

Abstract
We tested the idea that ancestral class I and II aminoacyl-tRNA synthetases arose on opposite strands of the same gene. We assembled excerpted 94-residue Urgenes for class I tryptophanyl-tRNA synthetase (TrpRS) and class II Histidyl-tRNA synthetase (HisRS) from a diverse group of species, by identifying and catenating three blocks coding for secondary structures that position the most highly conserved, active-site residues. The codon middle-base pairing frequency was 0.35 ± 0.0002 in all-by-all sense/antisense alignments for 211 TrpRS and 207 HisRS sequences, compared with frequencies between 0.22 ± 0.0009 and 0.27 ± 0.0005 for eight different representations of the null hypothesis. Clustering algorithms demonstrate further that profiles of middle-base pairing in the synthetase antisense alignments are correlated along the sequences from one species-pair to another, whereas this is not the case for similar operations on sets representing the null hypothesis. Most probable reconstructed sequences for ancestral nodes of maximum likelihood trees show that middle-base pairing frequency increases to approximately 0.42 ± 0.002 as bacterial trees approach their roots; ancestral nodes from trees including archaeal sequences show a less pronounced increase. Thus, contemporary and reconstructed sequences all validate important bioinformatic predictions based on descent from opposite strands of the same ancestral gene. They further provide novel evidence for the hypothesis that bacteria lie closer than archaea to the origin of translation. Moreover, the inverse polarity of genetic coding, together with a priori α-helix propensities suggest that in-frame coding on opposite strands leads to similar secondary structures with opposite polarity, as observed in TrpRS and HisRS crystal structures.
Keywords: sense/antisense double open reading frames, origin of translation, aminoacyl-tRNA synthetases, protein modularity, multiple sequence alignment, multiple structure alignment, ancestral gene reconstruction
Introduction
Aminoacyl tRNA synthetases (aaRS) occur in either of two structurally unrelated classes, I or II, according to the amino acid they activate (Cusack et al. 1990; Eriani et al. 1990; Carter 1993). Rodin and Ohno (1995) proposed that these two unrelated enzyme superfamilies descended from the same gene, one ancestor coded by each complementary ancestral strand. Although the evidence on which Rodin and Ohno based their proposal was quite strong, the concept has nevertheless proven difficult to embrace, because genetic complementarity between coding sequences severely constrains the sequence spaces that can be explored while simultaneously optimizing gene products translated from both strands.
The decisive selective advantage of sense/antisense coding (Pham et al. 2007) appears from the fact that amino acid specificities of the two aaRS classes are significantly skewed. Class I aaRS substrates have a favorable median free energy of transfer from water to cyclohexane, whereas substrates of class II aaRS are less favorable by approximately 4 kcal/mol and prefer water. Thus, ancestral class I and class II enzymes appear to have been required to create, respectively, the cores and solvent interfaces of primordial globular proteins. Genetic linkage implied by sense/antisense coding may then have ensured that activated amino acids of both types would be produced at the same time and place, increasing the likelihood of producing and selecting viable gene products.
tRNA aminoacylation is the defining reaction in codon-dependent translation. Primordial enzymes enabling the process were likely among the earliest catalysts. By virtue of that privileged position, it is also likely that their contemporary descendants include large portions of the proteome. It therefore seems of paramount importance to assess the validity of the hypothesis. For, if the hypothesis is correct, established paradigms must be revisited in light of the possibility of tracing so much of life as we know it to a single ancestral gene.
The Rodin–Ohno hypothesis makes testable biochemical and bioinformatic predictions. Segments corresponding to regions implicated in sense/antisense coding should exhibit catalytic activities characteristic of the full-length enzymes. To test this biochemical prediction, we developed procedures for stabilizing and expressing such constructs. We call them Urzymes, from Ur = primitive, original, earliest plus enzyme. A 130-residue class I tryptophanyl-tRNA synthetase (TrpRS) Urzyme from which the anticodon-binding domain and the long connecting peptide (CP1) separating the HIGH and KMSKS active-site signatures had been removed (Pham et al. 2007, 2010) accelerated tryptophan activation 109-fold. We subsequently demonstrated that 120–140-residue active-site fragments derived from the implicated region of class II histidyl-tRNA synthetase (HisRS; Li et al. 2011) have comparable catalytic activity, and that both Urzymes catalyze acylation of cognate tRNAs (Li L, Francklyn CS, Carter CW Jr, in preparation).
These catalytic activities make Urzymes important resources for both experimental and bioinformatic study of putative steps in the foundational evolution of catalytic activity and specificity from before the evolutionary era that is accessible through ancestral gene resurrection (Thornton 2004; Bridgham et al. 2006; Benner et al. 2007; Gaucher et al. 2008). The class I and II Urzymes both correspond to the most highly conserved and hence most ancient active-site components. Further, they are compatible with antiparallel alignment of their coding sequences, as envisioned by Rodin and Ohno (Pham et al. 2007). Their high, and comparable, catalytic activities imply that they are both stable and globular, at least in the presence of substrates, and therefore afford unexpectedly strong support for the Rodin–Ohno hypothesis.
This surprising biochemical validation invites further bioinformatic tests for possible sense/antisense coding relationships. Urzyme construction relies primarily on tertiary structural alignment and protein design, and hence is distinct from the study of nucleic acid sequences that might have encoded extinct enzyme precursors. Nonetheless, the comparable lengths and approximate antiparallel alignment of the TrpRS and HisRS Urzymes do suggest how to extend the bioinformatic approach described by Rodin and Ohno (1995) to include, in addition to their catalytic signatures, the secondary structural elements necessary to orient them. Although those authors were able to detect significant sense/antisense relationships in the full codons of the catalytic signatures, such information has long been stripped by gene duplication and speciation from the remaining parts of the presumably extinct ancestral aaRS sequences. Thus, the sense/antisense linkage between the two enzyme superfamilies, if it did exist, was abolished very early, possibly well before the genetic code even reached a canonical twenty amino acids with variable presence of synthetases for the amides, asparagine, and glutamine (Woese et al. 2000), let alone the idiosyncratic variation seen today with the extensions to selenocysteine and pyrrolysine (Ibba and Soll 2004).
Owing to the degeneracy of the genetic code and the wobble hypothesis (Crick 1966), we reasoned that the most persistent base-pairing remnant in sequences coding for descendants derived from ancestral aaRS genes on opposite strands would be residual middle- or second-base pairing between codons specifying the secondary structures that position active-site residues. We refer to this metric as <MBP>, for the “Middle (second) codon-Base Pairing” frequency. We examine here the overall <MBP> in multiple sense/antisense alignments derived from diverse contemporary TrpRS and HisRS sequences, its profile along the sequences, and its behavior in reconstructed ancestral sequences.
Global analysis of possible sense/antisense coding relationships between class I and II aminoacyl-tRNA synthetase sequences is a daunting task, because insertions and deletions have been possible along all independent trajectories throughout the biological era. We address here the more modest goal of assessing statistical evidence for ancestral sense/antisense coding derived from extant coding sequences only of the TrpRS and HisRS Urzymes.
The TrpRSċHisRS pair is not an obvious choice for comparison, for several reasons. TrpRS uses TIGN, an unusual variant of the class I HIGH signature. TrpRS belongs to class Ic, whereas HisRS belongs to class IIa; thus, they are probably more distantly related and may retain a weaker trace of their common complementary ancestry. Finally, both contemporary synthetases bind tRNA in the major groove (Yang et al. 2006), whereas most class I aaRS use a minor groove binding mode, suggesting that the ancestral states of class I and II aaRS likely bound to opposite grooves of the tRNA 3′ acceptor stem (Ribas de Pouplana and Schimmel 2001).
For various reasons, we nevertheless focus here on coding properties of the TrpRSċHisRS pair. Most importantly, these are the two aaRS for which we have demonstrated catalytically active Urzymes. Class Ia enzymes generally have both longer CP1 and additional insertions within the Rossmann fold. Thus, as the smallest of the class I aaRS, TrpRS poses the fewest difficulties in Urzyme design. Further, outside the TrpRS and HisRS families, gaps and insertions introduce additional difficulties in deciding which coding sequences from the class I and II superfamilies should be structurally aligned to evaluate the <MBP> metric in aligned codons. Finally, although it uses the TIGN catalytic signature, TrpRS contains the correct number of amino acids between the conserved proline and TIGN to align with both hydrophobic and charged residues in the class II motif 2 (Rodin and Ohno 1995).
