

Abstract
Noncoding genetic variation is known to significantly influence gene expression levels in a growing number of specific cases; however, the patterns of genome-wide noncoding variation present within populations, the evolutionary forces acting on noncoding variants, and the relative effects of regulatory polymorphisms on transcript abundance are not well characterized. Here, we address these questions by analyzing patterns of regulatory variation in motifs for 177 DNA binding proteins in 37 strains of Saccharomyces cerevisiae. Between S. cerevisiae strains, we found considerable polymorphism in regulatory motifs across strains (mean π = 0.005) as well as diversity in regulatory motifs (mean 0.91 motifs differences per regulatory region). Population genetics analyses reveal that motifs are under purifying selection, and there is considerable heterogeneity in the magnitude of selection across different motifs. Finally, we obtained RNA-Seq data in 22 strains and identified 49 polymorphic DNA sequence motifs in 30 distinct genes that are significantly associated with transcriptional differences between strains. In 22 of these genes, there was a single polymorphic motif associated with expression in the upstream region. Our results provide comprehensive insights into the evolutionary trajectory of regulatory variation in yeast and the characteristics of a compendium of regulatory alleles.
Keywords: adaptive evolution, evolution, regulatory variation, yeast
Introduction
Noncoding genetic variation makes a significant contribution to phenotypic diversity and disease susceptibility by modulating gene expression (Rockman and Kruglyak 2006; Skelly et al. 2009). Examples of noncoding variants causing phenotypic differences within and between species are rapidly accumulating in diverse lineages (Wray 2007). For example, noncoding variants have been identified that cause pigmentation differences in Drosophila (Wittkopp et al. 2002), skeletal reduction in stickleback fish (Shapiro et al. 2004), skin wrinkling in the domesticated dog (Akey et al. 2010; Olsson et al. 2011), and loss of neck feathers in chicken (Mou et al. 2011). Although the precise molecular mechanisms that causal noncoding variants act through remain poorly defined, many regulatory variants likely alter the binding of sequence-specific DNA-binding proteins. These proteins affect gene expression by interacting with the transcriptional machinery, cooperatively binding to other activating or repressing proteins, or modulating chromatin structure (Lee and Young 2000; Farnham 2009).
Yeast is an excellent system in which to study noncoding variation because of the availability of whole-genome sequences from diverse strains and species. For example, whole-genome sequences are available for 37 Saccharomyces cerevisiae strains, which are functionally and geographically diverse (Liti et al. 2009). In addition, sequence motifs for the majority of known DNA-binding factors in yeast have been characterized (Bryne et al. 2008). Motif usage across species has been studied extensively in yeast. Previous work on the evolution of noncoding regions has shown that motifs rapidly turn over between species, including yeast (Dermitzakis and Clark 2002; Moses et al. 2006; Borneman et al. 2007; Doniger and Fay 2007). In some cases, genes whose co-expression has been conserved across species may have acquired different regulators in different species, as in the case of ribosomal protein modules in yeast (Wapinski et al. 2010). Despite this frequent turnover, often the presence of specific motifs is conserved, if not the location (Doniger and Fay 2007). Motifs that are conserved within and between species are correlated with several characteristics, such as being upstream of essential genes, closer to transcription initiation sites, and within open chromatin regions (Francesconi et al. 2011).
More generally, previous analyses of noncoding regions in diverse species have found strong signatures of both positive and negative selection (Mustonen and Lassig 2005; Chen et al 2010; He et al. 2011). These studies have had several limitations, however; for example, they have focused on small collections of known binding sites (Mustonen and Lassig 2005), motifs involved in key developmental modules (He et al. 2011), or motifs ascertained based on their conservation (Chen et al. 2010).
Gene expression variation has also been studied extensively in yeast. These studies have revealed that specific classes of genes are more likely to diverge between species (Thompson and Regev 2009), and such loci share architectural features such as containing a TATA box in their promoter and harboring more binding sites for regulatory proteins (Tirosh et al. 2006). In addition, expression QTL (eQTL) studies have identified a significant role for cis-acting variation in gene expression differences between strains or species (Brem et al. 2002; Ronald and Akey 2007; Ehrenreich et al. 2009; Tirosh et al. 2009; Emerson et al. 2010). Differences in predicted motifs have been associated with expression differences for some of the genes with cis-linkages in a cross between two strains (Chen et al. 2010). In addition, Zheng et al. (2010) identified several hundred genes showing significant gene expression variation associated with differences in protein binding for the factor STE12 (Zheng et al. 2010). Thus, variation in DNA-binding motifs can be an important causal source of gene expression variation.
