Nitrososphaera detection in the samples (p = 0.014, Fisher’s exact test). The association, while significant, was not invariant, probably because of difficulties in detection due to the low level of Nitrosophaera in the samples, and possible presence of AmoA in other microbes or food materials. Inspection of published work showed detection of Nitrosophaera in metagenomic sequences from one of two individuals studied by [25], and in both 16S and metagenomic sequences in another cohort [41]. These data do not distinguish whether Nitrosophaera is replicating in the human gut or a transient present in food.
The Fungi of the Gut Microbiome
The fungal sequencing effort yielded 332,659 sequence reads, resulting in detection of 66 genera and 13 additional lineages that could not be classified to the genus level (Figure 1 and Figure S1 and S2). Fungal sequences were detected in every sample analyzed. Only 12 fungal genera were detected in 9 or more samples (Figure 1B, Figure S2). The phyla Ascomycota and Basidiomycota were mostly inversely correlated (Figure S1). The most prevalent genus in this sample set was Saccharomyces (present in 89% of the samples), followed by Candida (57%) and Cladosporium (42%) (Figure S1).
Co-occurrence Analysis using the Dice Index
Microbial communities in diverse settings have been shown to form syntrophic communities, in which metabolic waste products from one microbe provide nutrients for another. Thus an initial analysis of these samples was carried out to determine which microbes co-occur, as scored by the Dice index. For this, we only used data of relatively abundant genera (within Domain sample proportion of 0.01 or greater).
Numerous associations were detected. Figure 2 shows these interactions, incorporating data over all three Domains. Co-occurrence is indicated by the color code. Inspection of the figure shows several examples of co-occurrence involving a high proportion of samples (Figure 2, lower left corner, warmer colors). Bacteroides occurred commonly with Parabacteroides, Lachnospiraceae, and Ruminococcaceae. Lachnospiraceae occurred commonly with Faecalibacterium and Ruminococcaceae as well as Bacteroides. In contrast, co-occurrence of Methanobrevibacter and Nitrososphaera was low, as mentioned above.
Figure 2. Analysis of co-occurrence among microbial lineages scored using the Dice index.

Dice indexes across all genera pairs present at a proportion > = 0.01 are shown as a heatmap. Clustering was carried out using Ward’s criteria, based on the Euclidian distance between each genus pair using their Dice index across all other genera. Domain membership is color-coded on the left. Data are summarized in Table S9 and S10.
Candida and Saccharomyces were both associated with the group containing the above bacteria. Several other fungi achieved levels sufficient for inclusion in the analysis, but showed less clearcut co-occurrence with other community members. Methobrevibacter showed modest levels of association with the above group of bacteria, but was similarly also associated with Prevotella and several other bacterial groups. Nitrososphaera showed only modest associations with other lineages. The frequent co-occurrence of some of these microorganisms suggests candidate interactions among gut microbes for further investigation.
Covariation among Microbial Lineages
We next examined covariation among the three Domains, taking into account the relative abundance of each lineage in addition to presence-absence information. For the fungi and bacteria, multiple lineages were seen, and multiple different lineages were present in all samples, allowing use of correlation-based methods. However, for the archaea, only two lineages were detected with substantial frequency, Methanobrevibacter and Nitrosophaera, and these were mostly mutually exclusive. Thus for the archaea, samples were divided into three categories (containing one of the two archaea or no archaea), and co-occurring bacteria and fungi scored. For those few cases where both archaea were seen in a single sample, there was always a substantially greater abundance of one, so the sample was assigned based on the predominant lineage.
As a first global test of association among the three Domains, we conducted a Permanova test using the newly developed Generalized Unifrac distance [42], which showed significance (Tables S1 and S2). To characterize the lineages involved, we used a non-parametric Kruskal-Wallis test to determine which bacterial and fungal genera co-varied with the archaeal categories (Figure 3A).
Figure 3. Inter-generic relationships.

