

Graphical abstract
Highlights
► 89 genes encoding flavoproteins were identified in the human genome. ► Two thirds of human flavoproteins are linked to human diseases. ► Flavoenzymes are essential for the biosynthesis of other coenzymes and hormones. ► Flavoenzymes play a critical role in folate and cobalamin metabolism.
Keywords: Coenzyme A, Coenzyme Q, Folate, Heme, Pyridoxal 5′-phosphate, Steroids, Thyroxine, Vitamins
Abstract
Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoenzymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, β-oxidation and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyridoxal 5′-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-methylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. These flavoenzymes are discussed in detail to highlight their role in health and disease.
Introduction
Vitamin B2 (or riboflavin) is an essential dietary requirement for humans (Homo sapiens sapiens) because the biosynthesis of this compound is absent. The daily requirement for vitamin B2 was estimated to be 1.1 mg and 1.3 mg for adult females and males, respectively, but may vary depending on metabolic challenges and the efficiency of riboflavin uptake. Dietary riboflavin is taken up in the human gastrointestinal tract by poorly characterized transporters (Table 1, entry #74). Vitamin B2 is not used in any human enzyme per se but is chemically modified to the flavin mononucleotide (FMN)1 and flavin adenine dinucleotide form by riboflavin kinase (EC 2.7.1.26) and FAD synthetase (EC 2.7.7.2), respectively. FMN and FAD as well as excess riboflavin are excreted in the urine. Chastain and McCormick have also detected trace amounts of 7-hydroxy- and 8-hydroxy-flavins in human urine which are generated through as yet unknown catabolic reactions [1]. In addition, they have also found 8-sulfonyl-flavin which is supposedly released from enzymes bearing a covalent thioester linkage, e.g. monoamine oxidase A and B (MAOA and MAOB, Table 1).
Table 1.
Human flavoproteins.
| No. | E.C. | Enzyme | Cofactor | Structure clan (family)⁎ | Gene symbol | Gene location |
|---|---|---|---|---|---|---|
| 1 | 1.1.1.28 | d-lactate dehydrogenase | FAD | FAD_PCMH (FAD_binding_4) | LDHD | 16q23.1 |
| 2 | 1.1.1.204 | Xanthine dehydrogenase | FAD | FAD_PCMH (FAD_binding_5) | XDH | 2p23.1 |
| 3 | 1.1.3.15 | (S)-2-hydroxy-acid oxidase | FMN | TIM_barrel (FMN_dh) | HAO1 | 20p12.3 |
| HAO2 | 1p12 | |||||
| 4 | 1.1.5.3 | Glycerol 3-phosphate dehydrogenase | FAD | NADP_Rossmann (DAO) | GPD2 | 2q24.1 |
| 5 | 1.1.99.1 | Choline dehydrogenase | FAD | GMC_ oxred | CHDH | 3p21.1 |
| 6 | 1.1.99.2 | l-2-Hydroxyglutarate dehydrogenase | FAD | -------------------------------------- | L2HGDH | 14q21.3 |
| 7 | 1.1.99.- | d-2-Hydroxyglutarate dehydrogenase | FAD | -------------------------------------- | D2HGDH | 2q37.3 |
| 8 | 1.2.3.1 | Aldehyde oxidase | FAD | -------------------------------------- | AOX1 | 2q33.1 |
| 9 | 1.3.1.2 | Dihydropyrimidine dehydrogenase | FMN | TIM_barrel (DHO_dh) | DPYD | 1p21.3 |
| FAD | NADP_Rossmann (Pyr_redox_2) | |||||
| 10 | 1.3.1.72 | 3β-Hydroxysterol Δ24-reductase | FAD | -------------------------------------- | DHCR24 | 1p32.3 |
| 11 | 1.3.3.1 | Dihydroorotate dehydrogenase | FMN | TIM_barrel (DHO_dh) | DHODH | 16q22.2 |
| 12 | 1.3.3.4 | Protoporphyrinogen IX oxidase | FAD | NADP_Rossmann (Amino_oxidase) | PPOX | 1q23.3 |
| 13 | 1.3.3.6 | Acyl-CoA oxidase | FAD | Acyl-CoA_dh | ACOX1 | 17q25.1 |
| (ACOX, acyl-CoA_dh_1) | ACOX2 | 3p14.3 | ||||
| ACOX3 | 4p16.1 | |||||
| 14 | 1.3.3.- | Glutaryl-CoA oxidase | FAD | -------------------------------------- | C7orf10 | 7p14.1 |
| 15 | 1.3.5.1 | Succinate dehydrogenase | 8α-(N3-His)- | NAPH_Rossmann (FAD_binding_2) | SDHA | 5p15.33 |
| Flavoprotein subunit A | -FAD | (3q29)a | ||||
| 16 | 1.3.99.2 | Short-chain- (butyryl-) acyl CoA dehydrogenase | FAD | Acyl-CoA_dh (Acyl_CoA_dh_1) | ACADS | 12q24.31 |
| 17 | 1.3.99.3 | Medium-chain acyl-CoA dehydrogenase | FAD | Acyl-CoA_dh (Acyl_CoA_dh_1) | ACADM | 1p31.1 |
| 18 | 1.3.99.7 | Glutaryl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | GCDH | 19p13.2 |
| 19 | 1.3.99.10 | Isovaleryl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | IVD | 15q15.1 |
| 20 | 1.3.99.12 | 2-Methylbutyryl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACADSB | 10q26.13 |
| 21 | 1.3.99.13 | Long-chain-acyl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACADL | 2q34 |
| 22 | 1.3.99.- | Very long-chain acyl-CoA dh | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACADVL | 17p13.1 |
| 23 | 1.3.99.- | Isobutyryl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACAD8 | 11q25 |
| 24 | 1.3.99.- | Long-chain-unsaturated-acyl-CoA dh (molecular chaperone of complex I) | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACAD9 | 3q21.3 |
| 25 | 1.3.99.- | Long- and branched-chain-acyl-CoA dh | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACAD10 | 12q24.1 |
| 26 | 1.3.99.- | C22-long-chain-acyl-CoA dehydrogenase | FAD | Acyl-CoA_dh (acyl-CoA_dh_1) | ACAD11 | 3q22.1 |
| 27 | 1.4.3.1 | d-aspartate oxidase | FAD | NADP_Rossmann (DAO) | DDO | 6q21 |
| 28 | 1.4.3.2 | l-amino acid oxidase | FAD | NADP_Rossmann (Amino_oxidase) | LAO | 19q13.3-q13.4 |
| 29 | 1.4.3.3 | d-amino acid oxidase | FAD | NADP_Rossmann (DAO) | DAO | 12q24.11 |
| 30 | 1.4.3.4 | Monoamine oxidase | 8α-(Cys)-FAD | NADP_Rossmann (Amino_oxidase) | MAOA | Xp11.3 |
| MAOB | Xp11.3b | |||||
| 31 | 1.4.3.5 | Pyridoxal 5’-phosphate oxidase | FMN | FMN-binding (Pyridox_oxidase) | PNPO | 17q21.32 |
| Pyridoxine 5’-phosphate oxidase | ||||||
| 32 | 1.4.3.- | Catecholamine oxidase (renalase) | FAD | NADP_Rossmann (Amino_oxidase) | RNLS | 10q23.31 |
| 33 | 1.5.1.20 | Methylenetetrahydrofolate reductase | FAD | FAD_oxidored (MTHFR) | MTHFR | 1p36.22 |
| 34 | 1.5.3.7 | l-pipecolate oxidase | 8α-(Cys)-FAD | NADP_Rossmann (DAO) | PIPOX | 17q11.2 |
| 35 | 1.5.3.16 | Spermine oxidase | FAD | NADP_Rossmann (Amino_oxidase) | SMO | 20p13 |
| 36 | 1.5.5.1 | Electron-transferring flavoprotein-ubiquinone oxidoreductase | FAD | 4Fe-4S (ETF_QO) | ETFDH | 4q32.1 |
| 37 | -------- | Electron transferring flavoprotein | FAD | FAD_DHS (ETF_alpha) | ETFA | 15q24.2-q24.3 |
| HUB (ETF) | ETFB | 19q13.41 | ||||
| 38 | 1.5.99.1 | Sarcosine dehydrogenase | 8α-(N3-His)- | NADP_Rossmann (DAO) | SARDH | 9q34.2 |
| -FAD | ||||||
| 39 | 1.5.99.2 | Dimethylglycine dehydrogenase | 8α-(N3-His) | NADP_Rossmann (DAO) | DMGDH | 5q14.1 |
| -FAD | ||||||
| 40 | 1.5.99.- | Lysine-specific histone demethylase | FAD | NADP_Rossmann | KDM1A | 1p36.12 |
| 41 | 1.5.99.8 | Proline dehydrogenase | FAD | FAD_oxidored (Pro_dh) | PRODH | 22q11.21 |
| 42 | 1.6.2.2 | Cytochrome-b5 reductase | FAD | FAD_Lum_binding (FAD_binding_6) | CYB5R3 | 22q13.2 |
| 43 | 1.6.2.4 | NADPH-hemoprotein reductase | FMN | Flavoprotein (flavodoxin_1) | POR | 7q11.23 |
| (cytochrome P450 reductase) | FAD | FAD_Lum_binding (FAD_binding_1) | ||||
| 44 | 1.6.5.2 | NAD(P)H dehydrogenase (quinone) | FAD | Flavoprotein (Flavodoxin_2) | NQO1 | 16q22.1 |
| 45 | 1.6.5.3 | NADH-ubiquinone oxidoreductase of complex I, subunit UQOR1 | FMN | complex1_51K | NDUFV1 | 11q13.2 |
| 46 | 1.6.-.- | NADPH-dep. diflavin oxidoreductase 1 | FMN | Flavoprotein (Flavodoxin_1) | NDOR1 | 9q34.3 |
| FAD | FAD_Lum_binding (FAD_binding_1) | |||||
| 47 | 1.6.-.- | tRNA dihydrouridine synthase | FMN | TIM_barrel (Dus) | DUS2L | 16q22.1 |
| 48 | 1.8.1.4 | Dihydrolipoyl dehydrogenase | FAD | NADP_Rossmann (Pyr_redox_2) | DLD | 7q31.1 |
| 49 | 1.8.1.7 | Glutathione-disulfide reductase | FAD | NADP_Rossmann (Pyr_redox_2) | GSR | 8p12 |
| 50 | 1.8.1.9 | Thioredoxin-disulfide reductase | FAD | NADP_Rossmann (Pyr_redox_2) | TXNR1 | 12q23.3 |
| TXNR2 | 22q11.21 | |||||
| TXNR3 | 3q21.3 | |||||
| 51 | 1.8.1.- | ER flavoprotein associated with degr. | FAD | -------------------------------------- | FOXRED2 | 22q12.3 |
| 52 | 1.8.3.2 | Sulfhydryl oxidase | FAD | Erv1_Alr | GFER | 16p13.3 |
| 53 | 1.8.3.5 | Prenylcysteine oxidase | FAD | -------------------------------------- | PCYOX1 | 2p13.3 |
| 54 | 1.10.99.2 | Ribosyldihydronicotinamide | FAD | Flavoprotein (Flavodoxin_2) | NQO2 | 6p25.2 |
| dehydrogenase | ||||||
| 55 | 1.14.13.8 | Flavin-containing monooxygenases | FAD | -------------------------------------- | FMO1 | 1q24.3 |
| FMO2 | 1q24.3 | |||||
