

Abstract
Alzheimer’s disease (AD) is associated with a significant neuroinflammatory component. Mononuclear phagocytes including monocytes and microglia are the principal cells involved, and they accumulate at perivascular sites of β-amyloid (Aβ) deposition and in senile plaques. Recent evidence suggests that mononuclear phagocyte accumulation in the AD brain is dependent on chemokines. CCL2, a major monocyte chemokine, is upregulated in the AD brain. Interaction of CCL2 with its receptor CCR2 regulates mononuclear phagocyte accumulation in a mouse model of AD. CCR2 deficiency leads to lower mononuclear phagocyte accumulation and is associated with higher brain Aβ levels, specifically around blood vessels, suggesting that monocytes accumulate at sites of Aβ deposition in an initial attempt to clear these deposits and stop or delay their neurotoxic effects. Indeed, enhancing mononuclear phagocyte accumulation delays progression of AD. Here we review the mechanisms of mononuclear phagocyte accumulation in AD and discuss the potential roles of additional chemokines and their receptors in this process. We also propose a multi-step model for recruitment of mononuclear phagocytes into the brain. The first step involves egress of monocyte/microglial precursors from the bone marrow into the blood. The second step is crossing the blood-brain barrier to the perivascular areas and into the brain parenchyma. The final step includes movement of monocytes/microglia from areas of the brain that lack any amyloid deposition to senile plaques. Understanding the mechanism of recruitment of mononuclear phagocytes to the AD brain is necessary to further understand the role of these cells in the pathogenesis of AD and to identify any potential therapeutic use of these cells for the treatment of this disease.
Keywords: Microglia, mononuclear phagocytes, Alzheimer’s disease, chemokines
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the presence of β-amyloid (Aβ) deposits and neurofibrillary tangles in the brain [1, 2]. Various brain cells are found associated with Aβ deposits. These include neurons, astrocytes and microglia, the mononuclear phagocytes of the brain [1, 3-5]. The development of mouse models of AD that reproduce the accumulation of Aβ and associated microgliosis as observed in human disease led to an exponential increase in research investigating the roles of these cells in the pathogenesis of AD. These investigations are beginning to elucidate the functions of mononuclear phagocytes in AD and their mechanism(s) of recruitment and accumulation in the brain. This review discusses recent advances in the field.
MONONUCLEAR PHAGOCYTES IN AD
Evidence for the presence of microglia in senile plaques derives from earlier immunohistochemical studies that examined the brains of AD patients [6]. In normal brains, resident microglia are distributed uniformly throughout the gray and white matter [6]. In contrast, in human AD brains and in the brains of mouse models of AD, microglia are clustered in and around Aβ deposits [5]. The concentration of microglia increases in close proximity to Aβ deposits and is 2-5 fold higher than in neighboring brain regions that do not contain Aβ [7]. Microglia appear to interact with Aβ [8], and occasionally phagocytic macrophages containing Aβ were described in AD [9, 10] and in rare patients suffering from stroke with AD [11], suggesting a possible role for these cells in the removal or processing of Aβ. While it is tempting to conclude that microglia play a protective role in AD by mediating Aβ phagocytosis and clearance, the available data are far from conclusive. Indeed, some investigators did not detect phagocytosing microglia in AD transgenic mice [12]. Furthermore, it was not clear whether the cells containing intracellular Aβ are resident microglia or blood borne mononuclear phagocytes. More recently, published reports appear to support a role for blood-borne mononuclear phagocytes in the clearance of Aβ. A defect in mononuclear phagocyte recruitment in transgenic Tg2576 AD mice deficient for the chemokine receptor CCR2 was associated with higher perivascular Aβ levels, suggesting that early accumulation of mononuclear phagocytes in this AD model promotes Aβ clearance [13]. Furthermore, depletion of perivascular mononuclear phagocytes in the TgCRND8 mouse AD model significantly increased the number of thioflavin S-positive cortical blood vessels [14]. In support of this protective role, enhancing mononuclear phagocyte accumulation delays progression of AD. AD mice that constitutively express interleukin-1 in the brain [15], or that are deficient in peripheral transforming growth factor β-SMAD2/3 signaling [16], have increased mononuclear phagocyte recruitment and reduced AD-like pathology. Based on these human and animal data, the accumulation of microglia/mononuclear phagocytes at sites of Aβ deposition appears to be an integral part of the pathogenesis of AD. Understanding the mechanism(s) of their accumulation in AD is necessary to fully understand the pathogenesis of this disease, and will certainly increase the likelihood of identifying effective disease modifying therapies for AD.