Pham et al. (2007) described a step toward a putative Urgene by aligning three large contemporary coding blocks from catalytic domains of Bacillus stearothermophilus TrpRS and Escherichia coli HisRS sense and antisense opposite one another. That alignment revealed that middle bases in 44% of codons throughout a 94-residue sense/antisense alignment spanning the active sites (fig. 1) were base-paired with their complements. A naive random probability based simply on one in four bases is approximately 0.25. We previously found that the middle-base pairing frequency in alignments of simulated two-codon hexanucleotides encoding random dipeptides was 0.27 ± 0.044. The uncertainty of this estimate of the random frequency implied that the statistical significance of the frequency (0.44) observed in the B. stearothermophilus TrpRS:E. coli HisRS alignment was supported by a P value of 0.007 (Pham et al. 2007).
Fig. 1.

Modularity in class I and II aminoacyl-tRNA synthetase Urzymes. Complementary modularity within aminoacyl-tRNA synthetase active sites. (A) Summary of information derived from the respective TrpRS and HisRS multiple sequence alignments. The Bacillus stearothermophilus TrpRS and Escherichia coli HisRS sequences are aligned antisense to each other, consistent with the Rodin–Ohno hypothesis. The three modular fragments comprising the Urgene are indicated by a thin colored strip between sequence and secondary structure codes. Yellow blocks indicate the motifs used to anchor the three coding blocks, which were assembled end-to-end to form a single “gene.” Codon middle base pairs are magenta, unpaired middle bases are blue. Sequence entropies of class I (darker shading) and class II (lighter shading) are shown as histograms above and below the sequences. Conserved positions have low sequence entropies. Significant numbers of inserted residues in the respective MSAs occur at sites indicated by black arrows. Secondary structures from the crystal structures of the full-length enzymes are encoded as: H = α-helix, E = extended (β), T = turn, and S = loop. (B) Decomposition of tertiary structures inferred from crystal structures of the intact enzymes into the three modular coding blocks, colored as in (A). Aminoacyl-5′AMP intermediates are denoted by spheres. Note that the class I (TrpRS) active site encloses the indole moiety completely, while the imidazole moiety of histidine has extensive solvent-exposed surface area. (C) Relationship between the Urgene in (A) and the active Urzymes. Gaps between the excerpted fragments (dark gray additions) represent the remaining puzzle of reconstructing an alignment coding two intact active enzymes (see Discussion).
We analyze here sense/antisense alignments derived from a much broader sample of contemporary multiple sequence alignments for TrpRS and HisRS, to investigate further the statistical significance of the 94-residue antiparallel construction in figure 1. The statistical evidence for middle-base pairing frequency is robust, and extends to profiles of middle-base pairing versus residue number and to increased frequencies in ancestral sequences reconstructed from most probable phylogenetic trees. These results provide strong bioinformatic support for the Rodin–Ohno hypothesis.
New Approaches
This work entails several novelties, arising from the unusual purpose of presenting bioinformatic evidence for ancestral sense/antisense genetic coding. First, we examine the statistical behavior of relationships between contemporary sequences of two distinct protein families, to infer characteristics of ancestral genes from an era close to the advent of genetic coding. Second, owing to the pervasive problem of indels, we use three-dimensional structure superposition to identify and assemble mosaic “Urgenes” encoding conserved secondary structures, along with highly conserved active-site residues, for both families. Third, we develop and examine a variety of data sets representing the null hypothesis that the ancestral sequences were not complementary. Fourth, we use k-means clustering and a two-dimensional clustering vector based on average complementarity and its position sensitivity along the sequence to distinguish between the sense/antisense and null hypotheses. Finally, we estimate the complementarity metric in the time domain by reconstructing ancestral genes from most-probable phylogenetic trees of middle- (second-) codon base sequences. We find that neither estimated divergence times (Hedges et al. 2006) nor node-heights are free of complications arising, probably, from horizontal gene transfer. This problem is discussed further in the supplementary section C, Supplementary Material online.
Results
The Putative Urgene Described Here (fig. 1) Is Excerpted from the Intact Urzymes
The original observation of Rodin and Ohno (1995) was that coding sequences for the highly conserved, class-defining “signature” sequences of the two aaRS classes had a statistically significant sense/antisense relationship, based on Jumble tests with Z scores of approximately 5.7–8.8 (Rodin and Ohno 1995). Consensus HIGH/motif 2 and KMSKS/motif 1 sense/antisense homologies of these sequences comprise only 9 and 11 amino acids, respectively, and constitute only approximately 20% of the length of the TrpRS Urzyme described in figure 4 of Pham et al. (2007) and only 5–6% of the contemporary full-length TrpRS and HisRS enzymes.
Fig. 4.

Bin assignment for TrpRS (A) and HisRS (B) ancestral nodes. The same phylogenetic information represented in figure 2 is reproduced here in horizontal cladograms, to indicate assignments of nodes grouped into each bin for purposes of plotting in figure 5. Node heights associated with each bin are included at the bottom of each colored rectangle. Bold lines denote outgroups.
To optimize correspondence between an enhanced set of contiguous, antiparallel codons, and following Pham et al (2007), we built “Urgenes” from segments reliably linked to the class-defining sequences already identified in the figure 1 of Rodin and Ohno. Three coding blocks defining the respective active sites were assembled by a semi-automated procedure from multiple sequence alignments for diverse species. Two of these blocks included amino acids corresponding to the class I HIGH- (and class II Motif 2; 46 residues, blue fragment) and KMSKS- (class II Motif 1; 18 residues, magenta fragment) containing segments. A third was provided by a linking segment involved in amino acid specificity in the class I Urzyme (30 residues, amber fragment). This amber fragment is also associated with a consensus motif; we previously identified the comparably conserved sequence GxDQ in this segment in class I active sites (Carter 1993; Pham et al. 2007). This motif and the corresponding antisense sequence in HisRS, FKRA, provide an anchor for the central segment. Anchoring motifs and structural superposition helped in adjusting for insertions and deletions in all three coding blocks and in assuring appropriate alignments.
Coding and translated amino acid sequences for B. stearothermophilus TrpRS and E. coli HisRS are aligned in opposite directions in figure 1A. The relatively higher sequence identity within a single aaRS family, together with structural considerations, increase our confidence that all alignments correspond to that constructed previously (Pham et al. 2007) with respect to three-dimensional structures. Class I and II multiple sense/antisense sequence alignments are uncorrelated by several criteria, also illustrated in figure 1. Sequence entropies, Sj,position = ∑i,j,amino acids(pi,j) × log(pi,j), derived from multiple sequence alignments show that the relative conservation of amino acids in contemporary coding sequences is uncorrelated to that on the other strand (fig. 1A; R = 0.11). Nor is there evident correlation between sites on opposite strands at which insertions are observed, indicated by the arrows, in a small number (<20% in the blue fragment and <5% in the amber fragment) of the sequences.
The resulting Urgene increased by 5-fold the number of consecutive codons subject to statistical testing over the number examined by Rodin and Ohno, at the expense of examining only the codon middle bases. We initially assembled 94-residue Urgenes for 98 TrpRS and 99 HisRS coding sequences. Multiple sequence alignments for the two Urgenes from contemporary TrpRS and HisRS sequences in the initial data set gave rise to approximately 9,700 different homologous comparisons, which were analyzed in detail. This initial database strongly emphasized bacterial sequences for both enzymes. We therefore enlarged the database to include additional archaeal/eukaryotic species to ensure more diverse multiple sequence alignments of 211 TrpRS and 207 HisRS sequences (fig. 2) and approximately 44,000 homologous sense/antisense comparisons. Species names and phylogenetic trees in Newick format for full alignments of both families, which include representative bacteria, archaea, and eukarya, are included in the supplementary material, Supplementary Material online.
Fig. 2.

Phylogenies of the abstracted 94-base coding fragments derived from TrpRS (A) and HisRS (B). Alignments were compiled by MUSCLE (Edgar 2004a, 2004b) and the trees determined by JModelTest (Guindon and Gascuel 2003; Darriba et al. 2012) and renamed using Taxnameconvert (Schmidt 2004; http://www.cibiv.at/software/taxnameconvert/, last accessed April 22, 2013) and drawn as cladograms by FigTree (Rambaut 2010). Trees in Newick format are provided in the supplementary material, Supplementary Material online. Phyla are indicated around the circumference and highlighted by different colors. Blue colors denote bacteria; amber and red denote archaea and eukaryotes, respectively.
Contemporary Coding Sequences Exhibit <MBP> = 0.35 ± 0.0002
To test the hypothesis that ancestral coding sequences of the two proteins were complementary and arose on opposite strands of the same gene, we used the initial (smaller; 98 vs. 99) MSAs to construct all-by-all sense/antisense alignments of (DNA) coding sequences from each class against the other, (H; CI vs. CII′). The middle-base pairing frequency, <MBP> was then computed for each of the 9,702 sense/antisense alignments, table 1, and used to construct the histogram shown in figure 3B and in the clustering analysis described later, which was not repeated for the larger data set.
Table 1.