In this study, we describe a comprehensive genome-wide analysis of polymorphisms located in 177 DNA sequence motifs across 37 S. cerevisiae strains (Liti et al. 2009). We expand the number of motifs studied from previous studies, and identify motifs genome-wide in an unbiased manner without regard to conservation. We perform extensive population genomics analyses that reveal DNA sequence motifs are subject to purifying selection, and quantify the strength of selection for each motif. Furthermore, we used RNA-Seq data that were previously collected for 22 of these strains and performed association analyses between polymorphisms in motifs and differences in gene expression. We identified six polymorphic motifs associated with widespread and consistent changes in gene expression, 49 polymorphic motifs associated with transcriptional variation at individual genes, and a compendium of high confidence regulatory alleles.
Results
Regulatory Motif Variation across S. cerevisiae Strains
We first examined patterns of motif differences across 37 globally and functionally diverse S. cerevisiae strains whose genomes have been sequenced (Liti et al. 2009; supplementary fig. S1, Supplementary Material online), by independently calling motifs in all strains (see Materials and Methods). We found substantial divergence in motif content across strains. The average pairwise number of motif differences per intergenic region is 0.91 motifs (range 0–27; fig. 1A), and as expected pairwise motif differences recapitulate the known phylogeny (data not shown). Across all strains, a median of eight motifs were called in each intergenic region, and a median of four motifs per intergenic region were variable in at least one of the 37 strains (range 0–137). One example of a highly divergent region is the region upstream of AAH1, an adenine deaminase, which is regulated by nutrient levels (fig. 1B). Another highly variable region is upstream of FLO1, which is involved in flocculation, a phenotype known to have diverged between laboratory and wild strains (Liu et al. 1996). Interestingly, a cluster containing both lab and wild strains shows a divergent motif pattern in this region (fig. 1C). A list of additional genes with highly polymorphic motif patterns is provided in supplementary table S1, Supplementary Material online.
Fig. 1.
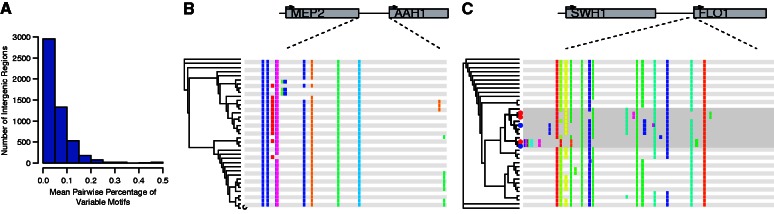
Examples of highly divergent regulatory regions across Saccharomyces cerevisiae strains. (A) Histogram of the mean pairwise percentage of variables motifs across 37 S. cerevisiae strains for each of the 5,468 intergenic regions. (B) Predicted motif calls for 37 S. cerevisiae strains are plotted for the intergenic region upstream of the gene AAH1. Each row is a strain, and colored boxes represent motif calls. Different colors represent distinct motifs. A phylogeny for the strains is shown to the left, as constructed from the motif calls for that region. (C) Predicted motif calls for a section of the intergenic region upstream of the gene FLO1. The region shown represents 1,000 bp upstream of the gene, out of 7,218 total upstream bases. A divergent clade is highlighted in gray, and within this clade wild strains are marked with a red dot in the phylogeny, whereas laboratory strains are marked with a blue dot.
