

Abstract
Although there are currently three generations of antifungal azoles on the market, even the third-generation agents show undesirable interactions with human cytochrome P450 (P450) enzymes. CYP46A1 is a cholesterol-metabolizing P450 in the brain that tightly binds a number of structurally distinct azoles. Previously, we determined the crystal structures of CYP46A1 in complex with voriconazole and clotrimazole, and in the present work we cocrystallized the P450 with posaconazole at 2.5 Å resolution. This long antifungal drug coordinates the P450 heme iron with the nitrogen atom of its terminal azole ring and adopts a linear configuration occupying the whole length of the substrate access channel and extending beyond the protein surface. Numerous drug-protein interactions determine the submicromolar Kd of posaconazole for CYP46A1. We compared the crystal structure of posaconazole-bound CYP46A1 with those of the P450 in complex with other drugs, including the antifungal voriconazole and clotrimazole. We also analyzed the accommodation of posaconazole in the active site of the target enzymes, CYPs 51, from several pathogenic species. These and the solution studies with different marketed azoles, collectively, allowed us to identify the determinants of tight azole binding to CYP46A1 and generate an overall picture of azole binding to this important P450. The data obtained suggest that development of CYP51-specific antifungal agents will continue to be a challenge. Therefore, structural understanding of the azole binding not only to CYPs 51 from the pathogenic species but also to different human P450s is required to deal efficiently with this challenge.
Introduction
Antifungal azoles are widely used for the treatment of superficial and invasive fungal infection and include the three generations of imidazole- and triazole-containing pharmaceuticals (Arnold et al., 2010). Compounds of the first generation (e.g., clotrimazole, bifonazole, econazole, and ketoconazole) contain imidazole in their ring system (Fig. 1) and are mainly applied topically because they have poor water solubility and limited oral bioavailability as well as unacceptable side effects if administered orally. The adverse effects are drug dependent and include gastrointestinal and endocrine disturbances, hepatotoxicity, and skin irritation (Maertens, 2004). Ketoconazole is the only first-generation azole that could be used systemically, but it is no longer the drug of choice because more effective and less toxic systemic azoles have been developed. These are the second (fluconazole and itraconazole) and third (voriconazole and posaconazole) generation azoles, which have the triazole ring instead of the imidazole structure. The triazole-containing antifungals display a broader spectrum of activity than the imidazole-containing azoles and have markedly improved safety profiles due to their increased affinities to the target enzyme in the invading pathogen along with a better oral bioavailability and pharmacokinetic/pharmacodynamic properties as compared with the antifungal imidazoles (Girmenia, 2009). The third-generation azoles also include ravuconazole, albaconazole, and isavuconazole, which are currently under investigation in clinical trials and are not yet approved for clinical use in the United States.
Fig. 1.

Chemical structures of the three generations of systemic azoles developed for use in humans. The structures and drug names in black and gray are of the marketed and experimental azoles, respectively. The numbering of some of the heteroatoms in clotrimazole, posaconazole, and voriconazole is also shown.
The mechanism of action of all antifungal azoles is based on the inhibition of the fungal cytochrome P450 (P450) from family 51 (CYP51 or sterol 14α-demethylase) that is essential for the biosynthesis of ergosterol, the major sterol of the fungal plasma membranes (White et al., 1998). Inhibition of fungal CYP51 leads to blockage of ergosterol synthesis, sterol depletion from the membranes, and accumulation of 14α-methylated precursors. Changes in the membrane sterol composition alter the fluidity and integrity of the fungal membranes and affect the activity of several membrane-bound enzymes. The ultimate result is cell lysis and death (White et al., 1998). CYP51, however, is present not only in fungi but in many other species, including humans (Lepesheva and Waterman, 2007), in which this enzyme is involved in the biosynthesis of cholesterol, the major sterol of mammalian membranes. Thus, one of the requirements for the antifungal azoles is to inhibit fungal CYP51 but avoid or minimize the inhibition of the human ortholog that metabolizes the same substrate lanosterol.
In addition, at therapeutic concentrations, antifungal azoles should also not inhibit other P450 isoforms present in humans that play important roles in the metabolism of endogenous and exogenous compounds. Currently, however, this is not the case and is one of the limitations of the antifungal azoles. Even the second- and third-generation antifungal azoles inhibit some of the host P450s [e.g., human CYP3A4, CYP2C9, and CYP2C19 (Lipp, 2010)], thus creating a potential for undesired drug-drug interactions.
To assist in development of more specific and potent antifungal azoles and gain insight into the mechanism of fungal azole resistance, another clinical limitation for this class of agents, crystal structures of the CYP51 enzymes from humans and a number of pathogenic species (Mycobacterium tuberculosis, Trypanosoma cruzi, Trypanosoma brucei, and Leishmania infantum) have been determined in complex with different marketed azoles (Podust et al., 2001; Chen et al., 2010; Lepesheva et al., 2010; Strushkevich et al., 2010; Hargrove et al., 2011). Yet only two human P450s from families other than 51 have been cocrystallized with the marketed azoles. They include ketoconazole-bound CYP3A4 (Ekroos and Sjogren, 2006) and clotrimazole- and voriconazole-bound CYP46A1 (Mast et al., 2010). CYP3A4 is the major drug-metabolizing P450 (Guengerich, 2005), with many antifungal azoles being the enzyme substrates (Lipp, 2010). In contrast, CYP46A1 is the endobiotic-metabolizing P450, which hydroxylates cholesterol in the brain and is responsible for cholesterol removal and turnover in this organ (Bjorkhem et al., 1997; Lund et al., 2003). Unexpectedly, CYP46A1 was found to have submicromolar Kd values for clotrimazole and voriconazole in vitro (Mast et al., 2008; Shafaati et al., 2010), and to be inhibited by voriconazole in vivo in mice (Shafaati et al., 2010). In subsequent studies, CYP46A1 was also found to tightly bind another azole posaconazole (Mast et al., 2012); hence, in the present work we cocrystallized CYP46A1 with this azole to gain insight into the features of the enzyme active site that enable binding of the three structurally very different antifungal azoles. The data obtained will aid in designing more specific antifungal azoles.