Statistics for Distributions of Middle Base Pairs for the Initial Alignment.
| Hypothesis | Distribution | N | <MBP> | SD | SE |
|---|---|---|---|---|---|
| H | TrpRS vs. HisRS′ | 9,702 | 0.376 | 0.049 | 0.00049 |
| N0,a | Random peptides vs. random peptides′ | 30,000 | 0.238 | 0.042 | 0.00024 |
| N0,b | Lectin C vs. PDZ′ | 20,001 | 0.215 | 0.055 | 0.00039 |
| N1,a | TrpRS vs. TrpRS′ | 4,753 | 0.257 | 0.047 | 0.00068 |
| N1,b | HisRS vs. HisRS′ | 4,851 | 0.255 | 0.037 | 0.00053 |
Fig. 3.

Middle-base pairing frequency distributions. The frequencies of middle base pairs in all-by-all sense/antisense coding alignments for class I (TrpRS) and class II (HisRS) Urzyme genes against each other (CI vs. CII′; center) are compared with distributions for two representations of the null hypothesis, in which there is no evidence for sense/antisense coding. The first (CII vs. CII′; top) is obtained by aligning each class II sequence antisense, in turn, against all other class II sequences in the sense direction. The second (Lectin vs. PDZ; bottom) is a similar comparison of the coding sequences for identical lengths of the genes for a lectin (pfam PF00059) against those for PDZ domains (pfam PF00595). Each is fitted to a normal distribution (solid line). The mean middle-base pairing frequency for all three distributions is shown by the vertical arrow.
The <MBP> value of the smaller compilation is 0.376; the standard deviation (SD) is 0.0224. However, as a relatively large number of different comparisons contribute to this value, we quote the standard error (SE) in estimating the mean value, SE = 0.0005. Alignments for the overall database (table 2; 211 vs. 207 sequences; 43,677 sense/antisense alignments) gave a corresponding <MBP> of 0.35 ± 0.0002. This lower value likely results from the increased representation of archaeal and eukaryotic sequences, as discussed later.
Table 2.
Statistics for Distributions of Middle Base Pairs for the Larger Data Set.
| Hypothesis | Distribution | N | <MBP> | SD | SE |
|---|---|---|---|---|---|
| H | TrpRS vs. HisRS′ | 43,677 | 0.349 | 0.0545 | 0.00026 |
| N1,a | TrpRS vs. TrpRS′ | 22,366 | 0.264 | 0.0428 | 0.00029 |
| N1,b | HisRS vs. HisRS′ | 21,528 | 0.254 | 0.0443 | 0.00030 |
| N+ | +1 Frame shifting | 43,677 | 0.258 | 0.0664 | 0.00032 |
| N− | −1 Frame shifting | 43,677 | 0.261 | 0.0474 | 0.00023 |
| Ns/s | TrpRS vs. HisRS | 43,677 | 0.252 | 0.0361 | 0.00017 |
Distributions under the Null Hypothesis All Have <MBP> Close to 0.25 ± 0.0005
The statistical significance of middle-base pairing observed in pairs of coding sequences aligned in opposite directions requires an estimate for the corresponding probability under the null-hypothesis, N, that contemporary coding sequences are unrelated and middle bases pair randomly. We report five levels of tests for the distribution of <MBP> under the Null hypothesis by testing:
After frame shifting and/or randomization of one of the two sequences.
Self-complementarity of each of the two aaRS MSAs.
Peptides drawn randomly from the PDB.
Homologous MSAs of random pairs of protein families from the PDB.
MSAs paralogous families.
Initial tests of the distribution under the null hypothesis were generated from the TrpRS and HisRS sequences themselves by accumulating <MBP> distributions for the same alignments shown in figure 1, but with +1 and −1 frameshifts and by randomizing one of the two sequences. These distributions all have means close to 0.25 with standard errors similar to those of other distributions.
Figure 3 shows distributions for sense/antisense alignments under the hypothesis (H; 9702 alignments) and for two mock sense/antisense coding sequence alignments under N1,b (CII vs. CII′; 4950 alignments, fig. 3A; and N0,b sequences either for random peptides (N0,z, not shown) or for Lectin C (pfam PF00059) versus PDZ domains (pfam PF00595 ∼20000 alignments, fig. 3C). The distribution for the Lectin C:PDZ domain alignments effectively rules out the possibility that high <MBP> in the TrpRS:HisRS sense/antisense alignment arises from sequence conservation within the two respective families, which are comparable in the case of H and N0,b. All three distributions have similar widths, consistent with comparable sampling diversity. Distributions of middle-base complementarity are summarized in table 1.
The H and Ni frequencies differ by approximately 200 times the root mean squared standard error, suggesting that they are very different. We performed one-sided, two-sample t tests for equal means but different variances for H versus N0,a,b and H versus N1,a,b. P values were ≪ 10−4 for each case, and in fact, were smaller than the computational rounding error. Similar P values were obtained when multiple regression models for middle-base complementarity were built using CI/CII′ sense/antisense alignment (H) and self sense/antisense alignment (N1,a,b) as predictors. Even considering the substantial homologies in the two multiple sequence alignments, the distribution under H is clearly distinct from all distributions representing the null hypothesis. Despite the fact that the larger data set produced a lower <MBP>, the difference between H and N hypotheses actually differ by approximately 350 times the root mean square standard error.
To validate confidence further, we tested for equal medians with the nonparametric Wilcoxon rank sum test for H versus N0,b and H versus N1,a and the P values (unsurprisingly) were ≪10−6. The last test performed was a two-sample one-sided Kolmogorov–Smirnov test with the null hypothesis that the two samples come from the same distribution again, with the H versus N0,a and H versus N1,a as the two pairs. Again, we obtained P values ≪10−3. Thus, we conclude with a very high degree of confidence that nonrandom effects lift the frequency of middle base pairs in the TrpRS HisRS sense/antisense Urgene alignment well above those of distributions for either N0,a,b and N1,a,b, distributions representing the null hypothesis. Table 2 updates tests of the null hypothesis based on the larger data set of 211 TrpRS and 207 HisRS sequences: N1a, N1b, together with N+ and N−, from frameshifting one of the two sequences, and Ns/s for the alignment of TrpRS and HisRS sequences in the same orientation.
For a more discriminating assessment of <MBP> values arising from using paralogous MSAs, we considered the behavior of distant (putative) and consensus paralogous sequences derived from other aaRS families. Close extant structural relatives of the TrpRS Urzyme are found in the Toprim domains that occur in primases, topoisomerases, and recombinases. We examined <MBP> in sense/antisense alignments between a small set of Toprim domains and the HisRS MSA. Crystal structures of six unique Toprim domains were aligned with the TrpRS Urzyme (supplementary fig. S2, Supplementary Material online) using the POSA server (Ye and Godzik 2005), and optimal choices were made for equivalent residues in the “blue” and “amber” fragments (Toprim domains lack the magenta fragment). Corresponding coding sequences were retrieved and used to form sense/antisense alignments with corresponding blue and amber fragments from all 207 HisRS sequences. The <MBP> for this alignment was 0.265 ± 0.0015, which is in the range of the other tests of the null hypothesis. Thus, either Toprim domains are unrelated to the class I Urzymes or the sequence divergence is too great to preserve evidence of sense/antisense ancestry.
There are sufficient numbers of sequences for the class I (TyrRS) and class II (ProRS) outgroups, used to root phylogenetic trees for TrpRS and HisRS, to afford a test of the <MBP> behavior of close, consensus paralogs (supplementary section A, Supplementary Material online). The outgroup Urgene middle bases share only 44% sequence identity with those of their homologs, which themselves have more than 60% identity. Having identified the three excerpts of the two Urgenes from these sequences, we carried out all-by-all <MBP> calculations for the four possible class I/class II alignments: TrpRS–HisRS, TrpRS–ProRS, TyrRS–HisRS, and TyrRS–ProRS. The resulting <MBP> values are shown in supplementary figure S1, Supplementary Material online. The four alignments have essentially equivalent high <MBP> values (0.336 ± 0.005), and the student t test probability for the difference between sense/sense and sense/antisense alignments within this group, which is derived from four independent aaRS, was less than 0.0001.
The only condition that yields a unique distribution with unexpectedly elevated <MBP> values is that proposed by Rodin and Ohno (1995). The middle-base pairing frequency for aligned TrpRS and HisRS active-site coding sequences, and indeed that for all four class I/class II comparisons, far exceeds what is expected under the null hypothesis. The three-block sense/antisense alignment described by (Pham et al. 2007) is therefore almost certainly drawn from a unique protein-coding subset consistent with a sense/antisense Urgene.