TRAFFICKING OF MONONUCLEAR PHAGOCYTES INTO THE NORMAL BRAIN
While the association of microglia with AD and other neurodegenerative diseases is widely accepted based on a large number of studies, the origin of these cells remains a subject of controversy in spite of decades of investigation. Earlier studies showed that peripheral monocytes labeled with carbon particles and transferred into syngeneic rats migrated into the brain in a model of stab wound injury where the blood brain-barrier (BBB) was disrupted, and these migrating cells started to transform into amoeboid microglia after 5 days of transfer [17, 18]. In addition, incubation of blood monocytes and spleen macrophages with astrocytes or astrocyte conditioned medium in vitro transformed these cells phenotypically into microglia [19]. These studies suggested that microglia, like other macrophages, can originate in the blood and migrate into the brain. More recent studies suggest that this pathway may only be relevant in conditions where there is breakdown of the BBB.
Sources of Microglia and Mononuclear Phagocytes in the Normal Brain
The ability of bone marrow derived-cells to populate the brain after irradiation and bone marrow transfer has been confirmed by several labs. Indeed, marrow-derived cells can be detected in the brain as early as three days after transplantation, and their numbers in the mouse brain continue to increase for several weeks afterwards [20]. These marrow-derived cells migrate to the cortex and hippocampus and various other areas of the brain stem and cerebellum [20-23]. We recently confirmed these findings and found that by 6 months post irradiation and transplant, ~60% of CD11b positive cells (i.e. microglia/mononuclear phagocytes) in the brain of mice that received bone marrow transplant were donor-derived [24]. While the ability to populate the brain with marrow-derived cells that assume microglial morphology and phenotype suggests that adult microglia can indeed be derived from the bone marrow, all studies involving bone marrow transfer required irradiation of the recipient mice before performing the transfer. To date, we could not find any published data showing engraftment of marrow-derived mononuclear cells in the brain without ablation of the recipient bone marrow by irradiation. Irradiation can alter the permeability of the BBB, change the gene expression profile of endothelial cells, and upregulate chemokines such as CCL2. For these reasons, it was suggested that irradiation by itself is sufficient to promote the influx of bone marrow-derived cells [25, 26] and that the source of microglia in the normal brain may not be the bone marrow. In addition, while proliferation of mononuclear phagocytes in the brain is limited [13], local self renewal can still sustain a steady number of mononuclear phagocytes under normal physiological conditions [25-27]. We propose that the pool of microglia/mononuclear phagocytes in the brain at any given point is derived from two sources: proliferation of resident cells, and recruitment of microglia/mononuclear phagocyte precursors from the bone marrow. It is possible that each one of these two sources contributes to a different subset of mononuclear cells in the brain. For example, bone marrow-derived cells may contribute preferentially to perivascular macrophages, while parenchymal microglia may be mostly derived from local proliferation. This possibility, while appealing, is not definitively proven yet.
MONONUCLEAR PHAGOCYTE TRAFFICKING TO THE BRAIN IN NEUROINFLAMMATORY CONDITIONS
There is increasing evidence that bone marrow-derived mononuclear phagocytes are important components of the neuroinflammatory response associated with many neurological disorders. Several infectious diseases of the central nervous system (CNS) provide compelling examples that support a myeloid origin for mononuclear cells in the brain. For example, early and rapid engraftment of bone marrow-derived IBA+ cells was observed in Scrapie, an example of prion infection that affects sheep and can infect mice [28]. Similarly, in a model of Streptococcus pneumoniae meningitis, recruited monocytes accumulate in the post-acute period of the infection and contribute to the pool of microglia [29]. In a model of infection with the pathogenic yeast Cryptococcus neoformans, circulating monocytes carrying intracellular organisms can cross the BBB, thereby allowing translocation of the yeast into the brain and facilitating intracerebral infection [30, 31]. In mouse models of viral encephalitis such as west Nile virus infections, failure of homing of peripheral mononuclear phagocytes into the brain and other organs was associated with increased susceptibility to lethal west Nile virus encephalitis [32]. In addition to the various infectious models mentioned, blood-borne monocytes can migrate into the CNS and initiate primary demyelination [33] and contribute to the inflammatory response in the 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine mouse model of Parkinson’s disease [34]. In AD, we and others found that when bone marrow-derived green fluorescent protein-labeled myeloid cells were transferred into irradiated AD mice, green fluorescent protein-labeled cells were found associated with sites of Aβ deposition in the recipient mice [24, 35, 36]. Based on this discussion, we conclude that there is extensive published data supporting a myeloid origin for mononuclear phagocytes including microglia in several important neuroinflammatory conditions.