| FMO3 | 1q24.3 | |||||
| FMO4 | 1q24.3 | |||||
| FMO5 | 1q21.1 | |||||
| 56 | 1.14.13.9 | Kynurenine 3-monooxygenase | FAD | -------------------------------------- | KMO | 1q43 |
| 57 | 1.14.13.39 | Nitric-oxide synthase | FMN | Flavoprotein (Flavodoxin_1) | NOS1 | 12q24.22 |
| FAD | FAD_Lum_binding (FAD_binding_1) | NOS2 | 17q11.2 | |||
| NOS3 | 7q36.1 | |||||
| 58 | 1.14.13.132 | Squalene monooxygenase | FAD | -------------------------------------- | SQLE | 8q24.13 |
| 59 | 1.14.99.- | Monooxygenase in coenzyme Q biosyn. | FAD? | -------------------------------------- | COQ6 | 14q24.3 |
| 60 | 1.16.1.2 | Ferrireductase (biliverdin IX beta red.) | FMN? | N-terminal domain (1-215) | STEAP3 | 2q14.2 |
| 61 | 1.16.1.8 | Methionine synthase reductase | FMN | Flavoprotein (Flavodoxin_1) | MTRR | 5p15.31 |
| FAD | FAD_Lum_binding (FAD_binding_1) | |||||
| 62 | 1.18.1.2 | Ferredoxin-NADP+ reductase | FAD | FAD_Lum_binding (FAD_binding_6) | FDXR | 17q25.1 |
| 63 | 1.-.-.- | NAD(P)H oxidase cytochrome b(558), beta subunit | FAD | -------------------------------------- | CYBB | Xp11.4 |
| 64 | 1.-.-.- | Thyroid oxidase / dual oxidase | FAD | -------------------------------------- | DUOX1 | 15q21.1 |
| DUOX2 | 15q21.1 | |||||
| 65 | 2.2.1.6 | Acetolactate synthase-like protein | FAD? | -------------------------------------- | - | 19p13.12 |
| 66 | 2.5.1.26 | Alkyldihydroxyacetone phosphate synthase | FAD | FAD_PCMH (FAD_binding_4) | AGPS | 2q31.2 |
| 67 | 4.1.1.36 | 4’-Phosphopantothenoylcysteine decarboxylase | FMN | Flavoprotein | PPCDC | 15q24.2 |
| 68 | 4.1.99.3 | Cryptochrome | FAD | FAD-binding of DNA-photolyase | CRY1 | 12q23.3 |
| (FAD_binding_7) | CRY2 | 11p11.2 | ||||
| 69 | -------- | Apoptosis inducing protein | FAD | NADP_Rossmann (Pyr_redox_2) | AIFM1 | Xq26.1 |
| 70 | -------- | Apoptosis inducing protein | 6-OH-FAD | NADP_Rossmann (Pyr_redox_2) | AIFM2 | 10q22.1 |
| 71 | -------- | Iodotyrosine deiodinase | FMN | Nitroreductase | IYD | 6q25.1 |
| 72 | -------- | Axon guidance protein | FAD | NADP_Rossmann (FAD_binding_3) | MICAL1 | 6q21 |
| Interacting with CasL | MICAL2 | 11p15.3 | ||||
| MICAL3 | 22q11.21 | |||||
| 73 | -------- | FAD-dependent oxidoreductase (molecular chaperone of complex 1) | FAD | -------------------------------------- | FOXRED1 | 11q24.2 |
| 74 | -------- | Riboflavin transporter | Riboflavinc | -------------------------------------- | SLC52A1 | 17p13.2 |
| SLC52A2 | 8q24.3 | |||||
| SLC52A3 | 20p13 | |||||
| 75 | 1.5.1.30 | Riboflavin / FMN reductase | FMNc | NADP_Rossmann (NmrA-like) | BLVRB | 19q13.2 |
| 76 | 2.7.1.26 | Riboflavin kinase | riboflavinc | Flavokinase | RFK | 9q21.13 |
| 77 | 2.7.7.2 | FAD-adenylyl transferase (synthetase) | FMNc | HUP (PAPS_reduct) | FLAD1 | 1q21.3 |
Abbreviations used: biosyn., biosynthesis; dh, dehydrogenase; degr., degradation; dep., dependent; ER, endoplasmic reticulum; red., reductase.
Pfam classification given in plain text is for the structure of human proteins and those in italics for structures of homologs.
Duplicated pseudogene.
Opposite orientation on chromosome.
Substrate of transporter, modifying enzyme or reductase (entries 74–77).
FMN and FAD possess a tricyclic heteroaromatic isoalloxazine ring that can reversibly accept and donate one or two electrons. Thus the majority of the enzymes utilizing FMN or FAD catalyze reduction–oxidation (“redox”) reactions in metabolic transformations. In fact, the majority of human flavoenzymes belongs to the oxidoreductases with only two example each for a transferase and lyase, respectively (Table 1, entries #65–68). Flavoenzymes may either use FMN or FAD as cofactor and most are specific for one or the other. In the case of human flavoproteins only twelve use FMN and 64 FAD as cofactor amounting to ca. 16% and 84% of the flavoenzymes, respectively (note that five enzymes utilize both FMN and FAD). In a global analysis of FMN and FAD usage in flavoproteins it was noted that 25% of flavoenzymes utilize FMN [2], hence the human flavoproteome has a clear bias towards FAD-dependent enzymes.
Most flavoenzymes bind FMN or FAD non-covalently (90%). In six human flavoenzymes the FAD cofactor is covalently linked via the 8-α-methyl group to either the nitrogen (N-3) of a histidine (succinate, sarcosine and dimethylglycine dehydrogenase) or the sulfur of a cysteine residue (MAOA, MAOB and l-pipecolate oxidase). Interestingly, monocovalent attachment to the 6-position of the isoalloxazine ring system as well as bicovalent attachment to the 8-α and 6-position observed in several bacterial, fungal and plant flavoenzymes are absent in human flavoproteins. This is also reflected by the scarcity of flavoenzymes adopting a topology similar to the p-cresolmethylhydroxylase (clan FAD_PCMH, family FAD_binding_4) with d-lactate dehydrogenase (EC 1.1.1.28) and alkalidihydroxyacetone phosphate synthase (EC 2.5.1.26) as sole examples for this type of structure. Interestingly, these two enzymes represent a subset of flavoenzymes in this clan that does not engage in covalent linkage of the FAD cofactor, which in fact appears to be the rule rather than the exception in this structure family [2]. Overall, it is encouraging that the structure of more than half of the human flavoproteins was solved by X-ray crystallography (Table 1). In addition, in 23 cases the structure of the human protein can be inferred from the known structure of a homologous flavoprotein. This leaves only fifteen flavoproteins where currently no structural information is available (Table 1). The prevalence of the Rossmann-fold in FAD-dependent flavoenzymes previously noted [2] is also seen in the human FAD-dependent proteins (see Fig. S1). In contrast to the global fold distribution the structural clan Acyl-CoA_dh takes second place and switches place with the clan FAD_PCMH. This is due to the relatively large number of acyl-CoA dehydrogenases (eleven) and acyl-CoA oxidases (three) compared to only three members in the FAD_PCMH clan. Flavoenzymes in the latter clan appear to be rare in mammals but are very prominent in metabolically active and diverse organisms such as bacteria, fungi and plants [2]. The most common structural clan found for human FMN-dependent proteins is Flavoprotein and TIM_barrel (five and four members, respectively, see Fig. S1) and reflects the general prevalence of these two clans in FMN-dependent flavoproteins [2].
The chromosomal location of the genes encoding flavoproteins is known for all flavoproteins (Table 1 and Fig. S2). They are uniformly distributed over the chromosomes with only chromosome 13, 18, 21 and Y lacking genes encoding flavoproteins (Table 1 and Fig. S2). Eleven flavoenzymes occur as isozymes encoded by multiple genes (2–5). In three cases (DUOX1–2, FMO1–5 and MAOA + B) the genes are on the same chromosome (15, 1 and X, respectively) whereas in all other cases (ACOX1–3, CRY1–2, HAO1–2, MICAL1–3, NOS1–3 and SLC52A1–3) the genes are on different chromosomes (Fig. S2).
Flavoenzymes in human diseases
A surprisingly large number of flavoproteins (ca. 60%) is associated with human disorders caused by mutations in the pertinent gene. Table 2 provides an overview of the 50 flavoproteins along with their corresponding record number in OMIM. Since most flavoproteins are localized in the mitochondria the diseases are connected with deficiencies in mitochondrial processes. Other compartments, most notably peroxisomes and the endoplasmic reticulum, are also affected by some flavoprotein deficiencies or dysfunctions. In several cases flavoprotein deficiencies occur in connected metabolic pathways and therefore give rise to similar clinical manifestations. For example, glutaric acidemia (OMIM 608801) can be caused by a deficiency of glutaryl-CoA dehydrogenase (type I), electron transferring flavoprotein (type IIA and IIB) or electron-transferring flavoprotein-ubiquinone oxidoreductase (type IIC). Similarly, Leigh syndrome (OMIM 256000) may arise from a defect in any of the respiratory electron transfer complexes in the inner mitochondrial membrane and thus deficiency of complex I (containing the FMN-dependent NDUFV1) or complex II (containing the FAD-dependent subunit A) as well as FOXRED1, an FAD-containing molecular chaperone of complex I, may constitute the molecular cause of the disease.