High <MBP> Values Occur at Consistent Positions across the MSAs
If high middle-base pairing is nonrandom, there should be some correlation between MBP values at the same position in different sequence pairs. To investigate this possibility with the smaller data set, we examined the ability of clustering algorithms to separate the 9,702 instances generated for TrpRS/HisRS alignments, H, from either 4,851 instances generated under N1,a or 4,950 instances from N1,b. If <MBP> values are well-enough separated, k-means clustering should group values accurately into two different clusters, according to the hypothesis under which they were generated. We assumed two initial centroids to cluster the <MBP> values. The algorithm was run separately for sets H versus N1,a and H versus N1,b. The overall accuracy was similar for both representations of the null hypothesis, 91% for H versus N1,a and 79% for H versus N1,b. Clustering on the basis only of <MBP> (table 3) yields higher sensitivity (fewer false negatives) but lower specificity (more false positives).
Table 3.
One- and Two-Dimensional Clustering.
| Clustering | 1D (<MBP>) |
2D (<MBP>, <cc>) |
||
|---|---|---|---|---|
| H | N1 | H | N1 | |
| H against N1,a | ||||
| H | 8,314 | 1,388 | 9,661 | 41 |
| N1,a | 184 | 4,667 | 3 | 4,848 |
| α/Specificity | 0.143 | 0.857 | 0.004 | 0.996 |
| β/Sensitivity | 0.038 | 0.962 | 0.001 | 0.999 |
| H against N1,b | ||||
| H | 6,667 | 3,025 | 9,696 | 6 |
| N1,b | 505 | 4,248 | 0 | 4,753 |
| α/Specificity | 0.312 | 0.688 | 0.001 | 0.999 |
| β/Sensitivity | 0.106 | 0.894 | 0.000 | 1.000 |
For nonrandom middle-base pairing across a pair of MSAs, information about middle-base pairing along each sequence should complement that in the <MBP> and enhance clustering. For each group of alignments, we therefore constructed a two-dimensional clustering vector by taking the average of all base pairs in four blocks, each with 25% of the alignments. Each profile consisted of the mean residue-by-residue correlation coefficient, <cc>, for the remaining 75% of the aligned sequences. The <cc> values for all three profiles were then averaged, implementing a re-sampling cross-validation (Picard and Cook 1984). Two-dimensional clustering using (<MBP>, <cc>) vectors increased both sensitivity and specificity of clustering essentially to unity (table 3). Positional information about the high middle-base pairing therefore contributes significantly to the clustering power of the two-dimensional vector.
<MBP> Increases for Multiple Nodes of Ancestral Sequence Reconstruction
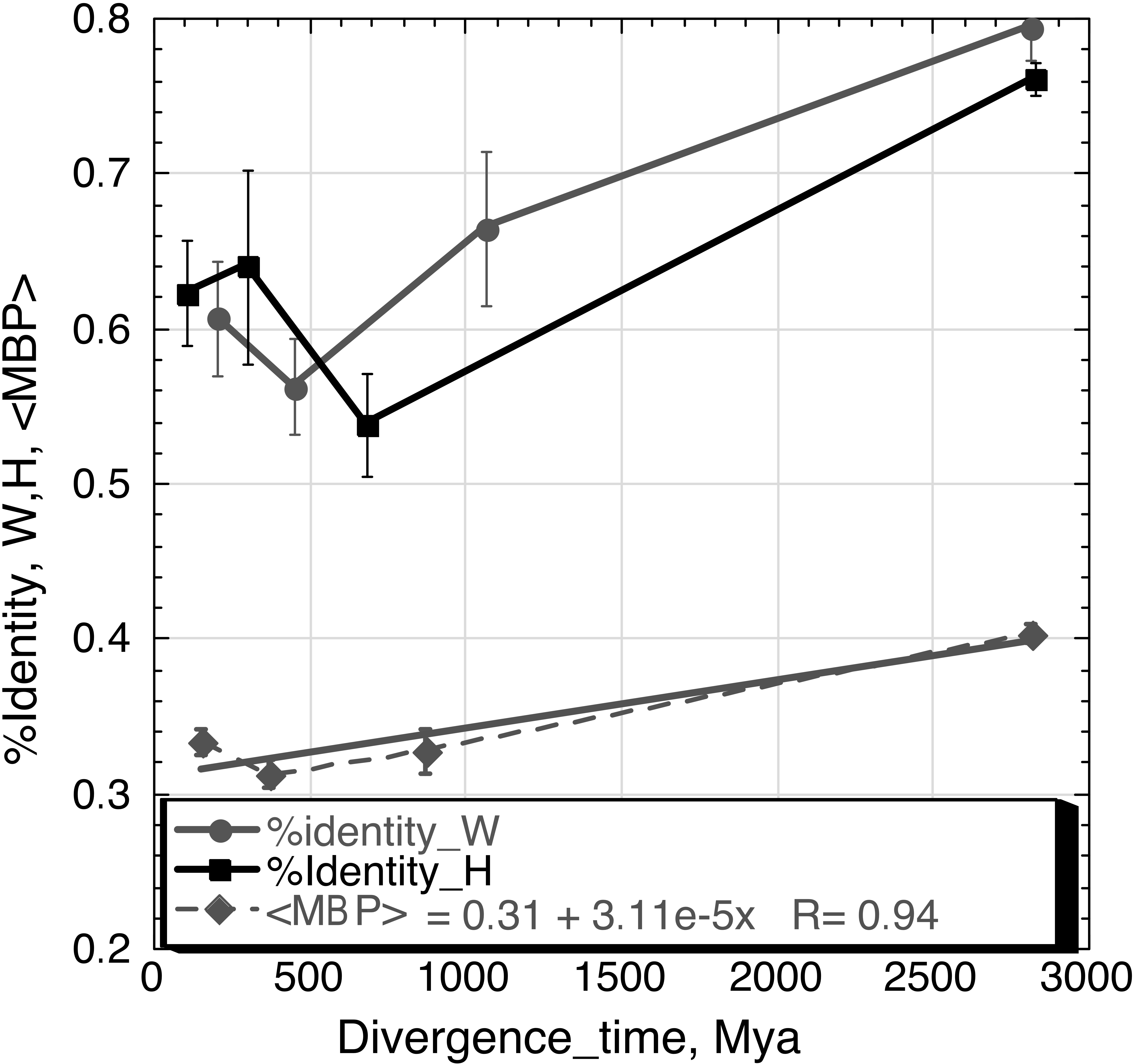
The behavior of <MBP> values in the time domain substantially strengthens our conclusion. Even in the contemporary sequences, bacterial TrpRS and HisRS sequences exhibit significantly higher <MBP> than archaeal sequences, which in turn have higher <MBP> than eukaryotic sequences (fig. 5A and table 4). Student t test probabilities are <0.0001 for the contribution to <MBP> of eukarya (−0.044), and are significant (0.01–0.007) for the contributions of the TrpRS bacterial (+0.017), and the joint presence of both TrpRS and HisRS bacterial sequences (+0.038), relative to the intercept (0.35). The regression shows that approximately 0.97 of the variation in <MBP> observed in the nine entries of figure 5A plus the full data set can be explained by the kingdom of origin. Student t test probabilities are computed on the basis of standard errors based on 6 degrees of freedom.
Fig. 5.

Sources of variation in <MBP>. (A) <MBP values for contemporary sequences, sorted according to protein family and domain of origin. Standard errors are indicated by suspended ellipses (drawn with Excel [Microsoft 2008]). Eukaryal, archaeal, and bacterial trees reconstructed separately suggest that contemporary bacterial sequences may be closer than archaeal sequences to a putative ancestral sense/antisense aaRS Urgene and hence to the origin of translation. (B) Time-dependence of <MBP> for sense/antisense alignments of reconstructed sequences for ancestral nodes (drawn with Kaleidagraph [Synergy 2005]). The expected increase in sequence complementarity was observed throughout all bins excepting the final bin, for which the reconstructions are least reliable as bacterial sequences are reconciled with archaeal in reconstructed nodes (not shown). <MBP> values for each bin except the final bin of the sense/antisense alignments of reconstructed TrpRS and HisRS 94-mer middle base sequences are plotted against the averaged node heights.
Table 4.
Regression Model for <MBP> in Contemporary TrpRS and HisRS 94-mer Sequences as a Function of Phylogenetic Domains.
| Term | Estimate | SE | t Ratio | P > |t| |
|---|---|---|---|---|
| Intercept | 0.35 | 0.0042 | 85.0 | <0.0001 |
| Eukarya | −0.044 | 0.0036 | −12.1 | <0.0001 |
| TrpRS_Bacteria | 0.017 | 0.0047 | 3.7 | 0.01 |
| TrpRS_Bact*HisRS_Bact | 0.038 | 0.0094 | 4.1 | 0.007 |
We constructed optimal phylogenetic trees for the 94-element middle base sequences from the TrpRS and HisRS sequences. Phylogenies derived for the abstracted 94-mer alignments are shown in figure 2. Despite the fact that the trees are based on middle bases of the excerpted Urgene (fig. 1), they are similar, to each other and to phylogenies derived from other kinds of MSA. Finally, although their overall appearances are approximately similar, comparison of the two trees reveals that it takes more steps for the HisRS tree to reach the root from the contemporary sequences, compared with the TrpRS tree. This is consistent with the consensus (Brown et al. 2001; O’Donoghue and Luthey-Schulten 2003; Andam and Gogarten 2011; Fournier et al. 2011) that TrpRS separated from TyrRS (used as the outgroup to root the tree) more recently than HisRS separated from other ProRS and other class IIa aaRS.