The heatmaps quantify the intergeneric relationships. (A) Normalized z-score of the bacterial and fungal proportions for samples grouped according to their archaeal status (Methanobrevibacter positive, Nitrososphaera positive, or archaea negative). Asterisks indicate Kruskall-Wallis significant comparisons after FDR adjustment (FDR of 25, 20, 15, and 10% are marked with 1, 2, 3 or 4 asterisks, respectively). Domain membership is color-coded on the left. (B) Spearman correlations between Fungi and Bacteria. Asterisks in red indicate FDR adjusted significant correlations (FDR 20%) and the remaining raw p-values are shown to illustrate general patterns within the data (p-values < = 0.05, 0.01, 0.005, 0.001 are marked with 1, 2, 3 or 4 asterisks, respectively).
Several lineages co-varied with Methanobrevibacter, including the commonly encountered bacteria Ruminococcus, and rarer lineages such as Oxalobacter and Papillibacter. Of the fungi, Candida and Saccharomyces were both positively associated with Methanobrevibacter. Both fungal genera were negatively associated with Nitrososphaera.
The Prevotella/Bacteriodes ratio was implicated as important for the gut microbiome structure in previous studies [10], [16], [41], so we assessed correlations with fungal and archaeal taxa. We performed a PermanovaG test using the Prevotella/Bacteroides ratio and the generalized Unifrac matrices obtained from the fungal composition data. A significant relationship was observed (p = 0.0146) indicating a potential influence of the Prevotella/Bacteriodes ratio on fungi. A post-hoc test with the individual weighted and unweighted unifrac matrices showed that there was a significantly correlation between the Prevotella/Bacteriodes ratio and the weighted Unifrac distances (p = 0.0133), but not with the unweighted Unifrac matrix. Thus the Prevotella/Bacteroides ratio correlated with the amounts of fungi present, but not the types. For the archaea, Bacteriodes was significantly negatively correlated with Methanobrevibacter, but positively correlated with both Nitrososphaera and no archaea. Prevotella showed a reciprocal pattern, but it did not achieve significance, probably because of the lower numbers of Prevotella-positive samples in the data set.
We also used a PermanovaG test to determine whether the proportion of fungal phyla (Ascomycota or Basidiomycota) correlated with the gut bacterial lineages. Both Ascomycota and Basidiomycota were significantly correlated with the bacterial lineages (p = 0.0202 and p = 0.0037, respectively). A post-hoc Permanova test with the individual weighted and unweighted Unifrac matrices was significant only when using the weighted Unifrac matrix (p = 0.0205 and p = 0.004, for the Ascomycota and Basidiomycota, respectively). A targeted analysis of the Fungi (Figure 3B) showed a negative association of Candida with Bacteriodes. Together these results indicate that the types of fungal species in the gut were not correlated with the bacterial taxa present, but rather their relative proportions.
Associations with Diet
We next investigated correlations between diet and the archaeal and fungal taxa. Dietary information was collected using diet inventories that scored usual (long-term) diet, and recent (short-term) diet. For each inventory, a clustering method was used to identify co-varying groups of dietary components, thereby reducing the number of variables and reducing penalties for multiple comparisons in the statistical analysis. We found that many of the clusters formed natural categories, such as carbohydrates, animal protein, and amino acids. Correlations between diet and bacteria have been reported previously for this data set [10] and were recapitulated here after using the pre-clustered dietary data (below). As a first step, Permanova tests were carried out using the nutrient cluster data and the weighted and unweighted Unifrac distance matrixes to determine whether the fungal and archaeal lineages were also associated with diet. Multiple significant associations were detected and are cataloged in Tables S3 and S4.
Relationships between archaea and nutrients were explored using a Kruskall-Wallis test on the Permanova-selected nutrient cluster measurements. As above, samples were separated into archaea-negative, Methanobrevibacter-positive or Nitrososphaera-positive groups. A higher intake of carbohydrates was correlated with Methanobrevibacter-positive samples. This trend was observed in comparisons to both the long-term and short-term dietary data. For the long-term diet, samples with Nitrososphaera or no archaea were enriched for clusters representing vegetable fat and poly-unsaturated fats. Samples with no archaea were enriched for a cluster representing total fat and total mono-unsaturated fats in the recent diet data (Figure 4).
Figure 4. Archaea-Diet relationships.

Heatmap of normalized average means for nutrient cluster measurements of the samples classified according to the dominant archaeal genus. Usual diet (A) and recent diet (B) relationships considered significant are marked with asterisks as described in Figure 2A.