Materials and Methods
Materials.
Posaconazole (POS), albaconazole, and ravuconazole were obtained from Toronto Research Chemicals (Toronto, ON, Canada). Cholesterol was obtained from Avanti Polar Lipids (Alabaster, AL). [1,2-3H]Cholesterol was obtained from PerkinElmer (Waltham, MA). Cymal 6 was from Affymetrix (Santa Clara, CA). PEG 8000 was from Hampton Research (Aliso Viejo, CA). Voriconazole (VOR), clotrimazole (CLO), ketoconazole, itraconazole, and other chemicals were from Sigma-Aldrich (St. Louis, MO).
CYP46A1 Expression and Purification.
All studies were performed on truncated, ∆(2–50), human CYP46A1, which was heterologously expressed in Escherichia coli as described elsewhere (White et al., 2008). The P450 was purified either as a substrate-free enzyme (Mast et al., 2008) or as POS-bound. The latter was obtained by saturating CYP46A1 with the drug during the last purification step, as described elsewhere (Mast et al., 2010).
Drug Binding to ∆(2–50)CYP46A1.
Apparent Kd values of different antifungal azoles for ∆(2–50)CYP46A1 were determined as described elsewhere (Mast et al., 2010, 2013b) on the basis of the spectral response induced in P450 upon addition of a drug. Briefly, titrations were performed using 0.25–0.4 μM CYP46A1 and 0.1–1.0 mM stocks of POS, CLO, or VOR in 50% aqueous methanol. The final concentrations of methanol were no more than 1%, and at these concentrations methanol did not cause any spin-state changes in CYP46A1. Spectral data were fit to either
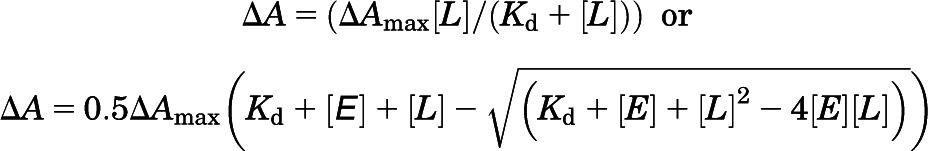 |
equations, in which ∆A is the spectral response at different ligand (drug) concentrations [L], and ∆Amax is the maximal amplitude of the spectral response. The former equation was used when Kd was higher than the total enzyme concentration [E], whereas the latter was applied when Kd was lower than the enzyme concentration, assuming 1:1 stoichiometry. The total CYP46A1 concentration was determined from the reduced CO difference spectrum, as described elsewhere (Omura and Sato, 1964).
Determination of the IC50 and Ki.
Purified 0.25 μM ∆(2–50)CYP46A1 was reconstituted with 0.5 μM NADPH cytochrome P450 oxidoreductase, 7.7 μM cholesterol, trace amounts of [3H]cholesterol (250,000 cpm), and varying concentrations of a drug under study (from 0.000001 to 100 μM). The enzymatic reaction was initiated by NADPH (final 1 mM) and performed for 8 minutes at 37°C in 1 ml of 50 mM KPi, pH 7.2, containing 100 mM NaCl, 0.02% Cymal 6, and 2 U of catalase. The reaction was terminated by vortexing 2 times with 2 ml of CH2Cl2. The organic phase was isolated, evaporated, dissolved in methanol, and subjected to high-pressure liquid chromatography, as described elsewhere (Mast et al., 2004). Two product peaks were seen in the high-pressure liquid chromatography profile: one (the major peak) corresponded to 24-hydroxycholesterol, and the other peak (the minor peak, ~1/10 of the major peak area) corresponded to the mixture of 24,25- and 24,27-dihydroxycholesterol produced as a result of 25- and 27-hydroxylations of 24-hydroxycholesterol. Data were fit for the inhibition of the main product, 24-hydroxycholesterol, using the following equation:
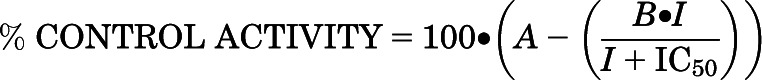 |
in which I is the inhibitor concentration, the IC50 is the concentration of the inhibitor at which the enzyme activity is reduced by 50%, and the value of 1 − (A − B) is the maximum inhibition observed at an infinite inhibitor concentration. We could also estimate the Ki for cholesterol 24-hydroxylation because the IC50 was determined at a cholesterol concentration equal to 1 Km (7.7 μM) and the azoles were competitive inhibitors of CYP46A1. In this case, the Ki = IC50/2.
Crystallization, Data Collection, and Structure Determination.