Evolutionary Forces Shaping Patterns of Polymorphism and Divergence of Regulatory Sequences
To quantify the strength of selection acting on intergenic regions more systematically, we used the McDonald-Kreitman framework to assess deviations from neutral expectations across intergenic regions (McDonald and Kreitman 1991). For measures of divergence, we used S. paradoxus as an outgroup. We initially characterized the evolutionary forces acting at four classes of sites: nonsynonymous, noncoding sites within predicted motifs, noncoding sites outside predicted motifs, and experimentally determined motifs (MacIsaac et al. 2006; see Materials and Methods). Specifically, we counted polymorphic and diverged sites across all intergenic and genic regions that could be aligned between S. cerevisiae and S. paradoxus (∼4,700 regions). As putatively neutral sites, we used synonymous sites. We found that purifying selection acts on all four classes of sites (P < 2.2 × 10−16). We next estimated the –log(Neutrality Index), denoted as -log(NI) (Rand and Kann 1996), to compare the magnitude of purifying selection across site types. A value for –log(NI) of zero is consistent with neutrality, negative values suggest negative selection, and positive values indicate positive selection. As expected, the −log(NI) was lowest for experimentally determined motifs, which appear to be under strong purifying selection. We also found that –log(NI) was lower at noncoding sites inside predicted motifs compared with noncoding sites outside of motifs and nonsynonymous sites, suggesting that a higher proportion of sites falling within predicted motifs are under purifying selection than in the other classes of sites (fig. 2A). The observation that −log(NI) at noncoding sites outside predicted motifs was similar to that at nonsynonymous sites is unexpected because noncoding sites outside motifs are generally thought to be subject to less functional constraint. However, this result may be due in part to the high threshold we used to call motifs; lowering the threshold resulted in the −log(NI) at noncoding sites outside motifs becoming closer to neutral expectations (supplementary fig. S2, Supplementary Material online).
Fig. 2.
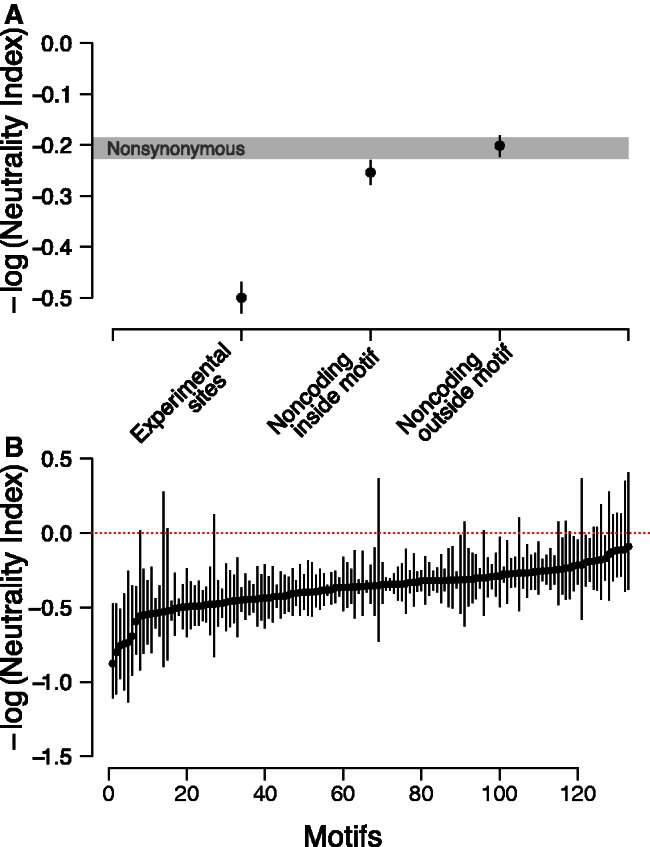
Evolutionary forces acting at intergenic regions. (A) –log(NI) scores for three classes of sites (experimentally determined sites, noncoding sites falling within predicted motifs, and noncoding sites falling outside predicted motifs) are plotted. –log(NI) scores were obtained by summing information across all sites of a particular class and using synonymous sites within genes as putatively neutral sites. Confidence intervals were obtained by bootstrapping (see Materials and Methods). 95% CI for nonsynonymous sites are shown as in gray. (B) –log(NI) scores for each of 133 motifs, sorted from lowest –log(NI) to highest –log(NI). –log(NI) scores were obtained by summing information across all sites genome-wide falling within a particular motif, and comparing with all synonymous sites. Motifs with low numbers of polymorphic and divergent sites were excluded due to low power to detect differences with such low counts (<15 total sites). Confidence intervals were obtained by bootstrapping (see Materials and Methods).
To identify heterogeneity of selective constraint across DNA-binding motifs, we calculated a motif specific estimate of the −log(NI). As shown in figure 2B, selective constraint varies widely across motifs, with some motifs under very strong purifying selection. Out of 133 motifs with sufficient data (see Materials and Methods), we identified 112 whose –log(NI) was significantly less than zero (supplementary table S2, Supplementary Material online). As expected from the earlier discussed analysis, a sizable number of motifs (63) had a –log(NI) significantly lower than that at nonsynonymous sites.