Fungal and bacterial proportions were also scored versus usual diet (Figure 5A) or recent diet (Figure 5B). Only diet categories with at least one significant association are shown. As reported previously, bacterial proportions were most strongly correlated with components of the long term diet–Bacteriodes was more abundant in individuals eating high levels of animal protein, amino acids, and fats, while Prevotella was higher among those eating higher proportions of carbohydrates. For fungi significant correlations were observed only with the recent diet inventory, differing from the observations with bacteria. In the recent diet data, Candida was positively correlated with carbohydrates and negatively with total saturated fatty acids. A trend in the same direction was seen for Candida in the usual long-term diet, but it did not achieve significance. Aspergillus was negatively correlated with short chain fatty acids in the recent diet data. No trends were seen with Saccharomyces. Thus, these data indicate that fungal abundance is particularly strongly associated with the composition of recently consumed foods.
Figure 5. Fungi-Diet relationships.

Heatmap of Spearman correlations between nutrient clusters and the bacterial and fungal genera detected in the dataset. Correlations which were considered significant using the Usual (A) and the Recent (B) diet data are marked with asterisks as in Figure 2A. Domain membership is color-coded on the bottom.
Discussion
Here we investigated the relationships of diet and the fungi and the archaea of the human intestinal microbiome. Previously we reported, for this same set of samples, that patterns in the bacterial part of the gut microbiome correlated with long-term diet. Here we characterized the sample set by sequencing marker genes of the archaeal rDNA 16S and the fungal ITS1 region. Many interactions among microorganisms and nutrients were identified. Methanobrevibacter and Candida were positively correlated with the ingestion of carbohydrates. This was most notable in short-term diet data for both groups, and in fact only achieved significance at all for Candida in the short-term diet data. These data support specific proposals for the interactions of members of the gut microbiome with dietary components and with each other.
We detected no fewer than 62 fungal genera and 184 species level OTUs, paralleling and extending a study of one subject, which also yielded a high number of fungal lineages [35]. Which of these lineages are true gut residents, and which are transients in food is unknown. Six individuals had sequences belonging to genus Agaricus, the white button mushroom, which is consumed as food. This genus was among those filtered out of the analysis due to low prevalence in the dataset, suggesting that fungal DNA in food may be mostly degraded during digestion. However, we cannot exclude the possibility that the high prevalence of Saccharomyces in the fungal data is due to the ingestion of yeast-containing foods such as bread and beer.
Nitrososphaera, which were encountered with unexpected frequency in our data, are different enough from other archaeal groups to be placed in their own phylum, the Thaumarchaeota. Members of the Nitrososphaera genus are able to oxidize ammonia and degrade urea, which presumably would also feed nitrogen into the gut microbial community. Nitrososphaera may have been previously underappreciated in microbiome studies due to its low abundance. Here, it was detected in 16% of the samples analyzed, though in low abundance, in a mutually exclusive pattern with Methanobrevibacter. The basis of possible antagonism between Methanobrevibacter and Nitrososphaera is unknown. Nitrososphaera also showed a positive association with the ingestion of proteins and amino acids, both for usual and recent diet. The correlation was not sufficient to survive correction for multiple comparisons, but may nevertheless indicate utilization of ammonia and or urea to meet their energy and carbon requirements. Alternatively, we cannot exclude the possibility that Nitrososphaera was ingested with foods, possibly associated with meats.
Previously, interactions among microbial lineages of the gut were proposed to separate human populations into “enterotypes”, leading to considerable controversy. Arumugam et al. [16] described three interacting networks of microbial lineages, centered on the presence of Prevotella, Bacteriodes, and Ruminococcus, together with other interacting taxa. However, subsequent work indicated that the most prominent feature of the data was an inverse correlation between the Prevotella and Bacteroides genera [41], [10]. Here we show a positive association of Methanobrevibacter and Candida with the Prevotella group, and each of these was further correlated with a diet high in carbohydrates. A negative correlation of Methanobrevibacter and Bacteroides was also observed, paralleling the original Arumugam et al. paper. However, we also observed a strong positive relationship between Methanobrevibacter and Ruminococccus, which were initially described to belong to distinct enterotypes. Thus our findings associate archaea and fungi with aspects of the enterotype concept.