The POS-bound ∆(2–50) CYP46A1 was crystallized at 18°C in sitting drops after mixing 1 μl of an ~40 mg/ml P450 preparation with 1 μl of precipitant solution (12% PEG 8,000, 50 mM KPi, pH 5.8, and 20% glycerol). The crystals obtained were then used for microseeding with a cat whisker into the 1:1 (v/v) P450-precipitant mixture containing PEG 8,000 at a reduced concentration (10%). X-ray diffraction data were collected from a single crystal at the Stanford Synchrotron Radiation Lightsource beam line 11-1 at 100 K (Soltis et al., 2008). The diffraction limit was 2.5 Å. The data set was processed with XDS (X-ray Detector Software; MPI for Medical Research, Heidelberg, Germany) (Kabsch, 2010) and the Collaborative Computational Project, Number 4 (1994) programs. The structure of POS-bound CYP46A1 was determined by molecular replacement using a cholesterol 3-sulfate-bound structure as the search model with cholesterol 3-sulfate omitted [Protein Data Bank (PDB) ID 2Q9F] and Phaser (McCoy et al., 2007). The model was refined using Refmac (Computational Crystallography Group) (Murshudov et al., 1997), and Coot (Crystallographic Object-Oriented Toolkit) for electron density fitting (Emsley and Cowtan, 2004). The POS complex crystallized in space group I4122 with one molecule per asymmetric unit. Data collection and refinement statistics are given in Supplemental Table 1; coordinates and structure factors have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB ID 4J14).
Results
Binding and Inhibitory Properties of the Azoles That Were Cocrystallized with ∆(2–50)CYP46A1.
POS was identified as a tight binder of CYP46A1 based on the interaction with the full-length enzyme (Mast et al., 2012). However, for crystallization we used the truncated ∆(2–50) variant; we determined the apparent Kd of POS as well as its Ki for ∆(2–50)CYP46A1. For comparison, we also evaluated two other antifungal azoles, VOR and CLO, which were previously co-crystallized with ∆(2–50)CYP46A1 (Mast et al., 2010). Similar to full-length CYP46A1, POS induced a type II spectral response in the truncated P450 and had the submicromolar apparent Kd (Table 1). This Kd (30 nM) was higher than that of CLO (~4 nM) but lower than the apparent Kd of VOR (220 nM). The Ki of the azole inhibitors correlated well with their Kd and was 17 nM for CLO, 64 nM for POS, and 226 nM for VOR. Thus, the Kd and Ki values of the three structurally distinct antifungal azoles for CYP46A1 are different, but they all fall in a range of tight binding and efficient inhibition of cholesterol 24-hydroxylation.
TABLE 1.
Binding and inhibition of ∆(2–50)CYP46A1 wild type and mutants by the antifungal azoles that were cocrystallized with the P450
The results represent the average of triplicate measurements ± S.D.
| Drug | Kd | Ki |
|---|---|---|
| μM | ||
| ∆(2–50)CYP46A1 Wild Type | ||
| CLO | ∼0.004 | 0.017 ± 0.003 |
| POS | 0.03 ± 0.01 | 0.064 ± 0.005 |
| VOR | 0.22 ± 0.02 | 0.226 ± 0.006 |
| T306A Mutant | ||
| CLO | ∼0.005 | ND |
| POS | 0.51 ± 0.03 | ND |
| VOR | 0.27 ± 0.02 | ND |
| A474V Mutant | ||
| CLO | 0.09 ± 0.01 | ND |
| POS | 0.04 ± 0.01 | ND |
| VOR | 5.4 ± 0.4 | ND |
ND, not determined.
Crystal Structure of ∆(2–50)CYP46A1 in Complex with POS.
The electron density for POS was well defined in the active site and substrate access channel, allowing unambiguous fitting of the drug (Fig. 2). This fitting revealed that the drug binds to CYP46A1 in a linear conformation with the branched-bulky end occupying the enzyme active site and the 2-hydroxypentanyl end facing the solvent. In a linear conformation, POS is longer than the channel connecting the heme group and protein surface, hence the terminal 2-hydroxypentanyl moiety hangs outside CYP46A1 (Fig. 3A) and has a less defined electron density. This is in contrast to the other POS moieties, which have multiple interactions with CYP46A1 (Fig. 3B). In the active site, the triazole ring coordinates the heme iron (the N4-Fe distance is 2.0 Å) and is positioned between the side chains of Ala302 and Thr306 by short van der Waals contacts. There could be a weak hydrogen bond between the N2 of this triazole, and the hydroxyl group of Thr306 located at a 3.4 Å distance.
Fig. 2.

Unbiased σA weighted 2|Fo|–|Fc| and |Fo|–|Fc| electron density for POS in the complex with CYP46A1 at 2.5 Å resolution. The 2|Fo|–|Fc| map (cyan) is contoured at 0.6 σ; the |Fo|–|Fc| map (green) is contoured at 3.0 σ. POS (PDB ligand X2N) carbon atoms are white, oxygen red, nitrogen blue, and fluorine purple; heme carbon atoms are orange, and iron purple.
Fig. 3.

Interactions of POS with CYP46A1. (A) Semitransparent representation of the CYP46A1 surface (in light blue) showing POS (carbon atoms are magenta) extending above the protein surface. (B) Amino acid residues of CYP46A1 within 4 Å from POS (except Leu229). The heme carbon atoms are red; the nitrogen, oxygen, fluorine, sulfur, and iron atoms are blue, red, black, yellow, and orange, respectively.