CHEMOKINES AND ACCUMULATION OF MONONUCLEAR PHAGOCYTES IN AD
Chemokines are chemotactic cytokines that form a large family (50 members) of secreted and membrane-bound, 8-10 kDa proteins that induce the recruitment of leukocytes to sites of acute and chronic inflammation [37]. Chemokines bind to specific G protein-coupled seven-transmembrane cell surface receptors on target cells [37]. Chemokines are divided into three subfamilies based on the relationship of conserved cysteine residues in their sequences [37]. The α chemokines have the first two cysteine residues of the active protein separated by one amino acid (CXC motif), while β chemokines have adjacent cysteine residues (CC motif) [37]. Fractalkine, the only member of the CXXXC subfamily, has 3 amino acids between the first two cysteines residues and is also unusual in that it is a transmembrane protein containing a chemokine domain attached to a mucin-like transmembrane stalk. Chemokine receptors that bind CC chemokines have been named CC chemokine receptors (CCR1-10), those that bind CXC chemokines have been named CXC chemokine receptors (CXCR1-6), and the fractalkine receptor has been named CX3CR1. In a manner analogous to the receptor nomenclature, chemokines have also been given standardized names, such as CC chemokine ligand (CCL)1-28, CXC chemokine ligand (CXCL)1-16, and CX3C chemokine ligand (CX3CL)1 [37]. Chemokines and their receptors expressed on monocytes and microglia are summarized in Fig. (1). Of note, all the chemokine receptors expressed on monocytes can also be found on microglia in vitro and/or in vivo (Fig. 1).
Fig. 1.

Chemokines and their receptors expressed on monocytes and microglia and their potential role in Alzheimer’s disease.
CCL2 is a Potent Monocyte Chemoattractant in Acute and Chronic Inflammation
Binding of CCL2 to its receptor CCR2 stimulates production of reactive oxygen species [38], up-regulates adhesion receptors [39], and induces monocyte arrest on endothelium [37]. CCR2 deficiency led to decreased recruitment of monocytes into the peritoneum in a model of acute inflammation [37], and reduced the size of atherosclerotic lesions in a mouse model for atherosclerosis due to reduction in the number of lesional monocytes (a chronic process) [40]. CCR2 was also critical for the recruitment of mononuclear phagocytes into the CNS in experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis [41]. Interestingly, mice deficient in CCL2 also had reduced monocyte recruitment into the CNS in experimental autoimmune encephalomyelitis [42], and into atherosclerotic lesions in a mouse model of atherosclerosis [40]. Thus, CCL2 interaction with CCR2 regulates the recruitment and/or retention of mononuclear phagocytes in inflammation.
In addition to monocytes, CCL2 is expressed in reactive microglia in senile plaques and in some neurons and astrocytes [43, 44]. Based on an immunohistochemical study of AD brains, logistic linear regression modeling determined that CCL2 was the most reliable predictor of disease [44]. CCL2 is also induced by Aβ in microglia and induces microglia cell chemotaxis in vitro. We and others showed that Aβ induced monocytes and microglia to secrete CCL2 [45]. Human astrocytoma cells also produce CCL2 upon stimulation with Aβ [46]; however, we were not able to detect CCL2 or CCR2 RNA in astrocytes isolated from the brains of mice with AD-like pathology by laser capture microdissection [13]. In addition, we found that CCL2 is a potent chemoattractant for human fetal microglia, suggesting that these cells express CCR2 [13, 45]. Based on the above, CCL2/CCR2 interactions appear to play a key role in recruitment and/or activation of mononuclear phagocytes to sites of Aβ deposition in AD.