Moreover, we examined constraint acting at the level of individual intergenic regions. To this end, we compared polymorphism and divergence at sites that fell within predicted motifs in each region with synonymous sites in the genes flanking each region. We found that many intergenic regions had negative –log(NI), as expected from the motif-specific results described earlier, although the power of this analysis is lower given the reduced number of polymorphisms and divergent sites within each region. Using the MK test, we identified 152 regions that have significant evidence for purifying selection at false discovery rate (FDR) = 0.10. Eleven of these regions were significant after a more stringent Bonferroni correction for multiple testing (supplementary table S3, Supplementary Material online). We did not find any regions significant for positive selection at FDR = 0.10 or after a Bonferroni correction; however, four regions had a suggestive P value (P ≤ 0.05, uncorrected). Three of these regions flanked genes of unknown function; the remaining region flanks ADH4, an alcohol dehydrogenase gene, which has been linked to increased ethanol production (Mizuno et al. 2006). Interestingly, many S. cerevisiae strains were domesticated for use in fermentation, and thus positive selection for changes in the regulation of ADH4 may have occurred between S. cerevisiae and S. paradoxus, which may have made S. cerevisiae favorable for use in domestication.
Patterns of Motif Polymorphism Are Significantly Correlated with Transcriptional Variation among Strains
To assess the relationship between motif and gene expression variation, we obtained RNA-Seq data that had been collected on a subset of the 22 strains of S. cerevisiae analyzed earlier (Skelly et al. submitted). We performed extensive normalization of the data to account for batch effects and unknown sources of variation (see Materials and Methods). By analyzing the complete distribution of P values using the positive false discovery rate approach of Storey and Tibshirani (2003), we estimate that 79.0% of genes are differentially expressed across the 22 strains. Of these, 5,472 genes are significantly differentially expressed at a FDR = 0.10.
We investigated the relationship between motif polymorphism and transcriptional variation using two complimentary approaches. First, we tested for associations between the presence or absence of motifs and expression levels at downstream genes. Specifically, we performed association tests correcting for population structure for 13,089 motifs located upstream of 3,505 distinct genes (Connelly and Akey 2012). We note that with a small sample size of 22 strains, we have limited power to detect variants, except those with large effect sizes (supplementary table S4, Supplementary Material online). We found 49 polymorphic motifs located upstream of 30 distinct genes that were correlated with significant changes in gene expression (FDR = 0.10). Of the 49 associated polymorphic motifs, 21 resulted in increased expression with the presence of the motif (i.e., acted as an activator) and 28 resulted in decreased expression with the presence of the motif. Interestingly, 22 of these genes contained only a single polymorphic motif associated with expression variation in the upstream region (table 1). In addition, one gene did not contain any additional promoter variants located outside of motifs that are in strong linkage disequilibrium (r2 > 0.8) with the polymorphic motif (fig. 3). Moreover, we found evidence for one case of a bidirectional promoter, where polymorphism in the REB1 motif was associated with changes in expression of both flanking genes.
Table 1.
High Confidence Regulatory Polymorphisms.
| Motif | Downstream Gene | Log(Difference in Expression) | Distance Upstream of Gene (bp) | q Value |
|---|---|---|---|---|
| HCM1 | YJL155C | −0.26 | 300 | 0.04 |
| HCM1 | YEL044W | −0.27 | 955 | 0.04 |
| MOT3 | YKL059C | 0.29 | 509 | 0.04 |
| PHO2 | YJR108W | 0.57 | 144 | 0.04 |
| PHO2 | YGL169W | −0.32 | 381 | 0.04 |
| REB1 | YNL239W | 0.47 | 239 | 0.04 |
| SPT2 | YEL001C | 0.24 | 675 | 0.04 |
| YAP5 | YOR108W | −0.59 | 436 | 0.04 |
| CRZ1 | YAL049C | 0.39 | 146 | 0.06 |
| HAL9 | YPL255W | −0.32 | 494 | 0.06 |
| HAP2 | YNR049C | 0.27 | 28 | 0.06 |
| HAP2 | YOR071C | −0.44 | 2,828 | 0.06 |
| PHO2 | YPR119W | −0.12 | 331 | 0.06 |
| RAP1 | YPL108W | 0.48 | 407 | 0.06 |
| REB1 | YNL240C | 0.59 | 352 | 0.06 |
| STE12 | YKL108W | −0.28 | 91 | 0.06 |
| FHL1 | YJL094C | −0.77 | 148 | 0.07 |
| HAP2 | YBR222C | 0.39 | 245 | 0.07 |
| HAP2 | YGL117W | −0.71 | 1,245 | 0.07 |
| MOT3 | YLR152C | −0.67 | 242 | 0.07 |
| ABF2 | YPL167C | −0.24 | 116 | 0.09 |
| YAP3 | YLR007W | −0.16 | 366 | 0.09 |
Fig. 3.