The adjacent difluorophenyl ring of POS is in the hydrophobic pocket formed by the methyl groups of Ala474, Thr475, and Thr306, and the side chains of Ile301, Phe121, and Val126. We will call this pocket the Phe121 pocket because Phe121 is one of the key residues for the binding interactions. The only polar atom in this pocket is the carbonyl oxygen of Thr298 at a 3.2 Å distance from one of the fluorines of the difluorophenyl ring. Because of its position over the I helix (Fig. 4A), the Phe121 pocket is the only place in the CYP46A1 active site that enables simultaneous accommodation of the bulky difluorophenyl functionality and coordination of the heme iron by the POS triazole ring. Although the difluorophenyl ring can rotate 180° along the long axis of POS, in CYP46A1 this alternate positioning is not possible because of the heme propionate group and side chain of Ala367, which are within less than 4 Å distance from the POS oxolane ring and the oxolane-triazole ring linker.
Fig. 4.

Binding of POS, VOR, and CLO to CYP46A1. (A) Superimposed views of POS-, CLO-, and VOR-bound CYP46A1 showing how the three structurally distinct azoles are accommodated in the enzyme active site. The azole carbon atoms are magenta (POS), wheat (CLO), and marine (VOR). Phe121 and the CYP46A1 secondary structural elements defining the Phe121 pocket are also shown. Their coloring is the same as that of the P450 carbon atoms in panel B. (B) Conformational flexibility of amino acid residues defining the Phe121 pocket (all drugs are omitted for clarity). The position of amino acid residues of the ligand-free CYP46A1 is shown for comparison. The CYP46A1 carbon atoms are colored light blue (POS-bound P450), cyan (CLO-bound P450), coral (VOR-bound P450), and yellow (ligand-free P450). The heme carbon atoms are red (POS-bound P450), salmon (CLO-bound P450), and brown (VOR-bound P450). Coloring of other atoms is the same as in Fig. 3.
The architecture of the CYP46A1 active site/access channel also determines the linear configuration of POS molecule because the flexible parts of the drug are surrounded by the amino acid side chains and atoms of the peptide backbone at a 3.4–5.0 Å distance, restraining the rotation of these parts (Fig. 3B). The carbonyl of Ala474 and the side chains of Leu112 and Tyr109 are near the flexible methoxy linker. The side chains of Leu112 and Tyr109 also define the position of the POS phenyl ring along with the side chains of Phe371 and Trp368. The Met108 carbonyl and side chains of Ala111 and Phe80 surround the POS piperazine ring. Phe80 is also involved in restraining the second phenyl ring of POS along with the Val79 carbonyl, backbone amide of His81, and side chain of Arg226. Finally, the Phe80 carbonyl is at a 3.6 Å distance from the second triazole ring of POS, and the carbonyl oxygen of Arg226 is at 3.1 Å from the keto group substitution in this ring. There are only two atoms in the 5 Å vicinity of the 2-hydroxylpentanyl group: the carbonyl oxygen of Asn227 at 4.0 Å and the methyl group of the Leu229 side chain at 4.9 Å. Collectively, the interactions of POS with CYP46A1 are consistent with the tight-binding inhibitor.
We tested whether Thr306 is involved in the hydrogen bonding with the N2 of POS by assessing the apparent Kd of POS for the Thr306Ala mutant. The Kd increased ~17-fold (Table 1), a significant change for the elimination of a weak hydrogen bond which also has poor geometry of the O-H…N angle. Not only the Thr306 hydroxyl but also its methyl group are perhaps important for the tight drug binding. This functionality is 2.9 and 3.6 Å from the POS N2 and F2, respectively, thus constraining the drug from mobility. The smaller-size Ala substitution eliminates both the weak hydrogen bond and spatial constrain and leads to a significant change of the POS Kd for CYP46A1.
Comparison of Azole Binding to CYP46A1.
We superimposed the crystal structure of POS-bound CYP46A1 with those of the CLO- and VOR-bound enzyme (PDB IDs 3MDT and 3MDV, respectively) determined by us previously (Mast et al., 2010). This superimposition (Fig. 4A) revealed that in VOR and POS the coordinating nitrogen occupies similar spatial positions and is almost orthogonal relative to the heme iron with the N–Fe distance being 2.0–2.1 Å. In CLO-bound CYP46A1, the CLO nitrogen closest to the Fe (N3) is at 4 Å, too far to ligate the heme Fe. Yet the N3 of CLO aligns well with the N1 of VOR and POS through which the azole ring is attached to the rest of the molecule.
Remarkably, for all three azole complexes, the long axis of the POS molecules serves as a border line for the positioning of all drug functionalities. This is due to the Phe121 pocket above the axis, which can accommodate the bulky difluorophenyl ring of POS, two rings of CLO, and two rings of VOR. The pocket is defined by Val297, Thr298, Ile301, Ala302, Glu305, and Thr306 from the I-helix; Phe121, Gly124, Val126, and Ser127 from the B′–C loop; Ala367 from the K-β1–4 loop; Ala474 and Thr475 from the β4-1–β4-2 loop; Tyr109 and Leu112 from the B′ helix; and Leu219 and Ile222 from the F helix (Fig. 4B).