CCR2 Is Required for the Accumulation of Mononuclear Phagocytes in APP Tg2576 Mice before Formation of Visible Aβ Deposits
To test the role of CCR2 in mononuclear phagocyte accumulation in an AD mouse model, we bred CCR2 null mice with transgenic mice expressing the human amyloid precursor protein (APP) with the Swedish mutation (APP Tg2576) and analyzed the resulting APP-CCR2−/− mice for AD-like pathology [13]. We found that CCR2 deficiency significantly reduced the number of CD11b positive cells that accumulated in the brains of APP mice early in the disease process, before formation of senile plaques. CCR2 deficiency also abolished the accumulation of mononuclear phagocytes at sites of intracerebral injection of Aβ. Analysis of the phenotype of these recruited cells by flow cytometry showed that they express high levels of CD45 in addition to CD11b, suggesting they are monocytes. This reduction in the number of mononuclear phagocytes was associated with increased mortality and higher Aβ levels in the brain, suggesting that early mononuclear phagocyte accumulation promotes the clearance of Aβ and protects mice from Aβ toxicity early in the disease process. Our data indicate that CCR2 plays a non-redundant role in the pathogenesis of AD and support the hypothesis that early accumulation of monocytes is protective and promotes Aβ clearance.
Recruitment of Monocytes into the AD Brain is a Multistep Process
Studies on the accumulation of monocytes in atherosclerosis suggested that in addition to the important role of CCL2 and CCR2, other monocyte chemokines and their receptors may be involved. Indeed, CXCR2 and its ligand CXCL1 (KC), CX3CR1 and its ligand CX3CL1 (fractalkine), and CCR5 and its ligand CCL5 (RANTES) have all been implicated [47-51]. While it is possible that all these chemokines are acting in concert, these studies suggest that they may be acting in series. Each individual chemokine/chemokine receptor pair may be guiding the cells through a different tissue barrier and check point required to migrate from the bone marrow into the blood, then from the blood into the tissue, and finally to find the site of inflammation within a tissue [52, 53]. Indeed, initial observations suggested that the CCL2/CCR2 pathway controlled monocyte trafficking from the blood into inflamed tissue. However, recent studies evaluating monocyte trafficking in more detail have challenged this initial assumption [52, 53]. In several models of inflammation such as sterile peritonitis, atherosclerosis, and Listeria infection, it appears that the CCL2/CCR2 pathway controls monocyte trafficking out of the bone marrow and into the blood, as opposed to a role in the egress of monocytes out of the blood into the tissue. In contrast, the CX3CL1/CX3CR1 pathway appears to control monocyte capture and firm adhesion at the blood vessel to tissue barrier [54, 55]. We propose that the same paradigm may also apply to monocyte trafficking from the bone marrow into the senile plaque in the AD brain. While it is not known whether the same chemokine/chemokine receptor pairs involved in atherogenesis are also involved in AD, we hypothesize that the migration of monocytes from the bone marrow → blood → brain → plaque is controlled by distinct chemokine-receptor pairs that act at one or more steps in this multistep process (Fig. 2).
Fig. 2.
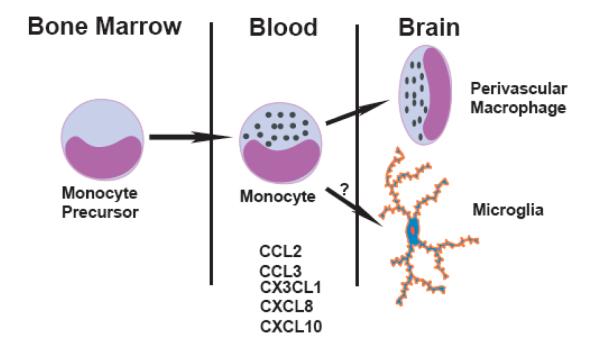
A proposed model for mononuclear phagocyte/microglia accumulation in Alzheimer’s disease highlighting the multiple steps involved in this process. Several chemokines that can recruit cells from the bone marrow → blood → brain → plaque may be involved. Distinct subsets of these chemokines and their receptors may act at one or more steps in this multistep process. The exact roles of these chemokines are only beginning to be elucidated.