Table 2.
Disease-related human flavoproteins.
| No. | E.C. | Enzyme | Disease | Metabolic function | Localisation | OMIM |
|---|---|---|---|---|---|---|
| 1 | 1.1.1.204 | Xanthine dehydrogenase | Xanthinuria type I | Purine degr. | Cytosol | 607633 |
| 2 | 1.1.5.3 | Glycerol 3-phosphate dehydrogenase | Diabetis mellitus type II | Electron transport | mito. i. membr. | 138430 |
| 3 | 1.1.99.1 | Choline dehydrogenase | tooth agenesis, cleft lip sperm motility | Choline degr. | mito. i. membr. | [78–82] |
| 4 | 1.1.99.2 | l-2-Hydroxyglutarate dehydrogenase | l-2-Hydroxyglutaric aciduria | “Metabolite repair” | mito. membr. | 609584 |
| 5 | 1.1.99.- | d-2-Hydroxyglutarate dehydrogenase | d-2-Hydroxyglutaric aciduria | “Metabolite repair” | Mitochondria | 605176 |
| 6 | 1.3.1.2 | Dihydropyrimidine dehydrogenase | Deficiency | Pyrimidine catab. | Cytosol | 612779 |
| 7 | 1.3.1.72 | 3β-Hydroxysterol Δ24-reductase | Desmosterolosis | Sterol biosyn. | ER membr. | 606418 |
| 8 | 1.3.3.1 | Dihydroorotate dehydrogenase | Miller syn. | Please specify the significance of footnote “a” cited in the Table 2, as a corresponding footnote text has not been provided. | mito. i. membr. | 126064 |
| 9 | 1.3.3.4 | Protoporphyrinogen IX oxidase | Variegate porphyria | Heme biosyn. | Mito. i. membr. | 600923 |
| 10 | 1.3.3.6 | Acyl-CoA oxidase | Deficiency | Lipid degr. | Peroxisomes | 609751 |
| 11 | 1.3.3.- | Glutaryl-CoA oxidase | Glutaric aciduria III | Glutaryl degr. | Peroxisomes | 231690 |
| 12 | 1.3.5.1 | Succinate dehydrogenase | Complex II deficiency | Citric acid cycle | mito. i. membr. | 600857 |
| Flavoprotein subunit A | Leigh syn. | |||||
| paraganglioma 5 | ||||||
| 13 | 1.3.99.2 | Short-chain- (butyryl-) acyl CoA dehydrogenase | Deficiency | β-Oxidation | mito. matrix | 201470 |
| 14 | 1.3.99.3 | Medium-chain acyl-CoA dehydrogenase | Deficiency | β-Oxidation | mito. matrix | 607008 |
| 15 | 1.3.99.7 | Glutaryl-CoA dehydrogenase | Glutaric acidemia | Lysine degr. | mito. matrix | 608801 |
| 16 | 1.3.99.10 | Isovaleryl-CoA dehydrogenase | Isovaleric acidemia | Leucine degr. | mito. matrix | 607036 |
| 17 | 1.3.99.12 | 2-Methylbutyryl-CoA dehydrogenase | Deficiency | Isoleucine degr. | mito. matrix | 600301 |
| 18 | 1.3.99.13 | Long-chain-acyl-CoA dehydrogenase | Deficiency | β-Oxidation | mito. matrix | 609576 |
| 19 | 1.3.99.- | Isobutyryl-CoA dehydrogenase | Deficiency | Valine degr. | mito. matrix | 611283 |
| 20 | 1.3.99.- | Long-chain-unsaturated-acyl-CoA dehydrogenase | Deficiency | β-Oxidation | mito. matrix | 611126 |
| 21 | 1.3.99.- | very long-chain acyl-CoA dehydrogenase | Deficiency | β-oxidation | mito. matrix | 201475 |
| 22 | 1.4.3.3 | d-amino acid oxidase | Schizophrenia? amyotrophic lateral sclerosis | Oxidation of d-serine | Peroxisomes | 124050 |
| 23 | 1.4.3.4 | Monoamine oxidase | Brunner syn. antisocial behaviour autism | Oxidation of neuro-transmitter | mito. o. membr. | 309850 |
| 24 | 1.4.3.5 | Pyridoxal 5’-phosphate oxidase | Encephalopathy | Vitamin B6 metab. | Cytosol | 603287 |
| Pyridoxine 5’-phosphate oxidase | ||||||
| 25 | 1.4.3.- | Catecholamine oxidase (renalase) | Hypertension? | Oxidation | Secreted (Blood) | 609360 |
| 26 | 1.5.1.20 | Methylenetetrahydrofolate reductase | Homocystinuria neural tube defects schizophrenia | Folate metab. | Cytosol | 607093 |
| 27 | 1.5.5.1 | Electron-transferring flavoprotein-ubiquinone oxidoreductase | Glutaric acidemia IIC | Electron transport | mito. i. membr. | 231675 |
| 28 | -------- | Electron transferring flavoprotein | Glutaric acidemia IIA | Electron transport | mito. matrix | 608053 |
| Glutaric acidemia IIB | Electron transport | 130410 | ||||
| 29 | 1.5.99.1 | Sarcosine dehydrogenase | Sarcosinemia | Choline degr. | mito. matrix | 604455 |
| 30 | 1.5.99.2 | Dimethylglycine dehydrogenase | DMGDH-deficiency | Choline degr. | mito. matrix | 605850 |
| 31 | 1.5.99.8 | Proline dehydrogenase | Hyperprolinemia type I schizophrenia | Amino acid metab. | 606810 | |
| 32 | 1.6.2.2 | Cytochrome-b5 reductase | Methemoglobinemia type I and II | Heme metab. | membr. soluble (erythro.) | 613213 |
| 33 | 1.6.2.4 | NADPH-hemoprotein reductase | Antley-Bixler syn. Disordered steroidogenesis | Electron donor to P450 enzymes | Microsomes (ER) | 124015 |
| (cytochrome P450 reductase) | ||||||
| 34 | 1.6.5.2 | NAD(P)H dehydrogenase (quinone) | Benzene toxicity breast cancer | Quinone detox p53 degr. | Cytosol | 125860 |
| 35 | 1.6.5.3 | NADH-ubiquinone oxidoreductase | Complex I deficiency | Electron transport | mito. i. membr. | 161015 |
| 36 | 1.8.1.4 | Dihydrolipoyl dehydrogenase | Leigh syn. Maple syrup urine dis., III | Energy metab. | mito. matrix | 238331 |
| 37 | 1.8.1.7 | Glutathione-disulfide reductase | Hemolytic anemia | detox. | Cytosol (erythro.) | 138300 |
| 38 | 1.8.3.2 | Sulfhydryl oxidase | Myopathy | Disulfide redox balance | mito. im. space | 600924 |
| 39 | 1.10.99.2 | Ribosyldihydronicotinamide dehydrogenase | Breast cancer susceptibility | Quinone detox. | Cytosol | 160998 |
| 40 | 1.14.13.8 | flavin-containing monooxygenases | trimethylaminuria | detox. | microsomes (ER) | 136132 |
| 41 | 1.14.13.39 | Nitric-oxide synthase | Hypertension | Vasodilation | Cytosol | 163729 |
| 163730 | ||||||
| 42 | 1.14.99.- | Monooxygenase in coenzyme Q biosyn. | Deficiency/nephrotic syn. | Coenzyme Q biosyn. | Golgi/mito. | 614647 |
| 43 | 1.16.1.8 | Methionine synthase reductase | Homocystinuria neural tube defects | Methionine biosyn. | Cytosol | 602568 |
| 44 | 1.-.-.- | NAD(P)H oxidase | Chronic granulomatous dis. | Generation of superoxide | Phagocytes | 300481 |
| Cytochrome b(558), beta subunit | Atypical mycobacteriosis | (membr.) | ||||
| 45 | 1.-.-.- | Thyroid oxidase/dual oxidase | Thyroid dishormonogenesis 6 | Thyroid biosyn. | membr. | 606759 |
| 46 | 2.5.1.26 | Alkyldihydroxyacetone phosphate synthase | Rhizomelic chondrodys-plasia punctata, type 3 | Lipid biosyn. | Peroxisomes | 603051 |
| 47 | -------- | Apoptosis inducing protein | Combinded oxidative phosphorylation deficiency | Redox control | mito./nucleus | 300169 |
| 48 | -------- | Iodotyrosine deiodinase | Thyroid dyshormonogenesis type 4 | Iodide salvage | Cytosol | 612025 |
| 49 | -------- | FAD-dependent oxidoreductase | complex I deficiency Leigh syn. | Molecular chaperone | mito. matrix | 613622 |
| 50 | -------- | Riboflavin transporter (member 3) | Brown-Vialetto-Van Laere syn. Fazio-Londe dis. | Riboflavin uptake | membr. | 613350 |
Abbreviations used: biosyn., biosynthesis; catab., catabolism; degr., degradation; detox., detoxification; dim., diminished; dis., disease; metabol., metabolism; mito., mitochondrium; mito. i. membr., inner membrane of mitochondria; mito. o. membr., outer membrane of mitochondria; mito. im. space, mitochondrial intermembrane space; ER, endoplasmic reticulum; membr., membrane; syn., syndrome; erythro., erythrocytes.