Maximum-likelihood ancestral sequence reconstruction using the Lazarus (Hanson-Smith et al. 2010) interface to PAML (Yang 2007a) for the larger database produced 210 and 206 reconstructed TrpRS and HisRS sequences, respectively. More than 75% of the reconstructed sequences had posterior probabilities more than 0.98, all were more than 0.89, and low values appeared randomly distributed. When these internal nodes were aligned sense/antisense, the <MBP>, 0.357 ± 0.002, was higher than that from contemporary alignments by 23 times the standard error.
Divergence times are difficult to assign consistently in our case because the time domain is both more extensive and hence somewhat more ill-defined than usual, and because we are comparing results derived for two different protein families, whose trees are nonisomorphous. Dating the ancestral nodes with node-height = 1 using the fraction (∼45%) of species represented in the TimeTree database (Hedges et al. 2006) gave divergence times between 2 and 3,200 Ma (supplementary section C, supplementary table S1, and fig. S3, Supplementary Material online). These reconstructed nodes are inferred from the most similar sequences, and should therefore be most recent, and yet the species from which they are derived range over the entire time period of cellular biology. Horizontal gene transfer therefore appears to be significant across the entire data set in ways we cannot track.
Under these circumstances, and as approximately half of the reconstructed nodes cannot be assigned divergence times with the present TimeTree database, it seems reasonable to use cladogram node-heights (i.e., the number of nodes connecting a node to a contemporary sequence) as a surrogate for divergence times, which is most nearly valid under the assumption of a constant molecular clock. Using node heights to form 10 bins related to the divergence times (fig. 4), we found that up to the most remote and least well-defined sequences, reconstructions more distant from the contemporary sequences exhibited higher <MBP> (fig. 5B). Further, <MBP> values for internal nodes of the bacterial trees increase to 0.42 ± 0.002. Thus, both by divergence time and by node-height, the independent ancestral sequence reconstructions of TrpRS and HisRS internal nodes suggest that both trees move toward progressively higher <MBP> in ancestral nodes, and over evolutionary time have diverged from sense/antisense complementarity.
Discussion
Questions posed here involve subtle distinctions between the Urzymes themselves and the coding blocks aligned in the putative Urgene. It should be noted that in this work, we have not aligned the entire coding sequences of the two active Urzymes, only three consecutive blocks spanning their active sites. These blocks encode all essential active-site defining residues and secondary structures, but they eliminate some portions of each Urzyme (fig. 1B and C). Conversely, neither have we shown that products of the putative Urgene are catalytically active.
Status of the Rodin–Ohno Sense/Antisense Coding Hypothesis for Class I and II aaRS
Common ancestry substantially reduces the number of independent observations (Felsenstein 1984). For this reason, Student t testing overestimates statistical significance. Moreover, the detailed procedures suggested by Felsenstein to estimate the number of independent observations are unwieldy. To establish appropriate estimates of middle-base pair frequency expected under the null hypothesis, we compiled such distributions after frame-shifting and randomization, and from actual protein coding sequences for both random and multiple sequence alignments for protein families that exhibit no evidence for coding sequence complementarity. Distant putative paralogs (Toprim domains) show no evidence of sense/antisense ancestry with class II aaRS, whereas analysis of closely paralogous class I and II aaRS outgroups exhibit essentially the same, elevated <MBP>.
Differences between the class I/II sense antisense alignments and all similar tests representing the null hypothesis are all more than 100 times the standard errors of the means. Thus, they imply with considerable certainty that sense/antisense alignments of TrpRS and HisRS Urgene sequences are drawn from a population distinct from those formed from most pairs of naturally occurring proteins, and hence that they are somehow linked. The positional sensitivity and robust increase in <MBP> with the node-height for reconstructed ancestral nodes (fig. 5B) strongly reinforce this conclusion in qualitatively different ways. The simplest explanation for the unique coding patterns illustrated in figure 3B is that the two classes of aaRS retain high middle-base complementarity because they actually did arise on opposite strands of the same gene, as proposed by Rodin and Ohno (1995). Our results therefore provide compelling bioinformatic evidence that the null statement corresponding to the Rodin–Ohno hypothesis should be rejected.
Bacteria Appear Uniformly Closer than Eukarya or Archaea to the Origin of Translation
Careful re-examination of the divergence of TrpRS from TyrRS (Brown et al. 1997) revealed that reciprocally rooted phylogenies of TrpRS and TyrRS sequences suggested a closer evolutionary relationship of Archaea to eukaryotes, placing the root of the universal tree in the Bacteria. Curiously, the highest <MBP> values between TrpRS and HisRS occur in bacterial sense/antisense alignments, leading to the approximate (diagonal) symmetry of figure 5A and the significantly higher <MBP> for reconstructed ancestral sequences (fig. 5B). Were the structural data for archaeal TrpRSs and HisRSs insufficient to correctly condition our choice of active-site fragments, so that unidentified errors in the resulting 94-residue Urgenes are responsible for their significantly lower <MBP> values? If not, the higher <MBP> of bacterial sequences affords novel evidence that bacteria are closer than other kingdoms to the origin of enzymatic aminoacylation, and hence to the origin of translation itself.
Our results also seem strongly to contradict the proposal raised by a recent detailed structural comparison of TrpRSs and TyrRSs from bacteria, archaea, and eukarya (Dong et al. 2010) that TrpRS diverged from TyrRS after archaea diverged from bacteria and that all bacterial TrpRSs arose by horizontal gene transfer. It seems unlikely that genes transmitted from archaea to bacteria would have experienced selective pressure to increase their coding complementarity with class II aaRS. Curiously, the TrpRS tree (fig. 2A) places eukaryotes within the crenarcheota (Williams et al. 2012), whereas HisRS trees do not. We cannot argue whether or not this too is a result of horizontal gene transfer.
TrpRS and HisRS Urzymes May Have had Modular, Functionally Active Precursors
Aspects of figure 1 suggest possible new insight into the modular nature of contemporary proteins. The three coding blocks suggest that the class I and II Urzymes may be mosaic structures with modular antecedents, corresponding to functional moieties suggested in figure 1B. Independent, prior functionality of the largest class I block, containing the HIGH signature has considerable support. It encodes the first β-α-β crossover of, and includes features broadly conserved in the Rossmann dinucleotide binding fold. The HIGH signature between the first β-strand and the α-helix is structurally and functionally homologous to the glycine-rich P-loop or Walker A sequences found many nucleotide triphosphate binding proteins, the N-terminus of the α-helix forms a phosphate binding site in most of these proteins and the hairpin linking the α-helix to the second β-strand contains a nonpolar core packing motif shared by Rossmannoid proteins (Cammer and Carter 2010).
TrpRS Crystal structures show that ATP binds initially to the HIGH sequence, prior to induced-fit active site assembly (Retailleau et al. 2003). The corresponding ∼50 residue peptides from some of these, ATPase (Chuang, Abeygunawardana, Gittis, et al. 1992; Chuang, Abeygunawardana, Pedersen, et al. 1992) and adenylate kinase (Fry et al. 1985, 1988), exhibit high affinity for ATP. We have recently shown that the TrpRS 46-mer containing TIGN has an ATP-binding affinity of approximately 90 µM (Li L, Weinreb V, Carter CW Jr, unpublished data). Further, the nonpolar packing motif at the C-terminus of the α-helix has a synergistic influence on the catalytic Mg2+ in full-length TrpRS (Weinreb et al. 2009, 2012). The nascent functionality and widespread, conserved occurrence of this motif in contemporary transducing proteins suggest that this ancient ancestral module functioned in nucleotide triphosphate utilization at a very early stage of protein evolution.
The antisense 46-mer derived from the HisRS Urzyme also encodes the site for ATP binding by class II aaRS (fig. 1B). It too has high ATP affinity (∼15 µM) when expressed separately (Li L, Weinreb V, Carter CW Jr, unpublished data). The possibility that the two peptides would have been aligned opposite one another under the hypothesis of Rodin and Ohno justifies further investigation into the possible simultaneous evolutionary origins of these two widespread ATP-binding motifs.