Additional Chemokines that may also Control Microglia Accumulation in AD
Monocytes and microglia express a similar chemokine receptor profile (Fig. 1). Furthermore, in addition to CCL2, other chemokines have been shown to be expressed in Aβ-stimulated monocytes and microglia and in AD brains. Indeed, we have shown that Aβ-stimulated mouse macrophages and microglia upregulate their mRNA levels of macrophage inflammatory protein (MIP)-1α/CCL3 and β/CCL4, IL-8/CXCL8 and RANTES/CCL5 [45]. Similarly, adult human microglia isolated post-mortem and incubated with Aβ upregulate CXCL8 and CCL2 and, to a lesser extent, CCL3 and CCL4 [56]. These chemokines induce the chemotaxis of adult rat brain microglia and a human fetal microglia cell line in vitro [57]. In support for a potential role for these chemokines in AD, CCL2 and CCL3 were detected in microglia, while CCL4 and IP-10/CXCL10 were seen in reactive astrocytes in AD brains [58]. Furthermore, microglia associated with senile plaques express CCR3, CCR5, CX3CR1, CXCR2, and CXCR3. CX3CR1 is highly expressed on microglia, while its ligand CX3CL1 is highly expressed on neurons [59]. The CX3CL1/CX3CR1 pathway has recently been shown to play a role in microglia neurotoxicity in vivo in mouse models of amyotrophic lateral sclerosis and Parkinson’s disease [59], raising the possibility that it may play a role in regulating microglial functions in AD. We propose that in addition to CCL2/CCR2, several additional chemokines/chemokine receptor pairs expressed on monocytes and microglia and induced in Aβ-stimulated microglia may also contribute to mononuclear phagocyte accumulation in AD.
In summary, understanding the mechanisms of mononuclear phagocyte accumulation in AD is an emerging field, and several important questions remain unanswered. Our proposed multistep model of mononuclear phagocyte accumulation in AD is attractive, but important definitive experiments need to be performed to show directly and unequivocally that blood-derived microglia are an important source of microglia in AD. Determining the exact chemokines/chemokine receptors involved in this process and the site of action of these chemokines/receptors will significantly enhance our understanding of the pathophysiology of AD, and may lead to novel therapeutic strategies to stop or delay the progression of AD by regulating the neuroinflammatory response that is characteristic of this disease.
ACKNOWLEDGEMENTS
This work was supported by NINDS grant NS 059005 (JEK) and a grant from the Dana Foundation Neuro-Immunology program (JEK).
ABBREVIATIONS
- Aβ
β-Amyloid
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- BBB
Blood-brain barrier
- CNS
Central nervous system
- CCL
Chemokine ligand
- CCR
Chemokine receptor
- CXCL
Chemokine ligand
- CXCR
Chemokine receptor
REFERENCES
- [1].Mott RT, Hulette CM. Neuropathology of Alzheimer’s disease. Neuroimaging Clin. N Am. 2005;15:755–765. doi: 10.1016/j.nic.2005.09.003. ix. [DOI] [PubMed] [Google Scholar]
- [2].Gomez-Isla T, Spires T, De Calignon A, Hyman BT. Neuropathology of Alzheimer’s disease. Handb. Clin. Neurol. 2008;89:233–243. doi: 10.1016/S0072-9752(07)01222-5. [DOI] [PubMed] [Google Scholar]
- [3].Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev. Neurosci. 2004;15:267–278. doi: 10.1515/revneuro.2004.15.4.267. [DOI] [PubMed] [Google Scholar]
- [4].Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- [5].Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- [6].McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- [7].Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- [8].Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 1990;119:32–36. doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- [9].Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- [10].D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol. Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- [11].Wisniewski HM, Barcikowska M, Kida E. Phagocytosis of beta/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol. 1991;81:588–590. doi: 10.1007/BF00310142. [DOI] [PubMed] [Google Scholar]
- [12].Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol. Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- [13].El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- [14].Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ling EA. Electron microscopic study of macrophages appearing in a stab wound of the brain of rats following intravenous injection of carbon particles. Arch. Histol. Jpn. 1979;42:41–50. doi: 10.1679/aohc1950.42.41. [DOI] [PubMed] [Google Scholar]
- [18].Imamoto K. Origin of microglia: cell transformation from blood monocytes into macrophagic ameboid cells and microglia. Prog. Clin. Biol. Res. 1981;59A:125–139. [PubMed] [Google Scholar]
- [19].Sievers J, Parwaresch R, Wottge HU. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: morphology. Glia. 1994;12:245–258. doi: 10.1002/glia.440120402. [DOI] [PubMed] [Google Scholar]