Since mutations are generally irreversible, adverse effects on the biological function of an encoded protein are untreatable and therapeutic interventions rely on protein substitution or, in the future, gene therapy. For several flavoproteins it was found that the mutation led to an amino acid exchange affecting the binding affinity of the flavin cofactor. In these cases it is conceivable that high-dose riboflavin supplementation may increase the concentration of flavin cofactors and this in turn may increase the fraction of active holo-enzyme. Ames and coworkers have compiled a list of flavoenzymes with decreased cofactor affinity where this strategy may be exploited successfully [3]. Among the flavoenzymes discussed are N-5,10-methylenetetrahydrofolate reductase (EC 1.5.1.20, MTHFR), NAD(P):quinone oxidoreductase (EC 1.6.5.2, NQO1), protoporphyrinogen IX oxidase (EC 1.3.3.4, PPOX), electron transferring flavoprotein (ETF), electron-transferring flavoprotein ubiquinone oxidoreductase (EC 1.5.5.1, ETFDH), glutaryl-CoA oxidase (EC 1.3.3.-, C7orf10), short-, medium-, long-chain acyl-CoA dehydrogenases (EC 1.3.99.2, ACADS; EC 1.3.99.3, ACADM; EC 1.3.99.13, ACADL) and complex I (EC 1.6.5.3, NDUFV1) [3]. More recently, it was shown that the neurological disorders Brown–Vialetto–Van Laere and Fazio-Londe syndrome were caused by mutations in the gene encoding an intestinal riboflavin transporter [4,5]. Since this transporter is largely unexplored, the effect of the mutation on riboflavin transport efficiency is currently unknown. However, it appears that at least in some patients riboflavin supplementation ameliorates the symptoms and positively affects disease progression [4,6,7]. Apart from a few flavoproteins with decreased affinity for cofactor mentioned by Ames and coworkers [3] it is largely unknown how mutations in genes encoding flavoproteins impact cofactor binding. In view of the large number of flavoproteins involved in human diseases it is worthwhile investigating the mutational effects on cofactor binding to thoroughly evaluate the benefits of high-dose riboflavin supplementation therapy (see also discussion below).
Flavoenzymes in cofactor biogenesis and metabolism
FMN and FAD are synthesized in the human organism from riboflavin (vitamin B2) by riboflavin kinase (EC 2.7.1.26) and FAD-adenylyl transferase (EC 2.7.7.2). Riboflavin is absorbed from nutrients as is the case for many other vitamins required to supply vitamin-derived coenzymes to the apo-forms of hundreds of enzymes in the human body. In this context, we were intrigued by the number of flavoenzymes that are involved in the biosynthesis of other cofactors such as coenzyme A, coenzyme Q (ubiquinone), heme and pyridoxal 5′-phosphate. Equally, flavoenzymes participate in the interconversion of various folate metabolites and hence play an important role in one-carbon (C1-unit) metabolism. Moreover, flavoenzymes catalyze essential reactions in biosynthetic pathways leading to cell signaling molecules such as the steroid and thyroid hormones. These topics are discussed in more depths in the next sections.
Flavoenzymes in folate and cobalamin metabolism
Tetrahydrofolate (THF) is a 6-methylpterin derivative that is widely used to shuttle C1-units in metabolic reactions. THF loaded with a C1-unit occurs in various oxidation states such as N-5,10-methylene-THF and N-5-methyl-THF. Two FAD-dependent enzymes, dimethylglycine dehydrogenase (EC 1.5.99.2, DMGDH) and sarcosine dehydrogenase (EC 1.5.99.1, SARDH), generate N-5,10-methylene-THF from THF which is then converted to N-5-methyl-THF by N-5,10-methylene-THF reductase (EC 1.5.1.20, MTHFR) at the expense of NADPH (Fig. 1). N-5-methyl-THF in turn is utilized by methionine synthase (EC 2.1.1.13, MS) yielding THF and methionine by transferring the methyl group to homocysteine. The cobalamine cofactor of MS is susceptible to oxidation and hence methionine synthase reductase (EC 1.16.1.8, MSR) is required to regenerate methylcob(I)alamin from cob(II)alamin. Hence, these four flavin-dependent enzymes play a role in the inter conversion of THF, N-5,10-methylene-THF and N-5-methyl-THF and are thus important to balance the pools of folate metabolites (Fig. 1).
Fig. 1.

Flavin-catalyzed reactions connected to folate metabolism: dimethlyglycine dehydrogenase (EC 1.5.99.1, DMGDH), sarcosine dehydrogenase (EC 1.5.99.2, SARDH), N-5,10-methylene-tetrahydrofolate reductase (EC 1.5.1.20, MTHFR) and methionine synthase reductase (EC 1.16.1.8, MSR). Structures depicted are from the human methionine synthase reductase (2qtl) as well as the N-5,10-methylene-tetrahydrofolate reductase from Thermus thermophilus HB8 (3apt).
DMGDH and SARDH also catalyze two consecutive reactions in choline catabolism (Fig. 1). DMGDH oxidizes dimethlyglycine to sarcosine, which is further oxidized to glycine. Both enzymes use THF to capture the C-1 fragment released by the oxidative demethylation of substrates yielding N-5,10-methylene-THF [8]. Since these reactions occur in the mitochondrial matrix, electrons extracted from the substrate are delivered to the mitochondrial electron transport chain via electron transferring flavoprotein (ETF) and electron transferring flavoprotein ubiquinone oxidoreductase (EC 1.5.5.1). DMGDH and SARDH were reported to contain a covalently linked 8α-(N3-His)-FAD cofactor [9,10] and hence belong to the small group of flavoproteins that bind the cofactor covalently (see Table 1). Unfortunately, the structure of these proteins is not known and therefore neither the mode of FAD binding nor the interaction with THF can be firmly established. Interestingly, a recently discovered lysine-specific histone demethylase (EC 1.5.99.-, KDM1A) related to flavin-dependent amine oxidases apparently does not utilize THF to capture the formyl group and hence the demethylation results in the generation of formaldehyde [11]. Since the lysine demethylation occurs in the nucleus and not in mitochondria, the enzyme donates the electrons to oxygen generating hydrogen peroxide as a by-product of the demethylation reaction. The fate of these two potentially harmful compounds in the nucleus is currently not known. A recent study showed that inhibitors of KDM1A selectively target cancer cells with pluripotent stem cell properties [12]. In light of these recent findings it can be concluded that aberrations in KDM1A regulation might lead to development of cancer.
Deficiency of DMGDH is apparently very rare and only a single case was reported so far (OMIM 605850, [13,14]). Binzak et al. identified a homozygous point mutation in a patient (326 A–G) resulting in a histidine to arginine exchange (H109R) in the encoded DMGDH [13]. It is noteworthy that the covalent linkage of the FAD cofactor is to H91 and hence the inactivity of the altered enzyme is probably not due to the inability to form the covalent linkage with the FAD cofactor. Detailed studies with the recombinant DMGDH H109R variant showed effects on both the specific activity (27 times lower) and Km (65 times higher) [15]. Interestingly, fish odor was reported as a salient symptom of DMGDH deficiency which is typically ascribed to impaired N-oxygenation of xenobiotics due to deficiency of flavin-containing monooxygenase isoform 3 (EC 1.14.13.8, FMO3) [16].
Sarcosinemia caused by a deficiency of SARDH [17] is also a rare metabolic disorder characterized by elevated levels of sarcosine in plasma and urine. The mutational frequency in the human SARDH gene apparently varies from 1:350000 to 1:3414. The clinical symptoms reported to be associated with SARDH deficiency include mental retardation and loss of speech (OMIM 268900). Recently, sarcosine was suggested to be a marker for the invasiveness of prostate cancer cell lines [18]. In the same study, it was also noted that reduced levels of SARDH activity led to the induction of an invasive phenotype in benign prostate epithelial cells. The connection of SARDH to cancer development and progression indicated by this study and the usefulness of sarcosine as a tumor marker is currently the subject of an intense scientific debate [19–21].
MTHFR connects the pool of N-5,10-methylene-THF, mainly derived from the glycine cleavage system (EC 2.1.2.10) and serine hydroxymethyltransferase (EC 2.1.2.1), with that of N-5-methyl-THF, which is used by MS to generate methionine from homocysteine (Fig. 1). This reaction not only provides an important building block for protein biosynthesis but also a substrate for the synthesis of S-adenosyl-methionine (SAM), an ubiquitous and powerful reagent in many biological methylation reactions. The MS reaction also regenerates THF, which then re-enters the reactions mentioned above capturing C1-units from glycine, serine, dimethylglycine and sarcosine. More than thirty deleterious mutations of the MTHFR gene are known as well as several common variants, like the 677C>T point mutation. MTHFR deficiency is connected to several serious diseases, such as neural tube defects, coronary heart disease and schizophrenia (OMIM 607093). Depending on the severity of the mutation hyperhomocysteinemia with homocystinuria or mild hyperhomocysteinemia is observed. The 677C>T polymorphism is of particular interest as it is recognized as the most frequent genetic cause of homocysteinemia [22,23]. This common C to T mutation gives rise to a conservative amino acid replacement in position 222 of MTHFR (A222V). Surprisingly, the A222V variant possesses reduced thermostability and weaker affinity to the FAD cofactor. In the same study it was also reported that folate (and adenosylmethionine) increases the affinity of FAD, which in turn increases the thermostability of the A222V variant [24]. This positive interplay suggests that the status of folate and riboflavin may be critical in cases where cofactor affinity is compromised by the amino acid exchange (see discussion above).
Mechanistically, MS uses N-5-methyl-THF to methylate its cob(I)alamin cofactor which in turn transfers the methyl group to the thiol group of homocysteine (Fig. 1). The cob(I)alamin state is highly sensitive to oxidation rendering the enzyme inactive [25]. In order to restore the reduced active form of the cofactor, cob(II)alamin is reductively methylated by MSR using NADPH as electron source and SAM as methyl group donor [26,27]. Since MSR is required to maintain MS activity, it is not surprising that allelic variants resulting in MSR deficiency present similar symptoms, such as homocystinuria, as seen in MS and MTHFR deficiency. Accordingly, megaloblastic anemia, an increased risk for neural tube defect and Down syndrome are among the disorders caused by inherited MSR deficiency (OMIM 602568). A common polymorphism found in the MSR gene (allele frequency 0.51) results in a single amino acid replacement, I23M, and increases the risk for neural tube defects [25,28].
Recently, Matthews and coworkers suggested that MSR also plays a role as a chaperone for MS and also acts as an aquacobalamin reductase [29]. They reported that MSR stabilizes the apo-form of MS and promotes the association with methylcobalamin. In addition, MSR catalyzes the NADPH-dependent reduction of aquacobalamin to cob(II)alamin and thereby accelerates the formation of holo-MS. Hence, MSR exhibits multiple beneficial effects on MS. In this context, it is interesting to note that MSR was also invoked as catalyst for the formation of adenosylcobalamin [25,30]. Recent evidence, however, suggests that reduction of Co2+ to Co+ occurs by free dihydroflavins when cob(II)alamin is bound to human adenosyltransferase [31]. It is currently unknown whether this process is driven solely by free dihydroflavins or involves a specialized flavoprotein reductase in vivo.