To test the pocket plasticity, we compared the three azole-bound CYP46A1 structures with that of the ligand-free CYP46A1 (Fig. 4B). This comparison reveals that most of the residues in this pocket adjust their position in response to the inhibitor binding with the pocket, changing its shape and size to better fit the drug molecule. The pocket plasticity, however, is not unlimited as indicated by the binding properties of the A474V mutant (Table 1). This larger size substitution significantly increased the apparent Kd of CLO and VOR, which place two of their rings in this pocket, and did not essentially alter the Kd of POS, which has to accommodate only one bulky substituent in this region.
Thus, the tight binding to CYP46A1 of the azole antifungals seems to depend on the ability of the drug to adopt a conformation that accommodates the Phe121 pocket in the CYP46A1 active site and places the drug’s main backbone along the imaginary axis that runs in the middle of the CYP46A1 substrate access channel. To test this notion, we used three different approaches: 1) we compared the Kd for CYP46A1 of the marketed azoles that are structurally related to VOR and POS, 2) we compared POS binding to different P450s, and 3) we compared the binding of azoles with the binding of the other CYP46A1 ligands.
Binding to CYP46A1 of the Structural Analogs of VOR and POS.
There are three marketed or experimental azoles—fluconazole, albaconazole, and ravuconazole—that similar to VOR; they have the triazole and difluorophenyl rings but the varying third functionality linked via the butanol linker (Fig. 1). VOR, albaconazole, and ravuconazole are all derivatives of fluconazole, in which this third functionality, the triazole ring, was replaced with the fluoropyrimidine ring (VOR), quinazoline ring (albaconazole), or the conjugated thiazole-benzonitrile ring system (ravuconazole). Based on the size of the third functionality, the four azoles could be ranked as follows: fluconazole < VOR < albaconazole < ravuconazole.
Similar to VOR, fluconazole and albaconazole induce a type II spectral response in CYP46A1, suggesting that the binding modes of the three antifungals to the CYP46A1 are similar, that is, through the coordination of the P450 heme iron by the triazole ring. Accordingly, the Kd of these triazoles for CYP46A1 likely reflects the goodness of fit of their varying third functionality to the Phe121 pocket above the heme iron (Table 2). Indeed, the triazole and quinazoline rings of fluconazole and albaconazole are either too small or too large for this pocket; hence, the Kd of these two antifungals is in a low micromolar range and more than 10 times higher than that of VOR, whose fluoropyrimidine ring fits the pocket well.
TABLE 2.
Binding to ∆(2–50)CYP46A1 of the structural analogs of POS and VOR
The order of the azoles reflects the increasing size/length of the varying functionality. The results represent the average of triplicate measurements ± S.D.
| Drug | Kd |
|---|---|
| μMa | |
| VOR and Its Analogs | |
| Fluconazole | 5.2 ± 0.3 |
| VOR | 0.22 ± 0.02 |
| Albaconazole | 2.0 ± 0.2 |
| Ravuconazole | 0.11 ± 0.01b |
| POS and Its Analogs | |
| Ketoconazole | 2.3 ± 0.2 |
| Itraconazole | NSRc |
| POS | 0.03 ± 0.01 |
All azoles, except itraconazole and ravuconazole, induced in CYP46A1 a type II spectral response.
Type I spectral response.
NSR, nondetectable spectral response (<0.002 absorbance units).
In contrast, the binding of ravuconazole to CYP46A1 is different because this antifungal induces a type I spectral response that occurs when the water molecule coordinating the heme iron in resting P450 is displaced by the added drug, leaving the sixth coordination position unoccupied. A different mode of binding is also consistent with the low Kd of ravuconazole, similar to that of VOR. Because the thiazole-benzonitrile ring system of ravuconazole is longer than the quinazoline ring of albaconazole, the Kd of ravuconazole should be higher than that of albaconazole, if the triazole of the drug coordinates the heme iron. It is, however, 20 times lower, which suggests that the nitrogen of the triazole ring is not oriented toward the heme iron.
We also compared the Kd of the two marketed azoles ketoconazole and itraconazole, both structural analogs of POS (Fig. 1). POS is a derivative of itraconazole, which in turn is a derivative of ketoconazole. As compared with ketoconazole, itraconazole is longer because it contains two additional rings, the phenyl and triazole, and a different terminal group. POS has the same number of rings as itraconazole but a different, more hydrophilic terminal group. Also, POS has the oxolane ring instead of the dioxolane ring, and the fluorine substitutions instead of chlorine substitutions in one of the phenyl rings.
Similar to POS, ketoconazole induces a type II spectral response in CYP46A1, suggesting the coordination of the heme Fe by the triazole ring. However, the apparent Kd of ketoconazole is >75-fold higher than that of POS (Table 2), likely because the shorter ring system provides a lesser number of interactions with CYP46A1 than the longer ring system of POS. Itraconazole did not induce any detectable spectral response in CYP46A1, likely because itraconazole is more hydrophobic than ketoconazole and POS and during titration precipitates upon addition to the aqueous buffer. This interpretation is consistent with the knowledge that itraconazole administered in a capsular form has the lowest oral bioavailability among the systemic azole (Maertens, 2004). Hence, we titrated CYP46A1 with itraconazole from the stock solution in dimethylsulfoxide. There was a weak type II spectral response in CYP46A1 that was insufficient to accurately determine the apparent Kd.
POS Binding to Different P450s.