The intergeneric relationships described here, together with the nutrient correlations, support models for specific interactions among microbes in the human gut. An example of syntrophism has been previously described for Ruminococcus and methanogens, where the methanogens consume H2, allowing Ruminococcus to produce twice as many ATP molecules from the same amount of substrate [43]. One possible syntrophic guild specified in our data includes Candida, Prevotella, Ruminococcus and Methanobrevibacter (Figure 6). Candida is able to degrade starches, especially after pre-treatment with amylases [44] such as the human-encoded amylases present in the mouth and small intestine. Thus, in one model, Candida may assist in breaking down starch in carbohydrate rich foods, which in turn liberates simpler sugars to be fermented by bacteria such as Prevotella and Ruminococcus. Fermentation byproducts produced would then be consumed by Methanobrevibacter with the subsequent production of CO2 and/or CH4 [43]. Alternatively, Prevotella might degrade starch (pre-treated or not by human alpha amylases) and mannan containing polysaccharides from food to smaller poly- and monosaccharides [45]. Prevotella would then take up the smaller mono and polysaccharides, catabolizing them to produce succinate and other byproducts [46], [47]. All such hydrolysis is extra cellular, which would provide Candida with simpler sugars for fermentation (potentially down to acetate). Ruminococcus might then consume the succinate produced by Prevotella and produce H2 or acetate−/H+ for consumption by Methanobrevibacter [44], [48], [46].
Figure 6. Possible syntrophic relationships in the human gut consistent with data reported in this study.
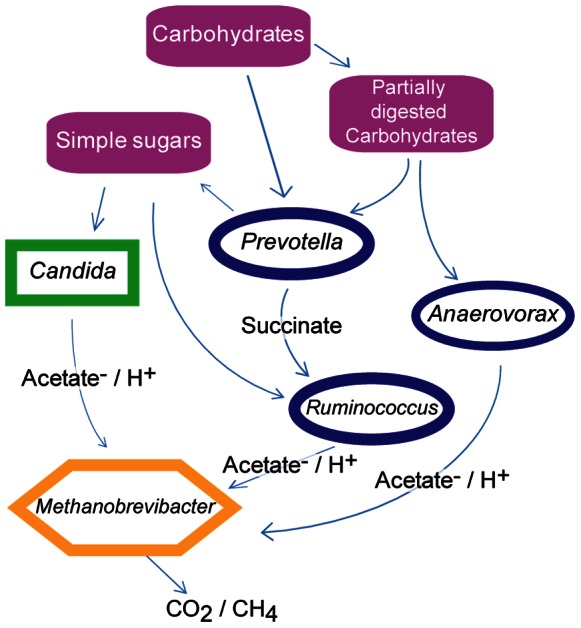
In summary, the findings presented here provide a broader picture of the human gut microbiome, integrating its full diversity with human diet. The associations presented here, together with other work [49], [41], [13], allow the proposal of specific relationships between nutrition and microbial consortia within the human gut.
Methods
In addition to the below, further information on methods can be found in the Supplemental Information, Figures S1–S3, and Tables S1–S10.
Ethics Statement
The Institutional Review Board of the University of Pennsylvania approved all study protocols and all participants provided written informed consent, or assent in the case of minors. Legal guardians provided written informed consent for minors (protocol #810009).
Samples
The samples described in [10] were used in this study. Bacterial 16S sequences data and diet data from [10] was used here. Briefly, healthy volunteers were screened to be free from any chronic gastrointestinal disease, cardiac disease, diabetes mellitus or immunodeficiency diseases, to have a normal bowel frequency (minimum once every 2 days, maximum 3 times per day), and body mass index (BMI) between 18.5 and 35. Demographic data was collect and analyzed as described previously for bacterial taxa ([10], Table S8). One stool sample was provided per subject and kept frozen at −80oC until processed for DNA extraction [10].
16S rDNA Gene, ITS1 Region and AmoA Gene PCR
Pyrosequencing was carried out using barcoded composite primers constructed as described in [50]. PCR reactions were carried out in triplicate using the Accuprime system (Invitrogen, Carlsbad, CA, USA). Each reaction contained 50 nanograms of DNA and 10 picoMol of each primer. Archaeal specific 16S rDNA primers and ITS1 fungal primers were adapted from the literature and used to amplify a rDNA 16S fragment; the final PCR cycling conditions were optimized to maximize specificity [51], [52], [38], [40],(Tables S5 and S6).