Heme biosynthesis
The biosynthesis of heme from succinyl-CoA and glycine is initiated in the mitochondria and then proceeds in the cytosol to generate coproporphyrinogen III. This intermediate is transported back into mitochondria to complete the oxidation of the macrocycle by the FAD-dependent protoporphyrinogen IX oxidase (EC 1.3.3.4, PPOX). This reaction involves the six-electron oxidation of the methylene groups linking the pyrrole rings to methenyl groups thereby generating an extensively conjugated π-electron system (Fig. 2). As the isoalloxazine ring system can only handle two electrons at a time, the cofactor needs to run three times through the catalytic cycle of flavin reduction and reoxidation to complete the oxidation of protoporphyrinogen IX to protoporphyrin IX. This catalytic cycling was suggested to proceed by substrate oxidation and release of the dihydro- and tetrahydro-intermediates rather than continuous processing of a constantly bound substrate [32]. PPOX is located in the inner mitochondrial membrane and anchored by acylation to the leaflet oriented towards the intermembrane space [33,34]. Diminished PPOX activity results in variegate porphyria (VP), which belongs to a group of metabolic disturbances caused by genetic defects affecting heme biosynthesis (collectively called as porphyrias). Symptoms of autosomal dominant VP include acute abdominal pain, neurological manifestations and/or cutaneous photosensitivity [35]. Apparently, the disease is characterized by severe, sometimes life-threatening, crisis triggered by external factors (e.g. medication, toxins) [36]. Perhaps the most controversial case of VP was postulated for King George III (1738–1820) who suffered long episodes of mental and physical illness culminating in the Regency crisis (1788–1789). According to Macalpine & Hunter, King George III was afflicted by VP, a claim supported by several pieces of evidence collected on ancestors and descendants [37,38]. More recently, Cox et al. suggested that high concentrations of arsenic in the medication administered to the king may have triggered the episodes of VP [39].
Fig. 2.

The penultimate reaction in heme biosynthesis involves the six-electron oxidation of protoporphyrinogen-IX to protoporphyrin-IX by the FAD-dependent protoporphyrinogen-IX oxidase (EC 1.3.3.4, PPOX) (human structure: 3nks).
The structure of the human enzyme was solved to 1.9 Å resolution (pdb code 3nks). Forty-seven variants of PPOX that were found to cause VP in humans were heterologously expressed in Escherichia coli and the properties of the variants studied in vitro [40]. Based on the observed effects and the locus of the amino acid in the structure of the enzyme the authors of that study classified the variants according to their proposed effect on either FAD- or substrate binding or structural alterations of the protein. Interestingly, the Human Gene Mutation Database (www.hgmd.cf.ac.uk) lists more than 130 mutations, which appear to be uniformly distributed over the entire length of the gene and concern invariant amino acids as well as regions of variability.
Pyridoxal 5′-phosphate biosynthesis
Pyridoxal 5′-phosphate (PLP) serves as a cofactor in more than 140 distinct enzymatic activities and is arguably one of the most utilized vitamin in nature. According to a recent analysis, the human genome contains 68 genes encoding proteins with a structural fold typical for PLP-dependent enzymes [41]. Although functional assignment of these enzymes was not possible in all cases (14 remained unassigned) this result indicates that the supply of PLP is critical for the maintenance of numerous metabolic activities especially in pathways involving biochemical transformations of amino acids, e.g. PLP-dependent decarboxylations of amino acids to generate active amines and neurotransmitters. In humans, PLP is mainly generated by enzymatic phosphorylation and oxidation of pyridoxine and pyridoxamine. These two vitamers are available either from nutritional sources or are produced in the course of the degradation of PLP-containing enzymes. In the first step, they are phosphorylated by pyridoxal kinase and then oxidized by pyridoxine 5′-phosphate oxidase (EC 1.4.3.5, PNPO) to the active cofactor PLP (Fig. 3). The three-dimensional structures of the enzyme from several bacteria as well as the recombinant human enzyme were determined by X-ray crystallography ([42], pdb code 1nrg). Interestingly, the structure of the E. coli enzyme features two PLP binding sites, one in the active site near the flavin’s isoalloxazine ring and a second closer to the surface of the dimeric protein ([43], pdb code 1g79). It is currently unclear whether the latter binding site is identical to the tight binding site identified by functional studies [43]. The human enzyme (261 amino acids) presumably also features a second tight binding site for PLP, however its exact location is currently not known [44]. Di Salvo et al. suggested that this second tight binding site might serve as transient storage for PLP, which is channeled to the apo-forms of PLP-dependent enzymes [44]. In support of this hypothesis, these authors reported that PNPO from E. coli interacts with several apo-forms of PLP-dependent enzymes with dissociation constants in the micromolar range (0.3–56 μM; [44]). Although this appears to be an attractive process for the delivery of PLP to its target enzymes, it is unlikely that this is applicable to all human PLP-dependent enzymes because it would require a common docking site between the single PNPO and the 68 predicted enzymes. On the other hand, PNPO’s cytosolic localization would enable the enzyme to deliver PLP directly to the apo-form of PLP-depenent enzymes in statu nascendi.
Fig. 3.

Oxidation of pyridoxamine and pyridoxine 5′-phosphate to pyridoxal 5′-phosphate by the FMN-dependent pyridoxamine/pyridoxine 5′-phosphate oxidase (EC 1.4.3.5, PNPO), the last step of the PLP-biosynthesis. The structure shown is that of the human enzyme (1nrg).
Several allelic variants of the PNPO gene were described (OMIM 603287). The reported mutations either lead to premature termination of protein biosynthesis (at position 174 [45]), extension of the protein by 28 amino acids (X262N) or splicing errors due to a mutation in intron 3 [46]. In addition, Mills et al., reported a point mutation in the PNPO gene resulting in the substitution of arginine in position 229 with tryptophan [46]. This R229W variant was recombinantly expressed and characterized by Musayev et al. [47]. They found that this variant is substantially compromised in its catalytic efficiency (ca. 850-fold) due to weaker binding of the substrate and decreased catalytic activity. Moreover, FMN possessed a 50-fold reduced affinity to the variant (see discussion above). These effects could be rationalized based on the involvement of the arginine residue in organizing important interactions in the active site of the enzyme. All of the reported mutations caused severe deficiency of PNPO activity resulting in neonatal epileptic encephalopathy. This disorder has a very poor prognosis postnatally and surviving children typically suffer from mental retardation.
PNPO expression in humans is prevalent in liver and kidney with most other tissues (brain, heart, muscle) having clearly reduced levels of mRNA [48]. Interestingly, abnormally low levels of PNPO activity were reported for some neoplastic cell lines, e.g. liver and neurally-derived tumors [49]. Whether and how these low PNPO activities are connected to tumorigenesis remains to be investigated.
Coenzyme A biosynthesis
Coenzyme A (CoA) biosynthesis from pantothenic acid (vitamin B5) is a five-step enzymatic process. The third reaction in this universal reaction sequence is the decarboxylation of phosphopantothenoylcysteine to 4′-phosphopantetheine by phosphopantothenoylcysteine decarboxylase (EC 4.1.1.36; PPCD) as shown in Fig. 4 [50]. The structure of the human enzyme shows a non-covalently bound FMN per protomer of the trimeric protein [51]. In eukaryotes PPCD occurs as a monofunctional enzyme whereas in most bacteria – with the exception of streptococci and enterococci – PPCD is fused with phosphopantothenoylcysteine synthase (PPCS) the second enzyme of CoA biosynthesis [50]. In contrast to the reactions described above, the decarboxylation does not involve a net redox change. PPCD is one of the few examples where the flavin is not used for a reduction–oxidation reaction in a human flavoprotein (Table 1, entry #67). However, the flavin apparently plays a role as a transient electron acceptor during the reaction involving the transfer of charge from the substrate thiolate group to the isoalloxazine ring [52]. Currently, the OMIM database does not list any diseases related to a deficiency of PPCD. This may be partly due to the fact that approximately 4% of all enzymes utilize substrates linked to coenzyme A (e.g. acyl-CoAs) [53] and hence a deficiency in coenzyme A seriously compromises the viability of cells. This notion is supported by the fact that only the initial enzyme of coenzyme A biosynthesis, pantothenate kinase (EC 2.7.1.33, PANK), is linked to inherited diseases (OMIM 606157) but none of the other enzymes required. Interestingly, four isoforms of PANK were discovered in the human genome and therefore it appears likely that a deficiency of one isoform may be compensated at least partially by the others [54].
Fig. 4.

Decarboxylation of N-[(R)-4′-phosphopantothenoyl]-L-cysteine to pantotheine 4′-phosphate by the FMN-dependent 4′-phosphopantothenoylcysteine decarboxylase (EC 4.1.1.36, PPCDC). The structure shown is that of the human enzyme (1qzu).
Coenzyme Q biosynthesis
Coenzyme Q or ubiquinone is an essential electron carrier in the mitochondrial electron transport chain shuttling electrons from complex I and II to complex III (see also section below). In contrast to vitamin-derived coenzymes, ubiquinone is synthesized de novo from aromatic precursors such as p-hydroxybenzoic acid. In 2011, Pierrel and coworkers identified a FAD-dependent monooxygenase encoded by coq6, for the required hydroxylation in 5-position of a ubiquinone precursor in the yeast Saccharomyces cerevisiae (Fig. 5) [55]. A human homolog of coq6 was recently discovered, which apparently catalyzes the hydroxylation in human ubiquinone biosynthesis (Table 1, entry #59) [56]. Flavin-dependent monooxygenases (hydroxylases) require a source of electrons for reduction of the flavin’s isoalloxazine ring in order to enable the generation of hydroxylating 4a-hydroperoxy intermediates. Based on the mode of flavin reduction, internal and external monooxygenases are distinguished which either use NAD(P)H or NADH directly for reduction or rely on the activity of an external NAD(P)H/NADH:FAD reductase to supply the reduced flavin cofactor [57]. In the case of yeast Coq6p, reduction of FAD is accomplished by ferredoxin reductase (termed Arh1) and ferredoxin (termed Yah1) at the expense of NADPH and thus has the character of an external reduction mode. It is currently unknown whether the human enzyme is reduced by the same mechanism. The human ortholog of coq6 was discovered in search of the genetic cause of nephrotic syndrome [56] and several reports of coenzyme Q deficiency are compiled in the OMIM (614647).