Examples of motifs effecting gene expression. (A) NAR1 expression in strains containing the two labeled sequences at the motif REB1 in the upstream intergenic region. Substitutions to the consensus motif sequence for REB1 are marked in blue. A sequence logo for REB1 representing the PSSM is shown in the upper left corner. (B) YER186C expression in strains containing the two labeled sequences at the motif AFT2 in the upstream intergenic regions. Substitutions to the consensus motif sequence are marked in blue. A sequence logo for AFT2 representing the PSSM is shown to the upper left of the plot.
In addition, we tested the hypothesis that levels of sequence conservation varied between polymorphic motifs that were or were not associated with differences in gene expression. Using phastCons scores from the 12-species yeast alignment (Siepel et al. 2005), we compared the mean conservation score at 1,039 polymorphic motifs nominally associated with expression differences among strains (P ≤ 0.05) with a null distribution constructed by drawing the same number of randomly chosen motifs not associated with gene expression differences. We found conservation was significantly higher at motifs associated with expression differences (P = 0.024). Thus, the statistical and bioinformatics data strongly suggest that these 22 polymorphic motifs are enriched for causal regulatory polymorphisms.
Second, we tested whether motifs were acting consistently as activators or repressors across a majority of genes upstream of which they were polymorphic. Specifically, for the ith motif, we identified all genes whose upstream intergenic region contained a variable motif i. We discarded genes where the variable motif was only observed in a single strain. Next, we converted gene expression values for this set of genes to a Z score and tested for differences between the distribution of expression values when motif i was present or absent (see Materials and Methods). At a FDR = 0.10, we found that polymorphisms in 9 out of the 148 motifs were significantly associated with consistent transcriptional differences (5 motifs were significantly associated with increased expression and 4 motifs were significantly associated with decreased expression; table 2 and fig. 4).
Table 2.
Motifs Associated with Consistent Expression Differences.
| Motif | Number of Genes Containing Upstream Motif | Number of Polymorphic Motifs | Average Effect Size (SD)a |
|---|---|---|---|
| GAL4 | 5 | 2 | 0.86 |
| SUT2 | 17 | 3 | 1.08 |
| LEU3 | 48 | 7 | 0.47 |
| RGT1 | 75 | 14 | −0.43 |
| UGA3 | 87 | 19 | −0.40 |
| MET4 | 88 | 28 | −0.32 |
| SWI4 | 104 | 17 | 0.58 |
| RDR1 | 106 | 25 | 0.25 |
| TOS8 | 265 | 70 | −0.28 |
aMeasured as the difference in expression Z scores between motif presence and absence averaged across genes.
Fig. 4.

Effects of variants at specific motifs on gene expression. For each motif, a box plot of the difference in expression Z scores between strains containing the motif and strains not containing the motif at all genes with a variable motif are plotted. Motifs are sorted by mean difference in expression. Motifs significant in our test for genome-wide differences in expression for showing lower expression when the motif is present are colored in blue, and motifs significant for showing greater expression when the motif is present are colored in magenta.