A nested PCR using specific primers for the AmoA gene was used to confirm the detection of Nitrosophaera sequences (Supplemental data. [53]). The nested PCR was performed to increase the sensitivity of the assay and improve the detection in these samples. Conventional PCR was not sufficiently sensitive to detect the 16S gene efficiently. PCR reactions were carried out using Invitrogen Accuprime. One µL of the total extracted DNA was used as template for the initial PCR. The nested PCR used 1 µL of PCR1 as template (Table S6). Blank extractions were used to control for environmental and reagent contamination. All PCR work was carried out in a laminar flow hood and all consumables and equipment were UV irradiated for a minimum of 30 minutes prior to use.
Pyrosequencing
Amplified 16S rDNA and ITS1 fragments were purified using 1∶1 volume of Agencourt AmPure XP beads (Beckman-Colter, Brea, CA). The purified PCR products from the stool samples were pooled in equal amounts prior to pyrosequencing using Roche/454 Genome Sequencer Junior. DNA pools were separated by amplicon type. All samples were submitted for sequencing, even control samples for which no visible amplicon was observed in agarose gels. For such samples, 40 out 50 µL of the bead-purified PCR product was pooled with the other samples for sequencing.
Sequence Analysis
Sequences obtained were decoded and quality controlled using the QIIME pipeline [54]. OUT’s were formed at 97% and 95% similarity for archaeal and fungal sequences respectively, and were considered for further analysis if they had a minimum of 5 sequences detected across all samples. Taxonomy was assigned to OTU representative sequences using the RDPclassifier [55] for archaeal sequences and BROCC [36] for fungal sequences. All taxonomy assignments were manually curated to check for accuracy and nomenclature using BLASTn against GenBank’s NR/NT database. For Archaea, the taxonomic assignment given by RDP to 2 out of the 12 archaeal OUT’s detected was corrected due to low coverage of those groups on RDP. For ITS, of the 290 OUT’s detected, 10 were missing mid-level (between phylum and genus) taxonomic information, which was filled in, and 5 yielded differing mid level taxonomies, of which only one of per genus was used. Two ITS OUT’s were automatically classified down to genus using BROCC, but had their taxonomy assignment brought up to Order level upon inspection of BLAST results (both OUT’s were present in one sample each). OTU sequence counts for each sample were aggregated at genus level. All downstream analysis was done at the Genus level using R unless otherwise noted. Genera were considered in the analysis if present in at least 9 out of the 96 available samples, and its absolute sequence count was equal to or greater than 10. For the ITS amplicon, samples were included in downstream analysis if they yielded at least 200 sequences. All novel sequence data was deposited at NCBI’s Sequence Read Archive under accession number SRP021021.
Beta Diversity
Taxonomic information was used to obtain the Taxonomic Distance between each genus using the R package Ade4, version 1.5 [56], [57]. The taxonomic distance matrix was used as input to calculate Unifrac distances using the R package GUniFrac, version 1.0 [42].
Inter-generic Relationships
Effects of bacteria on fungi, effects of the fungi on bacteria, and effects of archaea on bacteria and fungi were investigated using a Permanova test. Simulations of the effects of unequal variance in the different data sets compared indicated that differential variance was not a major confounder (Text S1). Within sample genus proportions were used to calculate Spearman correlations between bacterial genera and fungal genera. As only one or very few archaeal genera were detected in any sample, sequence proportions would be greatly skewed, invalidating any potential correlation results. Instead, samples were classified according to the archaea genera detected and bacterial and fungal proportions were used on as input for Kruskall-Wallis tests. P-values were considered significant using a FDR of 25%.
Co-occurrence
The Dice index [58] was used to determine the co-occurrence of genera across the entire dataset. Genera were considered present in a sample if its sequence proportion was at least 0.01.