Fig. 5.
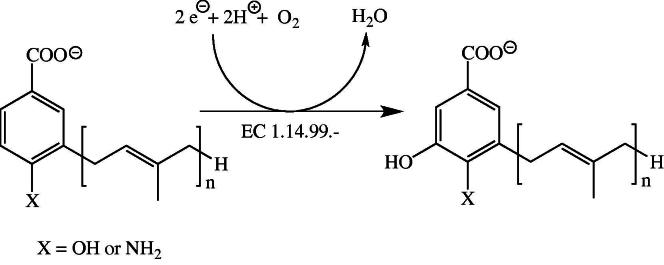
Hydroxylation of the ubiquinone precursors in 5-position of the aromatic system by COQ6 (EC 1.14.99.-). In yeast, the electrons required for reduction of dioxygen are supplied by an NADPH-dependent ferredoxin reductase-ferredoxin system.
Steroid biosynthesis
Steroid hormones play fundamental roles in developmental programs and homeostatic processes. Cholesterol, a central precursor for the biosynthesis of steroid hormones, bile acids and vitamin D, is synthesized from acetyl-CoA via the mevalonate pathway in a multistep pathway [58]. A crucial step in the biosynthesis is the cyclisation of the linear 2,3-oxidosqualene to the first tetracyclic ring system, lanosterol. The preceding reaction, catalyzed by squalene monooxygenase (EC 1.14.13.132, SQLE), enables this cyclisation by epoxidation of the 2,3-carbon–carbon double bond (Fig. 6, top) [59]. The oxygen atom introduced by SQLE remains in the molecule during processing to the steroid target structures and is important for the physical (amphiphilic character) and chemical properties (formation of esters). Although SQLE does not appear to be linked to an inherited disease, it was suggested earlier that the gene encoding SQLE is a candidate for Langer–Giedion syndrome, which is associated with mental retardation and microcephaly [60]. Interestingly, SQLE became a focus as a drug target for antimycotic compounds in the 1980s [61]. More recently it is also discussed as a potentially useful target in hypercholesterolemic therapy [62,63]. Currently, treatment of hypercholesterolemia is dominated by statins, which inhibit 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase (EC 1.1.1.34), a central and rate-limiting enzyme in the mevalonate pathway. A major drawback of HMG-reductase inhibition results from the adverse effects on the biosynthesis of non-steroidal isoprenoids (e.g. ubiquinone) since the enzyme catalyzes an “early” step in the pathway. To alleviate this problem steps occurring after the committing reaction of steroid biosynthesis, i.e. the synthesis of squalene from farnesylpyrophosphate catalyzed by squalene synthase (EC 2.5.1.21) might be potentially useful targets for the design of new cholesterol lowering compounds. Because SQLE is the next enzyme of this metabolic branch point it is a suitable point of intervention to reduce the biosynthesis of cholesterol [62,63].
Fig. 6.

Reactions of the two FAD-dependent enzymes in cholesterol biosynthesis. The reaction shown on top involves the insertion of an oxygen atom (blue circle) by SQLE (EC 1.14.13.132) and the reaction shown on the bottom the reduction of the side chain double bond (green circle) by DHCR24 (EC 1.3.1.72).
The ultimate reaction in cholesterol biosynthesis, the reduction of desmosterol, is catalyzed by 3β-hydroxysterol Δ24-reductase (EC 1.3.1.72, DHCR24). In contrast to SQLE, DHCR24 does not activate dioxygen for insertion into the substrate, but simply reduces the side-chain double bond at the expense of NADPH (Fig. 6, bottom). The gene encoding DHCR24 is a human homolog (also termed seladin-1) of the DIMINUTO/DWARF1 gene found in plants and Caenorhabditis elegans [64]. Several mutations in the human gene are known which result in reduced enzyme activity leading to desmosterolosis (OMIM 606418). Some of the mutations discovered are in or near the FAD binding site and hence may affect FAD binding. In other cases, however, the mutation is in less conserved areas and their effect on protein structure, folding or stability is unclear. Symptoms of the disease present at or shortly after birth and show a diverse range of developmental anomalies, such as failure to thrive, micro- or macrocephaly, psychomotor retardation, spasticity and seizures. The observed developmental and neurological defects correspond to the high expression of the gene in neuronal cells [64]. More recently it was also shown that DHCR24 is important for long bone growth in mice indicating that the enzyme’s activity is also critical for the development of other tissues [65].
Thyroxine biosynthesis and iodine salvage
The thyroid gland produces two iodinated tyrosine-derived hormones, triidothyronine (T3) and thyroxine (T4), which stimulate metabolism in most tissues. The initial biosynthetic reaction involves incorporation of iodine into tyrosine residues of thyroglobulin. In the next step, two neighboring iodotyrosine residues are oxidatively coupled and the active hormones, T3 and T4, are released by proteolytic cleavage of the precursor protein. The iodination and coupling reaction both require hydrogen peroxide, which is provided by two FAD-dependent thyroid oxidases (termed DUOX1 and 2) [66,67]. During turnover, FAD is reduced at the expense of NADPH and then reoxidized by dioxygen yielding hydrogen peroxide (Fig. 7). Several allelic variants were reported for DUOX2 leading to thyroid dyshormonogenesis 6 (OMIM 606759). How the resulting single amino acid exchange in the observed variants affect enzyme function is currently not known.
Fig. 7.

Role of DUOX1 and 2 in the biosynthesis of thyroxine. The iodination of tyrosine residues and the coupling of two iodinated tyrosines require hydrogen peroxide, which is provided by the oxidation of reduced FAD with molecular dioxygen.
During the production of T3 and T4 substantial amounts of mono- and diiodotyrosine are released [68]. Because iodine is a precious trace element, it is recycled in a single reductive step catalyzed by an FMN-dependent dehalogenase (IYD, Fig. 8 [69]). The released iodine can then be reused by thyroperoxidase for incorporation into thyroglobulin (see above). Several allelic variants were reported for IYD, which severely compromise the dehalogenase activity of the enzyme leading to hypothyroidism (dyshormonogenesis 4, OMIM 612025) [70,71].
Fig. 8.
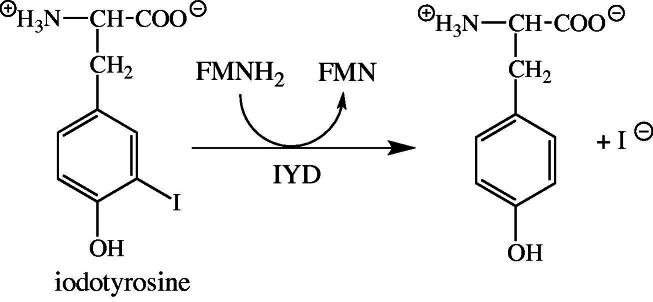
Dehalogenation of iodotyrosine catalyzed by IYD. The mono- and diiodotyrosine residues released from thyroglobulin are substrates of the enzyme. Reductively released iodine is then reused by thryoperoxidase for incorporation into tyrosine residues of thyroglobulin.
Flavoproteins providing assistance to other flavoproteins: Assembly of complex I
The human respiratory electron transport chain relies on complex I for electron transfer from NADH to ubiquinone coupled with proton translocation across the inner mitochondrial membrane. In the initial reaction NADH reduces the FMN cofactor in the NDUFV1 subunit (EC 1.6.5.3) of complex I, which in turn passes the electrons to iron-sulfur clusters. The formation of complex I, which consists of at least 36 nuclear- and seven mitochondrial-encoded subunits (OMIM 252010), requires several assembly factors, such as NDUFAF1 and NUBPL [72,73]. In addition, two FAD-dependent proteins recently emerged as critical factors for complex I assembly, ACAD9 and FOXRED1 (Table 1, entries #24 and #73, respectively). The exact role of the FAD-dependent OXidoREDuctase (FOXRED1) in assembly of complex I is not fully understood at the moment but it was clearly shown that it is essential for complex I activity in the inner mitochondrial membrane [72,74]. A BLASTp search identified two human FAD-dependent flavoproteins, SARDH and DMGDH, as related (22% and 21% identity, respectively) enzymes to FOXRED1. FOXRED1 comprises 486 amino acids and thus is much shorter than SARDH and DMGDH with 918 and 866 amino acids, respectively. The similarity of FOXRED1 to SARDH and DMGDH is found in the N-terminal part of the two dehydrogenases where FAD is covalently bound to a histidine. This amino acid residue is not conserved in FOXRED1 indicating that the FAD cofactor may not be covalently bound.
The discovery of FOXRED1 was closely associated with the search to identify the mechanism of complex I deficiency, leading for example to mitochondrial encephalopathy. Fassone et al. demonstrated that a point mutation in the FOXRED1 gene led to an amino acid exchange (R352 W) in the putative FAD binding site [74]. According to their molecular model of the protein this amino acid exchange could prevent FAD binding and hence compromises the assumed chaperone activity of the protein.
ACAD9 (EC 1.3.99.-) was first described as an acyl-CoA dehydrogenase specific for long-chain unsaturated fatty acids [75,76]. Recent evidence however questions the involvement of ACAD9 in the degradation of long-chain fatty acids in the mitochondrial matrix and suggests a role in biogenesis of complex I instead [77]. Although recombinant ACAD9 possesses dehydrogenase activity in vitro, mutations in ACAD9 result in complex I deficiency rather than a disturbance of long-chain fatty acid metabolism [77].
Concluding remarks
Our analysis of the human flavoproteome has led to the identification of several areas where further research is required. Because riboflavin must be supplied by the diet uptake in the human intestine is an important process to make the vitamin available for the synthesis of FMN and FAD. Three poorly characterized transporters seem to be responsible for riboflavin uptake in the human intestine. The relative contribution and role of these transporters is currently unclear and warrants further investigations. A better understanding of the uptake processes may also have therapeutic benefits as some inherited diseases feature reduced flavin affinity to the affected flavoproteins and thus are potentially treatable by riboflavin supplementation. Equally, little is known about flavin homeostasis and the processes leading to degradation and excretion of flavins.