Features Associated with Transcriptional Divergence
Finally, we investigated what characteristics were associated with high expression divergence. As a measure of expression divergence between strains, we calculated the average pairwise difference in expression between strains. We first tested whether the absolute value of the –log(NI) for motifs in each region was associated with expression divergence. We used the absolute value so that any region under either positive or negative selection would have a value greater than zero and regions under no selection would be closer to zero. We found a negative correlation (ρ = −0.07, P = 2.43 × 10−6, Spearman rank-sum test), demonstrating that regions under stronger selection showed less expression divergence. We also tested whether nucleotide diversity (π) within predicted motifs was associated with expression divergence. We found a positive correlation (ρ = 0.10, P = 6.84 × 10−14, Spearman rank-sum test), illustrating that higher nucleotide diversity was associated with higher expression divergence between strains. This correlation was still significant after controlling for the presence or absence of TATA box and for the nucleosome occupancy upstream of each gene (see Materials and Methods).
Discussion
Interpreting noncoding variation is challenging yet vital for identifying causal regulatory variation, delimiting the contribution of expression variation to phenotypic diversity and evolutionary diversification, and elucidating the molecular mechanisms through which noncoding variation acts. By focusing on interpretable noncoding variation, namely variants within known motifs for DNA-binding proteins, we were able to perform detailed evolutionary and statistical analyses on the evolutionary pressures acting at these motifs and the functional consequences of putative regulatory variation.
We first addressed the evolutionary pressures affecting motif diversity and divergence, and found that motifs are generally subject to purifying selection. These results are broadly consistent with previous analyses demonstrating purifying selection acting on yeast promoter and 3′-untranslated regions (Mustonen and Lassig 2005; Ronald and Akey 2007; Chen et al. 2010). Similarly, studies in humans have found decreased nucleotide diversity in open chromatin regions (Thurman et al. 2012; Vernot et al. 2012) and have correlated transcription factor occupied sites with higher conservation across multiple species (Neph et al. 2012). Similarly, we found that experimentally validated sites were subject to stronger purifying selection. Interestingly, we found that the level of purifying selection acting on all predicted motifs was still quite strong. We also found that the selection on experimentally determined sites and on predicted sites was stronger than that on nonsynonymous variants. One possible explanation for this is that a smaller proportion of nonsynonymous sites will actually affect gene function compared with the proportion needed to disrupt a motif. It is also possible that by testing only motifs within regions which could be aligned between the two species, we may be biased toward detecting conserved motifs. In comparison with other species, it is interesting that similar studies in Drosophila have found widespread evidence of adaptive evolution in noncoding regions (Andolfatto 2005), whereas we found little evidence for adaptive evolution. We speculate that these differences in the tempo and mode of noncoding evolution between species may be due, at least in part, to differences in effective population size. We also found that while a majority of motifs are under purifying selection, a subset is evolving neutrally. This may suggest that the position weight matrices for these motifs are ineffective at identifying functional binding sites or that, alternatively, these motifs are in general less constrained.
To investigate the effects of motif changes on transcriptional variation, we characterized gene expression differences among 22 strains. We identified nine motifs acting consistently as activators or repressors across a majority of genes they regulated. These transcription factors are involved in diverse processes, but it appears that they are broadly active in phosphate-limiting conditions. We also identified 30 genes where one or more motifs were associated with gene expression variation. Approximately one-third of these genes contained multiple motifs associated with expression variance at that gene, making it difficult to identify the causal variant, though it is also possible that there may be multiple motif changes contributing to gene expression differences at these loci, as observed previously (Prud’homme et al. 2006; Tao et al. 2006). In addition, we were able to identify 22 genes with only one motif associated with expression differences. Although for all but one of these there were other SNPs in strong LD with the associated motif in the intergenic region, SNPs that fall within motifs are a strong candidate for being a causal SNP because of their potential functional role.
We found that conservation scores across species were significantly higher at motifs associated with expression differences than at motifs not associated with expression differences, suggesting that cross-species conservation is useful for fine-scale mapping causal regulatory variation. In addition, measures of constraint within species that combine information across multiple motifs in a region were useful for predicting more general patterns of expression divergence. Specifically, we found that regions with less constrained motifs as measured by the −log(NI) and nucleotide divergence were more likely to have higher expression divergence, although the magnitude of the correlation was modest.
There are several limitations to our study design. Because we are using computationally predicted motifs, not all are actually used in vivo; however, by using stringent cutoffs for calling motifs (see Materials and Methods), we attempted to collate a high confidence set of predicted motifs on which to perform our analyses. The evolutionary analyses also suggest that we are identifying active sites that are under constraint. In addition, our study only tests the effects of motif variation on gene expression in one experimental condition. Finally, as our sample size was small, we are underpowered to detect associations attributable to rare variants or variants with small effect sizes (supplementary table S4, Supplementary Material online).