- [20].Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc. Natl. Acad. Sci. USA. 1997;94:4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- [22].Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, Ogawa M. Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 2004;186:134–144. doi: 10.1016/j.expneurol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- [23].Priller J, Flügel A, Wehner T, Boentert M, Haas CA, Prinz M, Fernández-Klett F, Prass K, Bechmann I, de Boer BA, Frotscher M, Kreutzberg GW, Persons DA, Dirnagl U. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- [24].El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol. Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- [25].Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- [26].Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- [27].Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29:227–234. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- [28].Priller J, Prinz M, Heikenwalder M, Zeller N, Schwarz P, Heppner FL, Aguzzi A. Early and rapid engraftment of bone marrow-derived microglia in scrapie. J. Neurosci. 2006;26:11753–11762. doi: 10.1523/JNEUROSCI.2275-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Djukic M, Mildner A, Schmidt H, Czesnik D, Brück W, Priller J, Nau R, Prinz M. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain. 2006;129:2394–2403. doi: 10.1093/brain/awl206. [DOI] [PubMed] [Google Scholar]
- [30].Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect. Immun. 2009;77:120–127. doi: 10.1128/IAI.01065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chretien F, Lortholary O, Kansau I, Neuville S, Gray F, Dromer F. Pathogenesis of cerebral Cryptococcus neoformans infection after fungemia. J. Infect. Dis. 2002;186:522–530. doi: 10.1086/341564. [DOI] [PubMed] [Google Scholar]
- [32].Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–253. doi: 10.1016/j.immuni.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Remington LT, Babcock AA, Zehntner SP, Owens T. Microglial recruitment, activation, and proliferation in response to primary demyelination. Am. J. Pathol. 2007;170:1713–1724. doi: 10.2353/ajpath.2007.060783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kokovay E, Cunningham LA. Bone marrow-derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson’s disease. Neurobiol. Dis. 2005;19:471–478. doi: 10.1016/j.nbd.2005.01.023. [DOI] [PubMed] [Google Scholar]
- [35].Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- [36].Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- [37].Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- [38].Zachariae CO, Anderson AO, Thompson HL, Appella E, Mantovani A, Oppenheim JJ, Matsushima K. Properties of monocyte chemotactic and activating factor (MCAF) purified from a human fibrosarcoma cell line. J. Exp. Med. 1990;171:2177–2182. doi: 10.1084/jem.171.6.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weber C, Alon R, Moser B, Springer TA. Sequential regulation of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity by CC chemokines in monocytes: implications for transendothelial chemotaxis. J. Cell Biol. 1996;134:1063–1073. doi: 10.1083/jcb.134.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barlic J, Murphy PM. Chemokine regulation of atherosclerosis. J. Leukoc. Biol. 2007;82:226–236. doi: 10.1189/jlb.1206761. [DOI] [PubMed] [Google Scholar]
- [41].Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Proudfoot AE, de Souza AL, Muzio V. The use of chemokine antagonists in EAE models. J. Neuroimmunol. 2008;198:27–30. doi: 10.1016/j.jneuroim.2008.04.007. [DOI] [PubMed] [Google Scholar]
- [43].Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin. Neurosci. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- [44].Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009;19:392–398. doi: 10.1111/j.1750-3639.2008.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Prat E, Baron P, Meda L, Scarpini E, Galimberti D, Ardolino G, Catania A, Scarlato G. The human astrocytoma cell line U373MG produces monocyte chemotactic protein (MCP)-1 upon stimulation with beta-amyloid protein. Neurosci. Lett. 2000;283:177–180. doi: 10.1016/s0304-3940(00)00966-6. [DOI] [PubMed] [Google Scholar]
- [47].Boisvert WA, Rose DM, Johnson KA, Fuentes ME, Lira SA, Curtiss LK, Terkeltaub RA. Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am. J. Pathol. 2006;168:1385–1395. doi: 10.2353/ajpath.2006.040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Braunersreuther V, Zernecke A, Arnaud C, Liehn EA, Steffens S, Shagdarsuren E, Bidzhekov K, Burger F, Pelli G, Luckow B, Mach F, Weber C. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:373–379. doi: 10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- [49].Zernecke A, Liehn EA, Gao JL, Kuziel WA, Murphy PM, Weber C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. 2006;107:4240–4243. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]
- [50].Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J. Clin. Invest. 2003;111:333–340. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Combadiere C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debré P, Tedgui A, Murphy PM, Mallat Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–1016. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- [52].Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- [53].Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- [56].Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol. Aging. 2001;22:957–966. doi: 10.1016/s0197-4580(01)00306-2. [DOI] [PubMed] [Google Scholar]
- [57].Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int. Rev. Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- [58].Xia MQ, Hyman BT. Chemokines/chemokine receptors in the central nervous system and Alzheimer’s disease. J. Neurovirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- [59].Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]