Although many human flavoprotein structures were solved experimentally by X-ray crystallography or modeled using homologous structures several flavoproteins still elude structural characterisation. Many of these are associated with cellular membranes and are therefore more difficult to obtain by recombinant expression in heterologous hosts. Moreover, membrane integral or associated proteins are challenging for structure elucidation by X-ray crystallography due to their lipophilic character. In view of the important roles of membrane-associated flavoproteins in humans, the potential as drug targets (e.g. SQLE) and the involvement in diseases (e.g. DUOX1/2, IYD) determination of the “missing” structures are certainly rewarding goals.
We found that many genes encoding flavoproteins occur as allelic variants with the potential to cause severe human diseases (Table 2). The number of allelic variants covers a wide range from only a few to more than one hundred. With the exception of a few systematic studies our knowledge of the effect of the mutation on stability, structure and activity of the flavoprotein is incomplete. This lack of insight should be improved by detailed analyses of mutational effects on flavoprotein properties. In light of many indications that flavoprotein variants suffer from a reduced affinity to their cognate flavin cofactor this seems to be of particular importance because riboflavin supplementation may help to remedy symptoms in affected individuals.
Methods
The names and gene abbreviations of human flavoproteins, compiled for a previously published review article [2], were used to search for inherited diseases in the Online Mendalian Inheritance in Man data base (OMIM; http://www.ncbi.nlm.nih.gov/omim). This search constituted the basis for Table 2 where the most relevant entry in OMIM is provided as a six-digit number (right column). The reader is referred to these entries for a complete list of references pertaining to the diseases mentioned in this article.
Acknowledgments
We thank the Austrian Research Fund (FWF) for financial support through project P22361 and the PhD program “Molecular Enzymology” (W901).
Footnotes
Abbreviations used: ACAD9acyl-CoA dehydrogenase isoform 9; ACADS, short-chain acyl-CoA dehydrogenase; ACADL, long-chain acyl-CoA dehydrogenase; ACADM, medium-chain acyl-CoA dehydrogenase; DHCR24, 3β-hydroxysterol Δ24-reductase; DMGDH, dimethylglycine dehydrogenase; ETF, electron transferring flavoprotein; ETFDH, electron-transferring flavoprotein ubiquinone oxidoreductase; FMO3, flavin-containing monooxygenase isoform 3; FOXRED1, FAD-dependent oxidoreductase; MSmethionine synthase; MTHFR, N-5,10-methylene-tetrahydrofolate reductase; MTRR, methionine synthase reductase; OMIM, Online Mendelian Inheritance in Man; MAOA, monoamine oxidase isozyme A; MAOB, monoamine oxidase isozyme B; MS, methionine synthase; MSR, methionine synthase reductase; MTHFR, N-5,10-methylenetetrahydrofolate reductase; NQO1, NAD(P):quinone oxidoreductase; PANK, panthothenate kinase; PDB, protein data base; PLP, pyridoxal 5′-phosphate; PNPO, pyridoxal 5′-phosphate oxidase; PPOX, protoporphyrinogen IX oxidase; PPCD, 4′-phosphopantothenoylcysteine decarboxylase; PPCS, phosphopantothenoylcysteine synthase; SARDH, sarcosine dehydrogenase; SQLE, squalene monooxygenase; THF, tetrahydrofolate; VP, variegate porphyria.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.abb.2013.02.015.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Chastain J.L., McCormick D.B. Am. J. Clin. Nutr. 1987;46:830–834. doi: 10.1093/ajcn/46.5.830. [DOI] [PubMed] [Google Scholar]
- 2.Macheroux P., Kappes B., Ealick S.E. FEBS J. 2011;278:2625–2634. doi: 10.1111/j.1742-4658.2011.08202.x. [DOI] [PubMed] [Google Scholar]
- 3.Ames B.N., Elson-Schwab I., Silver E.A. Am. J. Clin. Nutr. 2002;75:616–658. doi: 10.1093/ajcn/75.4.616. [DOI] [PubMed] [Google Scholar]
- 4.Bosch A.M., Abeling N.G., Ijlst L., Knoester H., van der Pol W.L., Stroomer A.E., Wanders R.J., Visser G., Wijburg F.A., Duran M. J. Inherit. Metab. Dis. 2011;34:159–164. doi: 10.1007/s10545-010-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green P., Wiseman M., Crow Y.J., Houlden H., Riphagen S., Lin J.P., Raymond F.L., Childs A.M., Sheridan E., Edwards S. Am. J. Hum. Gen. 2010;86:485–489. doi: 10.1016/j.ajhg.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anand G., Hasan N., Jayapal S., Huma Z., Ali T., Hull J., Blair E., McShane T., Jayawant S. Dev. Med. Child Neurol. 2012;54:187–189. doi: 10.1111/j.1469-8749.2011.04142.x. [DOI] [PubMed] [Google Scholar]
- 7.Koy A., Pillekamp F., Hoehn T., Waterham H., Klee D., Mayatepek E., Assmann B. Pediatr. Neurol. 2012;46:407–409. doi: 10.1016/j.pediatrneurol.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Wittwer A.J., Wagner C. J. Biol. Chem. 1981;256:4102–4108. [PubMed] [Google Scholar]
- 9.Cook R.J., Misono K.S., Wagner C. J. Biol. Chem. 1984;259:12475–12480. [PubMed] [Google Scholar]
- 10.Cook R.J., Misono K.S., Wagner C. J. Biol. Chem. 1985;260:12998–13002. [PubMed] [Google Scholar]
- 11.Forneris F., Battaglioli E., Mattevi A., Binda C. FEBS J. 2009;276:4304–4312. doi: 10.1111/j.1742-4658.2009.07142.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang J., Lu F., Ren Q., Sun H., Xu Z., Lan R., Liu Y., Ward D., Quan J., Ye T. Cancer Res. 2011;71:7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binzak B.A., Wevers R.A., Moolenaar S.H., Lee Y.M., Hwu W.L., Poggi-Bach J., Engelke U.F., Hoard H.M., Vockley J.G., Vockley J. Am. J. Hum. Gen. 2001;68:839–847. doi: 10.1086/319520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moolenaar S.H., Poggi-Bach J., Engelke U.F., Corstiaensen J.M., Heerschap A., de Jong J.G., Binzak B.A., Vockley J., Wevers R.A. Clin. Chem. 1999;45:459–464. [PubMed] [Google Scholar]
- 15.McAndrew R.P., Vockley J., Kim J.J. J. Inherit. Metab. Dis. 2008;31:761–768. doi: 10.1007/s10545-008-0999-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treacy E.P., Akerman B.R., Chow L.M., Youil R., Bibeau C., Lin J., Bruce A.G., Knight M., Danks D.M., Cashman J.R. Hum. Mol. Gen. 1998;7:839–845. doi: 10.1093/hmg/7.5.839. [DOI] [PubMed] [Google Scholar]
- 17.Eschenbrenner M., Jorns M.S. Genomics. 1999;59:300–308. doi: 10.1006/geno.1999.5886. [DOI] [PubMed] [Google Scholar]
- 18.Sreekumar A., Poisson L.M., Rajendiran T.M., Khan A.P., Cao Q., Yu J., Laxman B., Mehra R., Lonigro R.J., Li Y. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 19.Bohm L., Serafin A.M., Fernandez P., Van der Watt G., Bouic P.J., Harvey J. S. Afr. Med. J. 2012;102:677–679. doi: 10.7196/samj.5768. [DOI] [PubMed] [Google Scholar]
- 20.Issaq H.J., Veenstra T.D. J. Sep. Sci. 2011;34:3619–3621. doi: 10.1002/jssc.201100572. [DOI] [PubMed] [Google Scholar]
- 21.Lucarelli G., Fanelli M., Larocca A.M., Germinario C.A., Rutigliano M., Vavallo A., Selvaggi F.P., Bettocchi C., Battaglia M., Ditonno P. Prostate. 2012;72:1611–1621. doi: 10.1002/pros.22514. [DOI] [PubMed] [Google Scholar]
- 22.Frosst P., Blom H.J., Milos R., Goyette P., Sheppard C.A., Matthews R.G., Boers G.J., den Heijer M., Kluijtmans L.A., van den Heuvel L.P. Nat. Gen. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 23.Ueland P.M., Hustad S., Schneede J., Refsum H., Vollset S.E. Trends Pharmacol. Sci. 2001;22:195–201. doi: 10.1016/s0165-6147(00)01675-8. [DOI] [PubMed] [Google Scholar]
- 24.Yamada K., Chen Z., Rozen R., Matthews R.G. Proc. Natl. Acad. Sci. USA. 2001;98:14853–14858. doi: 10.1073/pnas.261469998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leclerc D., Wilson A., Dumas R., Gafuik C., Song D., Watkins D., Heng H.H., Rommens J.M., Scherer S.W., Rosenblatt D.S. Proc. Natl. Acad. Sci. USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig M.L., Matthews R.G. Annu. Rev. Biochem. 1997;66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 27.Olteanu H., Banerjee R. J. Biol. Chem. 2001;276:35558–35563. doi: 10.1074/jbc.M103707200. [DOI] [PubMed] [Google Scholar]
- 28.Wilson A., Platt R., Wu Q., Leclerc D., Christensen B., Yang H., Gravel R.A., Rozen R. Mol. Gen. Metab. 1999;67:317–323. doi: 10.1006/mgme.1999.2879. [DOI] [PubMed] [Google Scholar]
- 29.Yamada K., Gravel R.A., Toraya T., Matthews R.G. Proc. Natl. Acad. Sci. USA. 2006;103:9476–9481. doi: 10.1073/pnas.0603694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leal N.A., Olteanu H., Banerjee R., Bobik T.A. J. Biol. Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 31.Mera P.E., Escalante-Semerena J.C. J. Biol. Chem. 2010;285:2911–2917. doi: 10.1074/jbc.M109.059485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dailey H.A. Biochem. Soc. Trans. 2002;30:590–595. doi: 10.1042/bst0300590. [DOI] [PubMed] [Google Scholar]