In summary, our approach demonstrates the utility of using motif predictions in conjunction with functional genomics data for identifying functional noncoding sequence variation and DNA-binding proteins that have significant effects on gene expression. In the future, it will be important to integrate additional types of data, such as in vivo DNA-binding protein information and ChIP-Seq data, to facilitate the interpretation of noncoding variation, the identification of causal noncoding variants, and the correlation of transcriptional variation to phenotypic diversity. Such integrative genomics analyses are likely to play a key role in ultimately developing predictive models to distinguish functionally important noncoding variation from functionally and phenotypically benign variants.
Materials and Methods
Sequence Data and Alignments
We obtained sequence data and whole genome alignments for 37 S. cerevisiae strains and the S. paradoxus reference sequence (CBS432-0809) from the Saccharomyces Genome Resequencing Project (Liti et al. 2009). The alignment between S. cerevisiae and S. paradoxus was done by repeating masking the reference S. cerevisiae genome and CBS432. The programs LASTZ (Harris 2007) and TBA (Blanchette 2004) were used to construct the alignment. Substitution scoring parameters for LASTZ alignments were inferred using two S. cerevisiae strains (the reference strain and RM11_1A). For all further analyses, we excluded intergenic regions that aligned to more than one contiguous block in S. cerevisiae.
Motif Analysis
We searched all intergenic regions for the 37 strains and S. paradoxus for each of the 177 known DNA binding motifs on both strands using Position-specific site matrices obtained from JASPAR and converted to PWMs (Bryne et al. 2008). Note that these matrices come from experimental studies and are not ascertained based on conservation across species. In all further analyses, we did not include sites with missing data in 1 or more of the strains, or sites that were called due to indels to mitigate alignment errors. Motifs were called if they had 90% of the observed maximum weight matrix score.
For the experimentally determined sites, we used binding sites identified by ChIP-chip (MacIsaac et al. 2006) that were significant at P < 0.001 and not subject to conservation criteria. This list consists of 9,708 motif sites.
MK Test Measurements
We calculated the NI as: 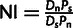 (Rand and Kann 1996). Here, D is the count of polymorphic sites between S. cerevisiae and S. paradoxus, and P is the count of polymorphic sites (frequency greater than 5%) between the S. cerevisiae strains, n = neutral sites (synonymous sites), and s = putative selected sites. When calculating the NI for each intergenic region, we used the synonymous sites from immediately flanking genes. For bootstrapping, we resampled 1,000 times from the data for each intergenic region.
(Rand and Kann 1996). Here, D is the count of polymorphic sites between S. cerevisiae and S. paradoxus, and P is the count of polymorphic sites (frequency greater than 5%) between the S. cerevisiae strains, n = neutral sites (synonymous sites), and s = putative selected sites. When calculating the NI for each intergenic region, we used the synonymous sites from immediately flanking genes. For bootstrapping, we resampled 1,000 times from the data for each intergenic region.
RNA Mapping and Normalization
Raw RNA reads were obtained from Skelly et al. (submitted). We mapped RNA-Seq reads to the S288c reference genome (UCSC sacCer2) using the program BFAST version 0.6.4e (Homer et al. 2009) with options –K 100 and –M 500 to bfast match. We aligned colorspace reads using a main index with mask 111111111111111111 (hash width 14) and secondary indexes with masks 1111101110111010100101011011111, 1011110101101001011000011010001111111, and 10111001101001100100111101010001011111 (all using hash width 14). We output the results in SAM format and converted to BAM format using samtools (Li et al. 2009). We computed read depth across genes using bedtools version 2.15.0 (Quinlan and Hall 2010).
We normalized counts for each gene by the number of total read counts for that strain. We then carried out a median normalization step to normalize across flow cells (Pickrell et al. 2010). After this step, we removed any genes that had no counts across any strain. Finally, we fit a linear model of the form log(normalized_counts) ∼ batch + flow cell + strain + significant surrogate variables. We used the R package sva to calculate surrogate variables, which revealed four significant surrogate variables (Leek and Storey 2007). Further tests used the residuals from this model.