Diet Analysis
Dietary information from [10] was used in this analysis. Usual diet was obtained using the Willett food frequency questionnaire [59]. Recent diet was obtained from 3 interviews recalling all consumed food on 3 days within the week preceding the sample acquisition (NHANES method, [60]). All interviews were carried out by trained nutritionists. Nutrient measurements across individuals obtained from the dietary questionnaires were used in a clustering procedure to reduce the number of comparisons to be made. First, Spearman correlations were calculated pairwise for all nutrient variables available and this correlation matrix was used as input in a clustering analysis, and 20 clusters (approximately 10% of the total number of nutrients available) were selected. Nutrients within each cluster were submitted to a Principal Component Analysis and the first principal component values were extracted and used as a surrogate dietary measurement (Nutrient Cluster Measurement). These nutrient cluster measurements were used in a Permanova analysis together with the taxonomy based, weighted and unweighted Unifrac distances calculated using the R packages Ade4 and GUniFrac. Clusters which were significant in the Permanova analysis were further used to calculate Spearman rank correlations using the proportion for each bacterial and fungal genus across all samples. The Nutrient Cluster Measurements for each sample were also classified according to their archaeal status and then submitted to a Kruskall-Wallis test to determine archaea/diet relationships. P-values with a False Discovery Rate of 25% or less were considered significant.
Supporting Information
The Fungal phyla detected are shown as sequence proportions within each sample (A). A Spearman rank correlation for the proportions of Ascomycota versus Basidiomycota across the samples was 0.7456. Care should be taken when interpreting this correlation as the proportional nature of sequencing data naturally yields inverse correlations. The prevalence of each fungal genera detected across all samples is depicted in (B). Genera are grouped by their phylum affiliation and only genera sequences that could be assigned to the genus level are shown.
(TIF)
Heatmap with all Fungal genera detected in the stool sample set used, and blank extraction controls. Colors indicate relative proportion within each sample.
(TIF)
Number of Fungal and Bacterial genera per sample. Samples were classified as Methanobrevibacter positive, Nitrososphaera positive, or Archaea negative. Difference between groups was tested using a Kruskal-Wallis test, followed by a post hoc Dunn’s multiple comparison test. Asterisks indicate significant comparisons (p<0.001).
(TIF)
Permanova of Archaea effects on the Bacterial and Fungal parts of the microbiome.
(XLSX)
Permanova of Fungal Phyla with Bacteria.
(XLSX)
Permanova analysis of the association between usual dietary groups and bacterial, fungal, and archaeal lineages.
(XLSX)
Permanova analysis of the association between recent dietary groups and bacterial, fungal, and archaeal lineages.
(XLSX)
Primers used in this study.
(XLSX)
PCR amplification conditions used in this study.
(XLSX)
Number of reads for each genus, in each sample analyzed.
(XLSX)
Permanova analysis of the association between demographic factors and bacterial, fungal, and archaeal lineages.
(XLSX)
Co-occurring genera according to the calculated Dice index.
(XLSX)
Positively co-varying genera. Archaeal covariation represents the positive relationship detected in Figure 3A. Fungi/Bacterial relationships are the same as the positive relationships represented in Figure 3B.
(XLSX)
Supporting information text.
(DOCX)
Acknowledgments
We thank members of the Bushman Lab for helpful discussions and Caitlin Greig for help in preparing the manuscript submission.