- 33.Arnould S., Takahashi M., Camadro J.-M. Proc. Natl. Acad. Sci. USA. 1999;96:14825–14830. doi: 10.1073/pnas.96.26.14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deybach J.C., da Silva V., Grandchamp B., Nordmann Y. Eur. J. Biochem. 1985;194:431–435. doi: 10.1111/j.1432-1033.1985.tb08943.x. [DOI] [PubMed] [Google Scholar]
- 35.Sassa S. Br. J. Haematol. 2006;135:281–292. doi: 10.1111/j.1365-2141.2006.06289.x. [DOI] [PubMed] [Google Scholar]
- 36.Puy H., Gouya L., Deybach J.C. Lancet. 2010;375:924–937. doi: 10.1016/S0140-6736(09)61925-5. [DOI] [PubMed] [Google Scholar]
- 37.Macalpine I., Hunter R. Br. Med. J. 1966;1:65–71. doi: 10.1136/bmj.1.5479.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Röhl J.C.G., Warren M.J., Hunt D.M. Bantam Press; London: 1998. Purple Secret, Genes, “Madness” and the Royal Houses of Europe. [Google Scholar]
- 39.Cox T.M., Jack N., Lofthouse S., Watling J. Lancet. 2005;366:332–335. doi: 10.1016/S0140-6736(05)66991-7. [DOI] [PubMed] [Google Scholar]
- 40.Qin X., Tan Y., Wang L., Wang Z., Wang B., Wen X., Yang G., Xi Z., Shen Y. FASEB J. 2011;25:653–664. doi: 10.1096/fj.10-170811. [DOI] [PubMed] [Google Scholar]
- 41.Percudani R., Peracchi A. EMBO Rep. 2003;4:850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musayev F.N., Di Salvo M.L., Ko T.-P., Schirch V., Safo M.K. Protein Sci. 2003;12:1455–1463. doi: 10.1110/ps.0356203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safo M.K., Musayev F.N., Di Salvo M.L., Schirch V. J. Mol. Biol. 2001;310:817–826. doi: 10.1006/jmbi.2001.4734. [DOI] [PubMed] [Google Scholar]
- 44.Di Salvo M.L., Contestabile R., Safo M.K. Biochim. Biophys. Acta. 2011;1814:1597–1608. doi: 10.1016/j.bbapap.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz A., Garcia-Villoria J., Ormazabal A., Zschocke J., Fiol M., Navarro-Sastre A., Artuch R., Vilaseca M.A., Ribes A. Mol. Gen. Metab. 2008;93:216–218. doi: 10.1016/j.ymgme.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Mills P.B., Surtees R.A., Champion M.P., Beesley C.E., Dalton N., Scambler P.J., Heales S.J., Briddon A., Scheimberg I., Hoffmann G.F. Hum. Mol. Gen. 2005;14:1077–1086. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- 47.Musayev F.N., Di Salvo M.L., Saavedra M.A., Contestabile R., Ghatge M.S., Haynes A., Schirch V., Safo M.K. J. Biol. Chem. 2009;284:30949–30956. doi: 10.1074/jbc.M109.038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang J.H., Hong M.L., Kim D.W., Park J., Kang T.C., Won M.H., Baek N.I., Moon B.J., Choi S.Y., Kwon O.S. Eur. J. Biochem. 2004;271:2452–2461. doi: 10.1111/j.1432-1033.2004.04175.x. [DOI] [PubMed] [Google Scholar]
- 49.Ngo E.O., LePage G.R., Thanassi J.W., Meisler N., Nutter L.M. Biochemistry. 1998;37:7741–7748. doi: 10.1021/bi972983r. [DOI] [PubMed] [Google Scholar]
- 50.Daugherty M., Polanuyer B., Farrell M., Scholle M., Lykidis A., de Crecy-Lagard V., Osterman A. J. Biol. Chem. 2002;277:21431–21439. doi: 10.1074/jbc.M201708200. [DOI] [PubMed] [Google Scholar]
- 51.Manoj N., Ealick S.E. Acta Crystallogr. 2003;59:1762–1766. doi: 10.1107/s0907444903016214. [DOI] [PubMed] [Google Scholar]
- 52.Steinbacher S., Hernandez-Acosta P., Bieseler B., Blaesse M., Huber R., Culianez-Macia F.A., Kupke T. J. Mol. Biol. 2003;327:193–202. doi: 10.1016/s0022-2836(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 53.Begley T.P., Kinsland C., Strauss E. Vitam. Horm. 2001;61:157–171. doi: 10.1016/s0083-6729(01)61005-7. [DOI] [PubMed] [Google Scholar]
- 54.Zhou B., Westaway S.K., Levinson B., Johnson M.A., Gitschier J., Hayflick S.J. Nat. Gen. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 55.Ozeir M., Muhlenhoff U., Webert H., Lill R., Fontecave M., Pierrel F. Chem. Biol. 2011;18:1134–1142. doi: 10.1016/j.chembiol.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 56.Heeringa S.F., Chernin G., Chaki M., Zhou W., Sloan A.J., Ji Z., Xie L.X., Salviati L., Hurd T.W., Vega-Warner V. J. Clin. Invest. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Berkel W.J., Kamerbeek N.M., Fraaije M.W. J. Biotech. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 58.Gibbons G., Mitropoulos K., Myant N. Elsevier Biomedical; Amsterdam: 1982. Biochemistry of Cholesterol. [Google Scholar]
- 59.Yamamoto S., Bloch K. J. Biol. Chem. 1970;245:1670–1674. [PubMed] [Google Scholar]
- 60.Nagai M., Sakakibara J., Wakui K., Fukushima Y., Igarashi S., Tsuji S., Arakawa M., Ono T. Genomics. 1997;44:141–143. doi: 10.1006/geno.1997.4825. [DOI] [PubMed] [Google Scholar]
- 61.Ryder N.S., Dupont M.C. Biochem. J. 1985;230:765–770. doi: 10.1042/bj2300765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Belter A., Skupinska M., Giel-Pietraszuk M., Grabarkiewicz T., Rychlewski L., Barciszewski J. Biol. Chem. 2011;392:1053–1075. doi: 10.1515/BC.2011.195. [DOI] [PubMed] [Google Scholar]
- 63.Chugh A., Ray A., Gupta J.B. Prog. Lipid Res. 2003;42:37–50. doi: 10.1016/s0163-7827(02)00029-2. [DOI] [PubMed] [Google Scholar]
- 64.Greeve I., Hermans-Borgmeyer I., Brellinger C., Kasper D., Gomez-Isla T., Behl C., Levkau B., Nitsch R.M. J. Neurosci. 2000;20:7345–7352. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mirza R., Qiao S., Tateyama K., Miyamoto T., Xiuli L., Seo H. J. Bone Miner. Metab. 2012;30:144–153. doi: 10.1007/s00774-011-0303-7. [DOI] [PubMed] [Google Scholar]
- 66.De Deken X., Wang D., Many M.C., Costagliola S., Libert F., Vassart G., Dumont J.E., Miot F. J. Biol. Chem. 2000;275:23227–23233. doi: 10.1074/jbc.M000916200. [DOI] [PubMed] [Google Scholar]
- 67.Dupuy C., Ohayon R., Valent A., Noel-Hudson M.S., Deme D., Virion A. J. Biol. Chem. 1999;274:37265–37269. doi: 10.1074/jbc.274.52.37265. [DOI] [PubMed] [Google Scholar]
- 68.Nunez J., Pommier J. Vitam. Horm. 1982;39:175–229. doi: 10.1016/s0083-6729(08)61137-1. [DOI] [PubMed] [Google Scholar]
- 69.Friedman J.E., Watson J.A., Jr., Lam D.W., Rokita S.E. J. Biol. Chem. 2006;281:2812–2819. doi: 10.1074/jbc.M510365200. [DOI] [PubMed] [Google Scholar]
- 70.Afink G., Kulik W., Overmars H., de Randamie J., Veenboer T., van Cruchten A., Craen M., Ris-Stalpers C. J. Clin. Endocrinol. Metab. 2008;93:4894–4901. doi: 10.1210/jc.2008-0865. [DOI] [PubMed] [Google Scholar]
- 71.Moreno J.C., Klootwijk W., van Toor H., Pinto G., D’Alessandro M., Leger A., Goudie D., Polak M., Gruters A., Visser T.J. N. Engl. J. Med. 2008;358:1811–1818. doi: 10.1056/NEJMoa0706819. [DOI] [PubMed] [Google Scholar]
- 72.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Gen. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fassone E., Duncan A.J., Taanman J.W., Pagnamenta A.T., Sadowski M.I., Holand T., Qasim W., Rutland P., Calvo S.E., Mootha V.K. Hum. Mol. Gen. 2010;19:4837–4847. doi: 10.1093/hmg/ddq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ensenauer R., He M., Willard J.M., Goetzman E.S., Corydon T.J., Vandahl B.B., Mohsen A.W., Isaya G., Vockley J. J. Biol. Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J., Zhang W., Zou D., Chen G., Wan T., Zhang M., Cao X. Biochem. Biophys. Res. Commun. 2002;297:1033–1042. doi: 10.1016/s0006-291x(02)02336-7. [DOI] [PubMed] [Google Scholar]
- 77.Nouws J., Nijtmans L., Houten S.M., van den Brand M., Huynen M., Venselaar H., Hoefs S., Gloerich J., Kronick J., Hutchin T. Cell. Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 78.Mostowska A., Biedziak B., Dunin-Wilczynska I., Komorowska A., Jagodzinski P.P. Birth Defects Res. A Clin. Mol. Teratol. 2011;91:169–176. doi: 10.1002/bdra.20771. [DOI] [PubMed] [Google Scholar]
- 79.Mostowska A., Hozyasz K.K., Biedziak B., Misiak J., Jagodzinski P.P. Eur. J. Oral. Sci. 2010;118:325–332. doi: 10.1111/j.1600-0722.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- 80.Mostowska A., Hozyasz K.K., Wojcicki P., Dziegelewska M., Jagodzinski P.P. J. Med. Genet. 2010;47:809–815. doi: 10.1136/jmg.2009.070029. [DOI] [PubMed] [Google Scholar]
- 81.Johnson A.R., Craciunescu C.N., Guo Z., Teng Y.W., Thresher R.J., Blusztajn J.K., Zeisel S.H. FASEB J. 2010;24:2752–2761. doi: 10.1096/fj.09-153718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson A.R., Lao S., Wang T., Galanko J.A., Zeisel S.H. PLoS ONE. 2012;7:e36047. doi: 10.1371/journal.pone.0036047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.