TrpRS, HisRS Urzyme Secondary Structures Are Consistent with Sense/Antisense Coding
A remarkable aspect of the genetic code (Zull and Smith 1990) reprised in figure 6A (see also Rodin et al. 2009) is that anticodons tend to exchange hydrophobic for hydrophilic side chains, and vice versa. These codon/anticodon exchange properties suggest that binary patterns will exhibit similar periodicities when read from the opposite strand, and hence will tend to code for similar secondary structures—a binary pattern encoding a helix on one strand will have a tendency to encode a helix when anticodons are read in the reverse direction from the opposite strand. Similarly a binary pattern repeating every two residues will likely code for extended β-structures from both strands. Thus, the genetic code itself suggests a tendency for proteins coded on opposite strands to exhibit approximate reflection symmetry in antiparallel secondary structure alignments. The inversion of chemical polarity associated with the genetic code suggests that the resulting folded proteins are in a sense “inside out”!
Fig. 6.

Inversion symmetry in the genetic code and in putative sense/antisense coding sequences. (A) Codons and anticodons encode amino acids with complementary physical chemistry (Zull and Smith 1990). Each black line denotes a codon–anticodon pair. Almost without exception, codons for hydrophobic core amino acids in the first two groups have anticodons for amino acids that define surfaces, and vice versa. Codons for proline and glycine include a sense/antisense pair, so that the sequence Pro-Gly, which frequently encodes a turn, is read as Pro-Gly in the reverse direction from the antisense strand. (B) Predictions based on helix-coil transition theory (Muñoz and Serrano 1994; Lacroix et al. 1998) for the two structures shown in figure 1. TrpRS 94-mer predictions (solid circles) are positive. HisRS 94-mer predictions have been multiplied by −1 and plotted in reversed order, C→N.
The observed secondary structures (Kabsch and Sander 1983) performed by Procheck (CCP4 1991) and tabulated in figure 1A are suggestive. Although coding instructions are reversed, secondary structures formed by the three modular fragments (fig. 1B) exhibit strong similarities between the two classes. Both 46-residue fragments consist of β-α-β secondary structures and amber fragments consist of two linked α-helices.
Tertiary structures doubtless modify the underlying secondary structure preferences dictated by the two coding strands. Thus, a more appropriate test for the expected reflection symmetry would compare helical preferences derived from helix-coil transition theory (Muñoz and Serrano 1994; Lacroix et al. 1998). When compared in this way, the reflection symmetry along the antiparallel coding sequences in figure 1A improves significantly (fig. 6B). Comparison between Toprim domain and HisRS blue and amber fragments serves equally well as a control here that that simply having “inside-out” secondary structures does not lead to the high <MBP> values observed for the class I/class II aaRS Urgenes.
Why Sense/Antisense Coding? Evolutionary Scenarios for Strand Specialization, Adaptive Radiation, and the Emergence of the Genetic Code
Sense/antisense genetic coding makes variation and evolutionary improvements substantially more difficult for such dual-function genes. An obvious question is as follows: what selective advantage compensated for such difficulty? A likely answer is that genetic linkage ensured simultaneous availability of both hydrophobic (class I) and hydrophilic (class II) aminoacylated tRNAs. Experimental work (Kamtekar et al. 1993; Moffet et al. 2003; Patel et al. 2009) has shown that as many as 50% of variants in combinatorial libraries with randomized binary patterns of hydrophobic and hydrophilic amino acids form molten globules, some of which exhibit catalytic activities.
Given that the chief selective advantage of a Rodin–Ohno Urgene was to ensure production of activated amino acids (or perhaps acylated tRNAs) with sufficient diversity to enhance the number of extant RNA molecules that could serve as instructions for globular statistical proteins, how might adaptive radiation of that gene have led to the contemporary genetic code?
The metaphor of a protein “Big bang” (Dokholyan and Shakhnovich 2001; Dokholyan et al. 2002; Koonin 2007) suggests the intuitive appeal of divergence as a dominant evolutionary paradigm. The high primary and tertiary structural homology of conserved segments in both class I and class II aaRS (O’Donoghue and Luthey-Schulten 2003), together with the TrpRS and HisRS Urzyme functional activities (Pham et al. 2010; Li et al. 2011) strongly imply that both classes diverged from ancestors each containing a distinct ∼120 amino acid core. It appears very unlikely to us that convergent evolution contributed significantly to the early development of translation.
Divergence requires that we assume some form of gene duplication if we are to outline how a single ancestral sense/antisense gene might have enriched a rudimentary genetic code. The simplest form of gene duplication would simply be replication to give statistically similar copies. We assume, then, that once established, a Rodin–Ohno gene would subsequently have replicated/duplicated many times with variation, giving rise to new pairs of synthetases. The Rodin–Ohno hypothesis does not specify the order in which daughters led subsequently either to linkage-breaking strand specialization of class I and II sequences on different gene copies, or to adaptive radiation that enlarged the Genetic Code. Both processes entail gene duplication (Ohno 1970), but the uses made of the daughter genes differ in the two cases.
A priori, the two processes may have occurred many different ways. Extreme scenarios are compared in figure 7A and B. In A, opposite strands specialize in descendants of the earliest copies, one becoming the ancestor to all class I, the other the ancestor to all class II aaRS. Adaptive radiation to the canonical set operated on specialized strands, and hence entailed independent class I and II trajectories. Scenario A has the advantage of greater flexibility in the adaptive radiation of only one coding strand. In B, descendants of the original sense/antisense ancestral gene retained strand-symmetric coding through some or all of the adaptive radiation, and strand specialization occurred subsequently to the emergence of a more elaborate Genetic Code.
Fig. 7.

Two extreme scenarios for generating strand-specialized genes and the adaptive radiation of the canonical 20 aminoacyl-tRNA synthetases. The two processes both entail gene duplication (Ohno 1970), but differ in the subsequent behavior of the daughter genes. (A) Strand specialization occurs first, releasing the constraint imposed by sense/antisense coding, and facilitating the adaptive radiation of aaRS for different amino acids. In the daughter genes, only the dark strand remains an active set of coding instructions, one for class I, the other for class II. (B) Adaptive radiation proceeds under the constraint imposed on both strands by the fact that they encode functional proteins. Both strands remain functionally important in the daughter genes until the sense/antisense constraint is relaxed later. Scenario B is somewhat more consistent with the observed near symmetric subclassification in classes I and II.
Scenario B places heavier constraints on the exploration of sequence space to find mutations that changed amino acid and tRNA specificities, but is more consistent with the rough equivalence of one large and two small subclasses in each class and with the equivalent <MBP> for the outgroups (supplementary fig. S1, Supplementary Material online). Notably, scenario B also affords a molecular implementation for the initial binary choices of Delarue’s (2007) stepwise expansion of the genetic code. Specifying pyrimidine middle bases defined a need for class I and II amino acids, and specifying purine middle bases led to two additional categories, signaling turns with glycine and serine and a stop signal. The relevance of such patterns in generating globularity underscores the elegance of the Rodin–Ohno scenario for the launch of precellular protein synthesis.
More broadly, how pervasive might descendants of a sense/antisense aaRS gene be in the contemporary proteome? Consensus holds that all class I aaRS share a common ancestor (Aravind et al. 2002) and belong to the Rossmannoid superfamily (Aravind et al. 1998; Wolf et al. 1999). The question arises: Are all Rossmannoid families paralogs, and hence descendants of ancestral class I aaRS? Or, did some Rossmannoid families converge from distinct ancestors? The low <MBP> values for the Toprim:HisRS comparison suggest that the Toprim domain may have resulted from convergent evolution.
On the other hand, the highly conserved switching motif (Cammer and Carter 2010) occurs with minimal variation in the N-terminal β-α-β crossover of more than 125 Rossmannoid protein families. The same supersecondary structure contains glycine-rich P-loop homologs at the N-terminus of the α-helix. Notably, this switching motif constitutes approximately 60% of the TrpRS Urgene. These observations argue that a substantial portion of Rossmannoid proteins probably belong to a divergent superfamily whose ancestor we suggest was the Rodin–Ohno gene.
It is less clear that class II aaRS belong to a similar superfamily. However, just as class I aaRS are monophyletic with several different families (Aravind et al. 2002), structural homologs identified for class II synthetases include the Bir1 family of biotin synthetases and asparagine synthetase (Artymiuck et al. 1994; Cusack 1994). A wider range of homologs, as is characteristic of the Rossomannoids, may be inherently more difficult to detect among proteins with large amounts of antiparallel β-structure. Our initial examination of sense/antisense coding Carter and Duax (2002) suggested homology between class II active sites and the ATP binding sites of HSP70 and actin, extending to the entire HisRS Urzyme and including Motif 3 (Carter CW Jr, unpublished). However, although the entire Motif I segment, including the same total number of α-helical and β-residues, occurs in HSP70, its orientation is different, suggesting that it may be “domain swapped.” These observations suggest that the ATP binding site of class II Urzymes, possibly including Motif 3, was an early ancestor of the actin superfamily. Thus, the TrpRS and HisRS Urzymes seem realistic ancestors for significant portions of the contemporary proteome.