CYP46A1 is not the only P450 crystallized in complex with POS. Previously, POS was cocrystallized with T. cruzi CYP51 (TcCYP51, PDB ID 3K1O) and T. brucei CYP51 (TbCYP51, PDB ID 2X2N), the two target enzymes from the pathogenic protozoan species (Chen et al., 2010; Lepesheva et al., 2010). In TbCYP51, POS adopts two conformations, hence two POS-bound structures of this P450 and POS-bound structures of each CYP46A1 and TcCYP51 were used for the P450s alignment based on protein α-carbons. This structural alignment revealed that binding conformations of POS in TcCYP51 and TbCYP51 are different from that in CYP46A1 (Fig. 5). In the three CYP51 structures, the methoxy linker connecting the POS oxolane and phenyl rings is bent, whereas in the CYP46A1 structure this linker has a linear configuration. Accordingly, there is at least one ∼100° turn in the POS conformation in the protozoan CYPs 51, and no such turn in POS complexed with CYP46A1. This difference in the conformations reflects how POS enters the P450 molecule and orients its triazole and difluorophenyl rings in the enzyme active site. In both TcCYP51 and TbCYP51, POS enters the P450 through the opening on the surface defined by the A′ and F′′ helices and the β10–β11 loop. In CYP46A1, the site of entry is different and defined by the helix B′, the β1–1–β1–2 loop and the junction of the helix F and F-G loop.
Fig. 5.

POS conformations in different P450s. POS carbon atoms are magenta (in CYP46A1 cocomplex), cyan and orange (in TbCYP51 co-complexes), and brown (in TcCYP51 cocomplex). The heme carbon atoms are red (CYP46A1), purple (TcCYP51), and dark salmon and brown (the two TbCYPs 51). Coloring of other atoms is the same as in Fig. 3.
Also different is the shape of the active site, including the shape and location of the pouch accommodating the POS difluorophenyl ring. In TcCYP51 and TbCYP51, this pouch represents a linear extension of the substrate access channel above the plane of the heme ring D and its methyl group. Consequently, POS piperazine, phenyl, and difluorophenyl rings form the linear chain with the triazole ring at an angle to this chain. In CYP46A1, however, this pouch is not the extension of the access channel over the heme plane but a pocket (the Phe121 pocket) above this extension, which also lies over the different heme ring (ring C and its vinyl group) as compared with the protozoan CYPs 51. Such active site topology places the difluorophenyl ring almost perpendicular to the long axis of the drug with the triazole ring being on the long axis.
Thus, to fit the differently positioned substrate access channels and differently shaped active sites, POS adopts different conformations in the protozoan CYPs 51 and CYP46A1, and this conformational adjustment seems to be the mechanism whereby the flexible POS exhibits undesired binding to CYP46A1, an enzyme that in turn also adjusts its conformation to better fit the drug molecules. Despite different binding conformations, the apparent Kd of POS for TcCYP51 is 60 nM (Lepesheva et al., 2010), similar to that for CYP46A1 (30 nM).
Binding to CYP46A1 of Ligands/Drugs Other Than Antifungal Azoles.
Besides VOR, CLO, and POS, CYP46A1 was cocrystallized with the substrate cholesterol sulfate (PDB ID 2Q9F), antidepressants tranylcypromine (PDB ID 3MDR) and fluvoxamine (PDB ID 4ENH), anticonvulsant thioperamide (PDB ID 3MDM), and anticancer agent bicalutamide (PDB ID 4FIA) (Mast et al., 2008, 2010, 2013b). Superimposition of these cocomplexes demonstrates that the positioning of all the ligands, except bicalutamide and fluvoxamine, follows a similar trend as the positioning of the antifungal azoles (Fig. 6, A and B). The phenyl ring of small tranylcypromine is in the same Phe121 pocket above the I helix, whereas the “long” thioperamide and cholesterol sulfate align well with the long axis of POS.
Fig. 6.

Binding of different ligands to CYP46A1. (A) Binding of POS (carbon atoms are magenta), VOR (carbon atoms are marine), and CLO (carbon atoms are wheat) as compared with that of tranylcypromine (carbon atoms are black), thioperamide (carbon atoms are lime), and cholesterol 3-sulfate (carbon atoms are gold). (B and C) Two views of POS binding as compared with that of bicalutamide (carbon atoms are green) and fluovoxamine (carbon atoms are brown). A and B also show Phe121 and the P450 secondary structural elements defining the Phe121-pocket. Panel C shows Trp368 and the CYP46A1 secondary structural elements defining the Trp368-pocket. The heme carbon atoms are red (POS-bound CYP46A1), brown (VOR-bound CYP46A1), salmon (CLO-bound CYP46A1), olive (tranylcypromine-bound CYP46A1), violet (thioperamide-bound CYP46A1), orange (cholesterol-sulfate-bound CYP46A1), light raspberry (bucalutamide-bound CYP46A1), and brown (fluvoxamine-bound CYP46A1). Coloring of other atoms is the same as in Fig. 3.
Bicalutamide and fluvoxamine are unusual drugs in terms of binding to CYP46A1 as they likely change their conformation once in the enzyme active site. Bicalutamide probably enters the CYP46A1 substrate access channel in the extended conformation, and then becomes folded inside the active site because of the conformational changes in CYP46A1 (Mast et al., 2013b). In contrast, fluovoxamine likely enters the substrate access channel in a folded conformation, and then unfolds when it reaches the active site (Mast et al., 2012). These specifics of binding make bicalutamide position its phenyl ring and cyano group along the median of the substrate access channel, the trifluoromethyl group in the Phe121 pocket above the I helix, and fluorophenyl ring in another pocket (the Trp368-pocket) in the active site (Fig. 6C). Likewise, fluvoxamine uses the Phe121 pocket above the I helix for its methoxy-containing arm and the Trp368-pocket for the CF3-phenyl ring. Thus, apparent differences in the positioning of fluvoxamine and bicalutamide represent the simultaneous use of the two pockets in the CYP46A1 active site.