Funding Statement
This work was supported by Project UH2/3 DK083981, the Penn Genome Frontiers Institute; National Institutes of Health (NIH) DK 089472 (GDW); Penn Digestive Disease Center (P30 DK050306); The Joint Penn-CHOP Center for Digestive, Liver, and Pancreatic Medicine; S10RR024525; UL1RR024134, and K24-DK078228. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, National Institutes of Health, or Pennsylvania Department of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ursell LK, Clemente JC, Rideout JR, Gevers D, Caporaso JG, et al. (2012) The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J Allergy Clin Immunol 129: 1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gevers D, Knight R, Petrosino JF, Huang K, McGuire AL, et al. (2012) The Human Microbiome Project: a community resource for the healthy human microbiome. PLoS Biol 10: e1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, et al. (2012) Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 151: 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minot S, Grunberg S, Wu GD, Lewis JD, Bushman FD (2012) Hypervariable loci in the human gut virome. Proc Natl Acad Sci U S A 109: 3962–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, et al.. (2011) The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. [DOI] [PMC free article] [PubMed]
- 6. Minot S, Wu GD, Lewis JD, Bushman FD (2012) Conservation of Gene Cassettes among Diverse Viruses of the Human Gut. PLoS One 7: e42342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, et al. (2012) Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol 13: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, et al. (2012) Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, et al. (2010) Direct sequencing of the human microbiome readily reveals community differences. Genome Biol 11: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, et al. (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science 334: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grice EA, Kong HH, Conlan S, Deming CB, Davis J, et al. (2009) Topographical and temporal diversity of the human skin microbiome. Science 324: 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science (New York, NY) 312: 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, et al. (2011) Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 108 Suppl 14578–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology 6: e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA (2012) The application of ecological theory toward an understanding of the human microbiome. Science 336: 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, et al. (2011) Enterotypes of the human gut microbiome. Nature 473: 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, et al. (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9: 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, et al. (2013) Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339: 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, et al. (2012) Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184. [DOI] [PubMed] [Google Scholar]
- 20. Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, et al. (2010) Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466: 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reyes A, Semenkovich NP, Whiteson K, Rohwer F, Gordon JI (2012) Going viral: next-generation sequencing applied to phage populations in the human gut. Nat Rev Microbiol 10: 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller TL, Wolin MJ, Conway de Macario E, Macario AJ (1982) Isolation of Methanobrevibacter smithii from human feces. Appl Environ Microbiol 43: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matarazzo F, Ribeiro AC, Faveri M, Taddei C, Martinez MB, et al. (2012) The domain Archaea in human mucosal surfaces. Clin Microbiol Infect 18: 834–840. [DOI] [PubMed] [Google Scholar]
- 24. Dridi B, Raoult D, Drancourt M (2011) Archaea as emerging organisms in complex human microbiomes. Anaerobe 17: 56–63. [DOI] [PubMed] [Google Scholar]
- 25. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, et al. (2005) Diversity of the human intestinal microbial flora. Science 308: 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart JA, Chadwick VS, Murray A (2006) Carriage, quantification, and predominance of methanogens and sulfate-reducing bacteria in faecal samples. Lett Appl Microbiol 43: 58–63. [DOI] [PubMed] [Google Scholar]
- 27. Dridi B, Henry M, El Khechine A, Raoult D, Drancourt M (2009) High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS ONE 4: e7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, et al. (2007) Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A 104: 10643–10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anderson HW (1917) Yeast-Like Fungi of the Human Intestinal Tract. The Journal of Infectious Diseases 21: 13. [Google Scholar]
- 30. Gumbo T, Isada CM, Hall G, Karafa MT, Gordon SM (1999) Candida glabrata Fungemia. Clinical features of 139 patients. Medicine (Baltimore) 78: 220–227. [DOI] [PubMed] [Google Scholar]
- 31. Murdoch TB, Xu W, Stempak JM, Landers C, Targan SR, et al. (2012) Pattern recognition receptor and autophagy gene variants are associated with development of antimicrobial antibodies in Crohn’s disease. Inflamm Bowel Dis 18: 1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seibold F, Stich O, Hufnagl R, Kamil S, Scheurlen M (2001) Anti-Saccharomyces cerevisiae antibodies in inflammatory bowel disease: a family study. Scand J Gastroenterol 36: 196–201. [DOI] [PubMed] [Google Scholar]
- 33. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, et al. (2012) Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336: 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scupham AJ, Presley LL, Wei B, Bent E, Griffith N, et al. (2006) Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microbiol 72: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamad I, Sokhna C, Raoult D, Bittar F (2012) Molecular detection of eukaryotes in a single human stool sample from Senegal. PLoS One 7: e40888. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Dollive S, Peterfreund GL, Sherrill-Mix S, Bittinger K, Sinha R, et al. (2012) A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol 13: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, et al. (2008) Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol 43: 831–841. [DOI] [PubMed] [Google Scholar]