Questions about the Rodin–Ohno hypothesis remain to be addressed: i) Can pairwise bioinformatic analyses of <MBP> between other class I and II aaRS superfamilies provide a phylogenetic tree describing their speciation to distinguish between scenarios A and B (fig. 7) and, by implication, better describe the development of the genetic code? Preliminary analysis of the two outgroups (supplementary fig. S1, Supplementary Material online) suggests that further clues to the existence and characteristics of a sense/antisense gene encoding ancestral class I and II aaRS likely will emerge from statistical reconstruction first of ancestral nodes for similar Urgenes for each family in both classes from contemporary multiple sequence alignments (Ronquist and Huelsenbeck 2003; Liberles 2007; Fournier et al. 2011), together with a comparison of all-by-all <MBP> statistics. ii) How were the excerpted blue, amber, and magenta fragments (fig. 1C) linked to form a functional Urgene? iii) Can structures comparable to those in figure 1 be coded with absolute complementarity of both coding strands? Remarkably, work described here suggests that each of these tasks may now be within reach.
Materials and Methods
Multiple sequence alignments of class I (TrpRS; CI) and class II (HisRS: CII) proteins were built using MUSCLE (Edgar 2004a, 2004b) with 211 and 207 samples, respectively, as indicated in figure 8. These alignments contain the original TrpRS and HisRS pair used in our previous studies to build the sense/antisense alignment in figure 1, which served as a reference. Multiple structure alignments were built using POSA (Ye and Godzik 2005) and were used to help curate the MSAs. New sense/antisense alignments are built for each fragment by replacing each protein’s coding sequence in the original sense/antisense alignment by the coding sequence of the same protein from another species from the multiple sequence alignment, thus helping to assure correct positioning of “anchors” from each class against each other. Anchors are derived from Rodin and Ohno (1995) for the 46-residue and 18-residue terminal segments, whereas the GxDQ segment (TrpRS) and its complement in figure 1A were used as anchors for the central segment of the alignment. The hypothesis, H, is computed from the middle base complementarities of CI versus CII′ alignments built in this way whereas N1,a and N1,b sets are built by computing the middle-base complementarities of CI and CII proteins against their own reverse complements. Additional null test sets in which members did and did not belong to multiple sequence alignments of two families were derived from the PDB. For the latter, N0,a, a set of a thousand peptides 94 residues long was chosen randomly from the PDB, aligning the middle bases of one against another’s reverse complement. The former, N0,b, was built from multiple sequence alignments of 50 samples of lectin (Pfam PF00059) and PDZ (Pfam PF00595) families, aligning the middle bases of aligned regions of one family against the other. That alignment serves as a control to rule out the possibility that the high middle-base complementarity of the CI versus CII′ alignment resulted from the fact that we built the alignments from multiple sequence alignments for homologs.
Fig. 8.

Schematic of computational procedures.
Clustering of each set in a pairwise manner was performed by using the k-means algorithm by specifying two clusters. In this process, middle bases are used to assemble a Euclidean distance measure, to separate two sets on one dimension. Alternately, positional distributions of matches in sense–antisense alignments are taken into account by representing each alignment with a string consisting of bits, where each Watson–Crick base pairing is represented with a 1 and a mismatch with 0. Prior to clustering, a profile of positional distribution of the matches in each set is built by averaging the bit strings representing the alignments. The distance of an alignment from a set’s positional distribution was measured by the Pearson correlation of the alignment bit string with the mean bit string of the set. Given this measure, clustering was performed again on two-dimensional data points (middle-base identity percentage and positional distances to each set).
Phylogenetic trees were constructed from the 94-base sequence alignments containing only codon-middle bases using jModelTest (Guindon and Gascuel 2003; Darriba et al. 2012), which generated the most probable tree based on different nucleotide substitution models. The optimum model for TrpRS was TVM + G and the optimum model for HisRS was GTR + G. Eukaryotic TrpRS sequences were identified as mitochondrial if they had a canonical KMSKS sequence in the magenta fragment, as eukaryotic cytoplasmic sequences lack the second K. Some ambiguity remains, however, about the lineages of eukaryotic HisRS sequences because for these several of the databases from which the sequences were obtained do not specify whether they are cytosolic or mitochondrial. These trees were input to the Lazarus interface (Hanson-Smith et al. 2010) to PAML (Yang 2007b) and used to generate all ancestral nodes. The TrpRS tree was rooted within Lazarus by an outgroup consisting of eight TyrRS sequences (Thermus thermophilus, Mimivirus, Escherichia coli, Staphylococcus aureus, Saccharomyces cerevisiae, Methanococcus jannaschi, Aeropyrum pernix, and Leishmania major); the HisRS tree was rooted by an outgroup consisting of six ProRS sequences (Enterococcus facaelis, Homo sapiens, Methanothermobacter thermoautotrophicus, Methanococcus jannaschi, Giardia lambia, and Rhodopseudomonas palustris).
The GC content was 0.37 ± 0.003 for the 94-residue base sequences of TrpRS Urgenes and 0.39 ± 0.003 for the HisRS sequences. Because these values are “typical” (S. cereviseae has 0.38 GC), we used the HKY85 evolutionary model (Hasegawa et al. 1985) in PAML to reconstruct sequences. The GC content remained much the same over all reconstructed nodes.
Supplementary Material
Supplementary material, figures S1–S3, and table S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank J. Thorne, M. Giddings, V. Hansen-Smith, E. Gaucher, and S. Rodin for discussion and helpful comments. We followed a detailed outline provided by J. Thornton for ancestral sequence reconstruction. The authors also thank all the anonymous referees for numerous suggestions that strengthened the paper. This work was supported by the National Institutes of Health grant GM78227.
References
- Andam CP, Gogarten JP. Biased gene transfer in microbial evolution. Nat Rev Microbiol. 2011;12:543–555. doi: 10.1038/nrmicro2593. [DOI] [PubMed] [Google Scholar]
- Aravind L, Anantharaman V, Koonin EV. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implication for protein evolution in the RNAWorld. Proteins. 2002;48:1–14. doi: 10.1002/prot.10064. [DOI] [PubMed] [Google Scholar]
- Aravind L, Leipe DD, Koonin EV. Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymiuck P, Rice D, Poirrette AR, Willet P. A tale of two synthetases. Nat Struct Mol Biol. 1994;1:758–760. doi: 10.1038/nsb1194-758. [DOI] [PubMed] [Google Scholar]
- Benner SA, Sassi SO, Gaucher EA. Molecular paleoscience: systems biology from the past. Adv Enzymol Relat Areas Mol Biol. 2007;75:9–140. doi: 10.1002/9780471224464.ch1. [DOI] [PubMed] [Google Scholar]
- Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312:97–101. doi: 10.1126/science.1123348. [DOI] [PubMed] [Google Scholar]
- Brown JR, Robb FT, Weiss R, Doolittle WF. Evidence for the early divergence of tryptophanyl- and tyrosyl-tRNA synthetases. J Mol Evol. 1997;45:9–16. doi: 10.1007/pl00006206. [DOI] [PubMed] [Google Scholar]
- Brown P, Berge JM, Hamprecht DW, Jarvest RL, McNair DJ, Mensah L, O'Hanlon RJ, Pope AJ. Synthetic analogues of SB-219383. Novel C-glycosyl peptides as inhibitors of tyrosyl tRNA synthetase. Bioorg Med Chem Lett. 2001;11:715–718. doi: 10.1016/s0960-894x(01)00040-3. [DOI] [PubMed] [Google Scholar]
- Cammer S, Carter CW., Jr Six rossmannoid folds, including the class I aminoacyl-tRNA synthetases, share a partial core with the anticodon-binding domain of a class II aminoacyl-tRNA synthetase. Bioinformatics. 2010;26:709–714. doi: 10.1093/bioinformatics/btq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CW., Jr Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu Rev Biochem. 1993;62:715–748. doi: 10.1146/annurev.bi.62.070193.003435. [DOI] [PubMed] [Google Scholar]
- Carter CW, Jr, Duax WL. Did tRNA synthetase classes arise on opposite strands of the same gene? Mol Cell. 2002;10:705–708. doi: 10.1016/s1097-2765(02)00688-3. [DOI] [PubMed] [Google Scholar]
- CCP4. The SRC(UK) collaborative computing project no. 4: a suite of programs for protein crystallography. Daresbury (UK): Daresbury Laboratory; 1991. [Google Scholar]