Discussion
The present work is a part of our ongoing crystallographic studies (Mast et al., 2010, 2012, 2013b) aimed at the understanding of why some of marketed drugs bind tightly to CYP46A1 in vitro and thus have a potential for undesired interactions with the P450 in vivo. Previously, we established the structural basis of the tight binding to CYP46A1 of the branched but compact VOR and bulky but small CLO (Mast et al., 2010). Herein, we determined the crystal of CYP46A1 in complex with POS, the longest antifungal azole. The determination of this structure enabled the structural alignment of the three azole-CYP46A1 cocomplexes and revealed the features of the azole binding that were not evident during the alignment of the VOR- and CLO-bound CYP46A1.
Specifically, both VOR and CLO have the asymmetric positioning of their rings in the CYP46A1 active site which are clustered in one region, the Phe121 pocket above the I-helix. Yet only the POS-CYP46A1 structure with the drug difluorophenyl ring in the same pocket and the rest of the molecule forming a linear sequence made it more obvious that the tight binding of different antifungal azoles to CYP46A1 is perhaps determined by their conformational flexibility and the ability to accommodate one or two of their rings in this pocket and arrange the other rings linearly similar to those in POS. The Kd of fluconazole and albaconazole, structural analogs of VOR, support this notion as are the different POS conformations in CYP46A1 and the CYP51 enzymes. The analysis of the azole binding to CYP46A1 also helped to better understand how other drugs interact with this enzyme and identify the preferred sites for their active site accommodation. Thus, one of the determinants of the tight azole (or other drugs) binding to CYP46A1 is the ability of a pharmaceutical to adopt a conformation that places with a good fit its noncoordinating functionalities into the pocket above the P450 I helix.
The other determinant of the tight binding could be the length of an azole as indicated by a comparison of the CYP46A1 Kd for POS with that of ketoconazole. However, this feature is also a determinant of the tight azole binding to the pathogenic CYPs 51 (Katragkou et al., 2012). The substrates for CYPs 51 and CYP46A1 are the structurally similar sterols, and both P450 families require a long access channel to accommodate their substrates. Hence, longer azoles will have additional interactions with both CYPs 51 and CYP46A1.
Structural similarity of cholesterol and the sterol substrates of CYPs 51 likely represents the third determinant of the undesired azole binding to CYP46A1. The CYP51 substrates have the same as cholesterol structural scaffold represented by the tetracyclic ring system flanked by a hydroxyl group at C3 and a branched iso-octyl side chain at C17 (Lepesheva and Waterman, 2007). The difference from cholesterol is in the substitutions in the ring system and the presence and position of the double bond(s). Thus, tight azole binding to CYP46A1 is likely not a coincidence but a reflection of the similarity of the metabolized substrates. This notion is consistent with available information on another structurally characterized sterol (cholesterol) metabolizing P450 CYP11A1, which also has a long substrate access channel (Mast et al., 2011; Strushkevich et al., 2011) and binds ketoconazole and POS with the low micromolar Kd (Mast et al., 2013a).
The superimposition of the three CYP46A1-azole co-complexes and the drug-free P450 indicates that not only the structure and conformational flexibility of the azoles and similarity of cholesterol to the CYP51 substrates but also the plasticity of the CYP46A1 active site is a determinant of the tight azole binding. This plasticity is in contrast to the structural conservation of the rigid substrate binding cavity in eukaryotic CYPs 51 as indicated by the structural analysis of the enzymes from humans and pathogenic T. cruzi, T. brucei, and L. infantum (Lepesheva and Waterman, 2011). In bacterial CYP51 from M. tuberculosis, the azole (fluconazole) enters the active site through a different channel yet occupies a position relative to the heme group similar to that of fluconazole in TbCYP51 and TcCYP51. Furthermore, a comparison of fluconazole binding to CYP51 from M. tuberculosis (PDB ID 1EA1) with that of ketoconazole to human CYP51 (PDB ID 3LD6) indicates that ketoconazole could bind to this bacterial CYP51 using the same space for the dihalogenated phenyl ring as the fluconazole and fit the rest of the molecule in the channel corresponding to that in human CYP51 (Strushkevich et al., 2010). The conservation of the azole binding to CYPs 51 as opposed to the plasticity of the active site of CYP46A1 along with the substrate similarity suggests that design of inhibitors for CYPs 51 that will not bind to CYP46A1 will be a challenge.
In summary, many, if not all, of the antifungal azoles that are currently on the market have been developed empirically, without the knowledge of the P450 crystal structures that have become available only within the last decade. Currently, we have structural information on a number of CYPs 51 from different species, yet only a limited number of human P450s (CYPs 51, 3A4 and 46A1) were cocrystallized with the marketed azoles. Among these P450s, CYP46A1 is the enzyme crystallized with the largest number of the marketed azoles. The present work demonstrates that only the third CYP46A1–azole complex made it possible to combine all the previous structural information on different CYP46A1–drug complexes and develop a general picture of the tight azole–drug binding. This example suggests that more human P450s as well as human CYP51 and CYP3A4 with additional azoles should be crystallized to facilitate the development of specific antifungal agents.