- 38. DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89: 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heuer H, Krsek M, Baker P, Smalla K, Wellington EM (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63: 3233–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, et al. (2010) Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog 6: e1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, et al. (2012) Human gut microbiome viewed across age and geography. Nature 486: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, et al. (2012) Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28: 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stams AJ, Plugge CM (2009) Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol 7: 568–577. [DOI] [PubMed] [Google Scholar]
- 44. Iannotti EL, Kafkewitz D, Wolin MJ, Bryant MP (1973) Glucose fermentation products in Ruminococcus albus grown in continuous culture with Vibrio succinogenes: changes caused by interspecies transfer of H 2. J Bacteriol 114: 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Downes J, Tanner AC, Dewhirst FE, Wade WG (2010) Prevotella saccharolytica sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol 60: 2458–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kovatcheva-Datchary P, Egert M, Maathuis A, Rajilic-Stojanovic M, de Graaf AA, et al. (2009) Linking phylogenetic identities of bacteria to starch fermentation in an in vitro model of the large intestine by RNA-based stable isotope probing. Environ Microbiol 11: 914–926. [DOI] [PubMed] [Google Scholar]
- 47. van Gylswyk NO (1995) Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int J Syst Bacteriol 45: 297–300. [DOI] [PubMed] [Google Scholar]
- 48. Purushe J, Fouts DE, Morrison M, White BA, Mackie RI, et al. (2010) Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol 60: 721–729. [DOI] [PubMed] [Google Scholar]
- 49. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, et al. (2008) Evolution of mammals and their gut microbes. Science (New York, NY) 320: 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, et al. (2008) The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS pathogens 4: e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Mol Ecol 2: 113–118. [DOI] [PubMed] [Google Scholar]
- 52. Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, et al. (2004) Methanogenic Archaea and human periodontal disease. Proc Natl Acad Sci U S A 101: 6176–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tourna M, Stieglmeier M, Spang A, Konneke M, Schintlmeister A, et al. (2011) Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci U S A 108: 8420–8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, et al. (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lozupone CA, Knight R (2008) Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev 32: 557–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. Journal of Statistical Software 22: 20. [Google Scholar]
- 58. Dice LR (1945) Measures of the Amount of Ecologic Association Between Species. Ecology 26: 6. [Google Scholar]
- 59. Willett WC, Stampfer MJ, Underwood BA, Speizer FE, Rosner B, et al. (1983) Validation of a dietary questionnaire with plasma carotenoid and alpha-tocopherol levels. Am J Clin Nutr 38: 631–639. [DOI] [PubMed] [Google Scholar]
- 60.Centers for Disease Control and Prevention (CDC). National Center for Health Statistics (NCHS). (2011) National Health and Nutrition Examination Survey Questionnaire. Hyattsville, MD. Available: http://www.cdc.gov/nchs/nhanes/nhanes2009-2010/questexam09_10.htm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Fungal phyla detected are shown as sequence proportions within each sample (A). A Spearman rank correlation for the proportions of Ascomycota versus Basidiomycota across the samples was 0.7456. Care should be taken when interpreting this correlation as the proportional nature of sequencing data naturally yields inverse correlations. The prevalence of each fungal genera detected across all samples is depicted in (B). Genera are grouped by their phylum affiliation and only genera sequences that could be assigned to the genus level are shown.
(TIF)
Heatmap with all Fungal genera detected in the stool sample set used, and blank extraction controls. Colors indicate relative proportion within each sample.
(TIF)
Number of Fungal and Bacterial genera per sample. Samples were classified as Methanobrevibacter positive, Nitrososphaera positive, or Archaea negative. Difference between groups was tested using a Kruskal-Wallis test, followed by a post hoc Dunn’s multiple comparison test. Asterisks indicate significant comparisons (p<0.001).
(TIF)
Permanova of Archaea effects on the Bacterial and Fungal parts of the microbiome.
(XLSX)
Permanova of Fungal Phyla with Bacteria.
(XLSX)
Permanova analysis of the association between usual dietary groups and bacterial, fungal, and archaeal lineages.
(XLSX)
Permanova analysis of the association between recent dietary groups and bacterial, fungal, and archaeal lineages.
(XLSX)
Primers used in this study.
(XLSX)
PCR amplification conditions used in this study.
(XLSX)
Number of reads for each genus, in each sample analyzed.
(XLSX)
Permanova analysis of the association between demographic factors and bacterial, fungal, and archaeal lineages.
(XLSX)
Co-occurring genera according to the calculated Dice index.
(XLSX)
Positively co-varying genera. Archaeal covariation represents the positive relationship detected in Figure 3A. Fungi/Bacterial relationships are the same as the positive relationships represented in Figure 3B.
(XLSX)
Supporting information text.
(DOCX)