- Chuang W-J, Abeygunawardana C, Gittis AG, Pedersen PL, Mildvan AS. Solution structure and function in trifluoroethanol of PP-50, an ATP-binding peptide from F1ATPase. Arch Biochem Biophys. 1992;319:110–122. doi: 10.1006/abbi.1995.1272. [DOI] [PubMed] [Google Scholar]
- Chuang W-J, Abeygunawardana C, Pedersen PL, Mildvan AS. Two-dimensional NMR, circular dichroism, and fluorescence studies of PP-50, a synthetic ATP-binding peptide from the β-subunit of mitochondrial ATP synthase. Biochemistry. 1992;31:7915–7921. doi: 10.1021/bi00149a024. [DOI] [PubMed] [Google Scholar]
- Crick FHC. Codon-anticodon pairing: the Wobble hypothesis. J Mol Biol. 1966;19:548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- Cusack S. Evolutionary implications. Nat Struct Mol Biol. 1994;1:760. [Google Scholar]
- Cusack S, Berthet-Colominas C, Härtlein M, Nassar N, Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature. 1990;347:249–255. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M. An asymmetric underlying rule in the assignment of codons: possible clue to a quick early evolution of the genetic code via successive binary choices. RNA. 2007;13:1–9. doi: 10.1261/rna.257607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan NV, Shakhnovich EI. Understanding hierarchical protein evolution from first principles. J Mol Biol. 2001;312:289–307. doi: 10.1006/jmbi.2001.4949. [DOI] [PubMed] [Google Scholar]
- Dokholyan NV, Shakhnovich B, Shacknovich EI. Expanding protein universe and its origin from the biological big bang. Proc Natl Acad Sci U S A. 2002;99:14132–14136. doi: 10.1073/pnas.202497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Zhou M, Zhong C, Yang B, Shen N, Ding J. Crystal structure of Pyrococcus horikoshii tryptophanyl-tRNA synthetase and structure-based phylogenetic analysis suggest an archaeal origin of tryptophanyl-tRNA synthetase. Nucleic Acids Res. 2010;38:1401–1412. doi: 10.1093/nar/gkp1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004a;5:113–132. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004b;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. Am Naturalist. 1984;125:1–15. [Google Scholar]
- Fournier GP, Andam CP, Alm EJ, Gogarten JP. Molecular evolution of aminoacyl tRNA synthetase proteins in the early history of life. Orig Life Evol Biosph. 2011;41:621–632. doi: 10.1007/s11084-011-9261-2. [DOI] [PubMed] [Google Scholar]
- Fry DC, Byler DM, Sisu H, Brown EM, Kuby SA, Mildvan AS. Solution structure of the 45-residue MgATP-binding peptide of adenylate kinase as examined by 2-D NMR, FTIR, and CD spectroscopy. Biochemistry. 1988;27:3588–3598. doi: 10.1021/bi00410a009. [DOI] [PubMed] [Google Scholar]
- Fry DC, Kuby SA, Mildvan AS. NMR studies of the MgATP binding site of adenylate kinase and of a 45-residue peptide fragment of the enzyme. Biochemistry. 1985;24:4680–4694. doi: 10.1021/bi00338a030. [DOI] [PubMed] [Google Scholar]
- Gaucher EA, Govindarajan S, Ganesh OK. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature. 2008;451:704–707. doi: 10.1038/nature06510. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hanson-Smith V, Kolaczkowski B, Thornton JW. Robustness of ancestral sequence reconstruction to phylogenetic uncertainty. Mol Biol Evol. 2010;27:1988–1999. doi: 10.1093/molbev/msq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T-A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Ibba M, Soll D. Aminoacyl-tRNAs: setting the limits of the genetic code. Genes Dev. 2004;18:731–738. doi: 10.1101/gad.1187404. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Protein design by binary patterning of polar and non-polar amino acids. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- Koonin EV. The biological big bang model for the major transitions in evolution. Biol Direct. 2007;2: 21. doi: 10.1186/1745-6150-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix E, Viguera AR, Serrano L. Elucidating the folding problem of a-helices: local motifs, long-range electrostatics, ionic strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- Li L, Weinreb V, Francklyn C, Carter CW., Jr Histidyl-tRNA synthetase urzymes: class I and II aminoacyl-tRNA synthetase urzymes have comparable catalytic activities fo cognate amino acid activation. J Biol Chem. 2011;286:10387–10395. doi: 10.1074/jbc.M110.198929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberles DA. Ancestral sequence reconstruction. Oxford: Oxford University Press; 2007. [Google Scholar]
- Microsoft. Excel. Version 12.2.8. Belleview (WA): Microsoft; 2008. [Google Scholar]
- Moffet DA, Foley J, Hecht MH. Midpoint reduction potentials and heme binding stoichiometries of de novo proteins from designed combinatorial libraries. Biophys Chem. 2003;105:231–239. doi: 10.1016/s0301-4622(03)00072-3. [DOI] [PubMed] [Google Scholar]
- Muñoz V, Serrano L. Intrinsic secondary structure propensities of the amino acids, using statistical ϕ-ψ matrices: comparison with experimental scales. Proteins. 1994;20:301–311. doi: 10.1002/prot.340200403. [DOI] [PubMed] [Google Scholar]
- O’Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67:550–573. doi: 10.1128/MMBR.67.4.550-573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer; 1970. [Google Scholar]
- Patel SC, Bradley LH, Jinadasa SP, Hecht MH. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Protein Sci. 2009;18:1388–1400. doi: 10.1002/pro.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham Y, Kuhlman B, Butterfoss GL, Hu H, Weinreb V, Carter CW., Jr Tryptophanyl-tRNA synthetase Urzyme: a model to recapitulate molecular evolution and investigate intramolecular complementation. J Biol Chem. 2010;285:38590–38601. doi: 10.1074/jbc.M110.136911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham Y, Li L, Kim A, Weinreb V, Butterfoss G, Kuhlman B, Carter CW., Jr A minimal TrpRS catalytic domain supports sense/antisense ancestry of class I and II aminoacyl-tRNA synthetases. Mol Cell. 2007;25:851–862. doi: 10.1016/j.molcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Picard RR, Cook RD. Cross-validation of regression models. J Am Statist Assoc. 1984;79:575–583. [Google Scholar]
- Rambaut A. Figtree. Version 1.4.0. Edinburgh (UK): University of Edinburgh; 2010. [Google Scholar]
- Retailleau P, Huang X, Yin Y, et al. (11 co-authors) Interconversion of ATP binding and conformational free energies by trptophanyl-tRNA synthetase: a closed, pre-transition-state ATP complex at 2.2 Å resolution. J Mol Biol. 2003;325:39–63. doi: 10.1016/s0022-2836(02)01156-7. [DOI] [PubMed] [Google Scholar]
- Ribas de Pouplana L, Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104:191–193. doi: 10.1016/s0092-8674(01)00204-5. [DOI] [PubMed] [Google Scholar]
- Rodin A, Rodin SN, Carter CW., Jr On primordial sense-antisense coding. J Mol Evol. 2009;69:555–567. doi: 10.1007/s00239-009-9288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin SN, Ohno S. Two types of aminoacyl-tRNA synthetases could be originally encoded by complementary strands of the same nucleic acid. Orig Life Evol Biosph. 1995;25:565–589. doi: 10.1007/BF01582025. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Schmidt HA. TAXNAMECONVERT. Version 2.4. 2004. Center for Integrative Bioinformatics, Vienna. [Google Scholar]
- Synergy. Kaleidagraph. Version 4.01. Reading (PA): Synergy Software; 2005. [Google Scholar]
- Thornton JW. Resurrecting ancient genes: experimental analysis of extinct molecules. Nat Rev Genet. 2004;5:366–375. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- Weinreb V, Li L, Carter CW., Jr A master switch couples Mg2+-assisted catalysis to domain motion in B. stearothermophilus tryptophanyl-tRNA synthetase. Structure. 2012;20:128–138. doi: 10.1016/j.str.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb V, Li L, Kaguni LS, Campbell CL, Carter CW., Jr Mg2+-assisted catalysis by B. stearothermophilus TrpRS is promoted by allosteric effects. Structure. 2009;17:952–964. doi: 10.1016/j.str.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TA, Foster PG, Nye TMW, Cox CJ, Embley TM. A congruent phylogenomic signal places eukaryotes within the Archaea. Proc R Soc B. 2012;279:4870–4879. doi: 10.1098/rspb.2012.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Olsen GJ, Ibba M, Soll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases—analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710. [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007a;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007b;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Ye Y, Godzik A. Multiple flexible structure alignment using partial order graphs. Bioinformatics. 2005;21:2362–2369. doi: 10.1093/bioinformatics/bti353. [DOI] [PubMed] [Google Scholar]
- Zull JE, Smith SK. Is genetic code redundancy related to retention of structural information in both DNA strands? Trends Biochem Sci. 1990;15:257–261. doi: 10.1016/0968-0004(90)90048-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}