Supplementary Material
Acknowledgments
The authors thank Dr. M. Shimoji for generating the Thr306Ala CYP46A1 mutant, and the staff at the Stanford Synchrotron Radiation Lightsource for assistance in data collection.
Abbreviations
- CLO
clotrimazole
- P450
cytochrome P450
- POS
posaconazole
- PDB
Protein Data Bank
- VOR
voriconazole
Authorship Contributions
Participated in research design: Mast, Stout, Pikuleva.
Conducted experiments: Mast, Zheng, Stout.
Performed data analysis: Mast, Stout, Pikuleva.
Wrote or contributed to the writing of the manuscript: Mast, Stout, Pikuleva.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM62882]. I.A.P. is a recipient of the Jules and Doris Stein Professorship from the Research to Prevent Blindness.
Portions of this research were performed at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research, and by the National Institutes of Health National Institute of General Medical Sciences (NIGMS) [Grant P41GM103393] and the National Center for Research Resources (NCRR) [Grant P41RR001209]. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR, or NIH.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Arnold TM, Dotson E, Sarosi GA, Hage CA. (2010) Traditional and emerging antifungal therapies. Proc Am Thorac Soc 7:222–228 [DOI] [PubMed] [Google Scholar]
- Björkhem I, Lütjohann D, Breuer O, Sakinis A, Wennmalm A. (1997) Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24(S)-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J Biol Chem 272:30178–30184 [DOI] [PubMed] [Google Scholar]
- Chen CK, Leung SS, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. (2010) Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl Trop Dis 4:e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763 [DOI] [PubMed] [Google Scholar]
- Ekroos M, Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA 103:13682–13687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- Girmenia C. (2009) New generation azole antifungals in clinical investigation. Expert Opin Investig Drugs 18:1279–1295 [DOI] [PubMed] [Google Scholar]
- Guengerich FP.(2005) Human cytochrome P450 enzymes, in Cytochrome P450 (Ortiz de Montellano PR. ed) pp 377–530, Kluwer Academic/Plenum, New York [Google Scholar]
- Hargrove TY, Wawrzak Z, Liu J, Nes WD, Waterman MR, Lepesheva GI. (2011) Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (CYP51) from Leishmania infantum. J Biol Chem 286:26838–26848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr D Biol Crystallogr 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katragkou A, Tsikopoulou F, Roilides E, Zaoutis TE. (2012) Posaconazole: when and how? The clinician’s view. Mycoses 55:110–122 [DOI] [PubMed] [Google Scholar]
- Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. (2010) Structural insights into inhibition of sterol 14α-demethylase in the human pathogen Trypanosoma cruzi. J Biol Chem 285:25582–25590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. (2007) Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta 1770:467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. (2011) Structural basis for conservation in the CYP51 family. Biochim Biophys Acta 1814:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp HP. (2010) Clinical pharmacodynamics and pharmacokinetics of the antifungal extended-spectrum triazole posaconazole: an overview. Br J Clin Pharmacol 70:471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. (2003) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 278:22980–22988 [DOI] [PubMed] [Google Scholar]
- Maertens JA. (2004) History of the development of azole derivatives. Clin Microbiol Infect 10 (Suppl 1):1–10 [DOI] [PubMed] [Google Scholar]
- Mast N, Andersson U, Nakayama K, Bjorkhem I, Pikuleva IA. (2004) Expression of human cytochrome P450 46A1 in Escherichia coli: effects of N- and C-terminal modifications. Arch Biochem Biophys 428:99–108 [DOI] [PubMed] [Google Scholar]
- Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, Pikuleva IA. (2011) Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J Biol Chem 286:5607–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD. (2010) Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem 285:31783–31795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Clark M, Wiseman J, Stout CD, Pikuleva IA. (2012) In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol Pharmacol 82:824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Pikuleva IA. (2013a) Inhibition and stimulation of activity of purified recombinant CYP11A1 by therapeutic agents. Mol Cell Endocrinol 371:100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci USA 105:9546–9551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Zheng W, Stout CD, Pikuleva IA. (2013b) Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J Biol Chem 288:4613–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. (2007) Phaser crystallographic software. J Appl Cryst 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255 [DOI] [PubMed] [Google Scholar]
- Omura T, Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. J Biol Chem 239:2370–2378 [PubMed] [Google Scholar]
- Podust LM, Poulos TL, Waterman MR. (2001) Crystal structure of cytochrome P450 14α -sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci USA 98:3068–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaati M, Mast N, Beck O, Nayef R, Heo GY, Björkhem-Bergman L, Lütjohann D, Björkhem I, Pikuleva IA. (2010) The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase in vitro and in vivo. J Lipid Res 51:318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis SM, Cohen AE, Deacon A, Eriksson T, González A, McPhillips S, Chui H, Dunten P, Hollenbeck M, Mathews I, et al. (2008) New paradigm for macromolecular crystallography experiments at SSRL: automated crystal screening and remote data collection. Acta Crystallogr D Biol Crystallogr 64:1210–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park HW. (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc Natl Acad Sci USA 108:10139–10143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strushkevich N, Usanov SA, Park HW. (2010) Structural basis of human CYP51 inhibition by antifungal azoles. J Mol Biol 397:1067–1078 [DOI] [PubMed] [Google Scholar]
- White MA, Mast N, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. (2008) Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr D Biol Crystallogr 64:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TC, Marr KA, Bowden RA. (1998) Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin Microbiol Rev 11:382–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.