Assessing Differential Expression across Strains
We used a random effects model to test for a strain effect using the R package lme4, using the Maximum Likelihood method and calculating P values using the χ2 distribution (R Development Core Team 2011). We assigned q values by permuting the strain assignments 1,000 times and repeating the analysis, calculating the empirical P value from this distribution, then using the R package q value to assign q values (Storey and Tibshirani 2003).
Testing for Motif Effects on Expression at One Gene
For each gene with a variable motif, we tested the hypothesis that there was a difference in expression between strains with the motif present and strains with the motif absent, using the program EMMA to control for population structure (Kang et al. 2008). P values were again assigned by permuting the motif presence/absence labels 1,000 times, and calculating q values as earlier.
Testing for Motif Effects on Expression across All Genes
For each gene, we converted the normalized expression values to Z scores. For each motif, we then identified genes that had a variable motif upstream. We tested for differential expression by combining expression Z scores from all strains with the motif present, and compared them to Z scores from strains with the motif absent, combining these Z scores across the genes identified earlier. We tested for differential expression using a t test, and determined q values by permuting the labels of present/absent for each gene 1,000 times.
Simulations
The simulations were done as previously described (Connelly and Akey 2012). Briefly, we chose 1,000 random SNPs, which fell within genes or 1,000 bp up- or downstream of genes and which had a minor allele frequency of at least 3 out of 22 as causal SNPs and simulated data based on the genotype at each SNP. We generated simulated data of three effect sizes, 25% of variance in phenotype explained by the genotype, 50% of variance explained by the genotype, and 75% of the variance explained by the genotype. This was equal to a fixed effect of k = 1.64, 2.85, and 4.885 times the standard deviation, respectively, solving for k using the formula percent variance explained = p(1 − p)k2/(p(1 − p)k2 + 1 − 1/n) = ∼1/(1 + 1/(p[1 − p]k2) where k is the fixed effect of x times the standard deviation, p is the frequency of the polymorphism with the fixed effect, and n is the number of individuals (Yu et al. 2006). To assess power, we tested for association between the simulated data and the genotype at the causal variant for each of the 1,000 simulations using EMMA (Kang et al. 2008). To assess the type I error rate, we chose 1,000 random SNPs and asked how often they showed association with any of the 1,000 simulated data sets.
Motif Conservation
We obtained phastCons scores from the UCSC genome browser for each position in the S288c genome (Siepel et al. 2005). We used the P values from the gene-specific test above to identify motifs nominally associated with expression differences among strains (P ≤ 0.05, n = 1,039). To assess significance of polymorphic motif conservation scores, we generated a null distribution by calculating mean conservation from 1,000 randomly selected motifs that are not associated with expression differences (P > 0.05).
Nucleotide Diversity within Motifs and Expression Divergence
We obtained calls for the presence or absence of a TATA box upstream of each gene (Tirosh et al. 2006). For a measurement of nucleosome occupancy, we used the genome-wide nucleosome occupancy data (Lee et al. 2007) and calculated nucleosome occupancy 100 bp upstream of transcription start sites (Zhang and Dietrich 2005), a similar approach to that taken by Tirosh and Barkai (2008). We used a linear model to test for the effect of nucleosome occupancy, TATA box presence, and nucleotide diversity within motifs on expression divergence.
Supplementary Material
Supplementary figures S1 and S2 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank the Yeast Resource Center. This work was supported by National Institute of Health grants R01GM094810 and R01GM098360 to J.M.A. M.J.D. is a Rita Allen Scholar.
References
- Akey J, Ruhe A, Akey D, Wong A, Connelly C, Madeoy J, Nicholas T, Neff M. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107:1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- Blanchette M. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, Seringhaus MR, Wang LY, Gerstein M, Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–D106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, van Nimwegen E, Rajewsky N, Siegal ML. Correlating gene expression variation with cis-regulatory polymorphism in Saccharomyces cerevisiae. Genome Biol Evol. 2010;2:697–707. doi: 10.1093/gbe/evq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CF, Akey JM. On the prospects of whole-genome association mapping in Saccharomyces cerevisiae. Genetics. 2012;191:1345–1353. doi: 10.1534/genetics.112.141168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG. Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol. 2002;19:1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- Doniger SW, Fay JC. Frequent gain and loss of functional transcription factor binding sites. PLoS Comput Biol. 2007;3:e99. doi: 10.1371/journal.pcbi.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Gerke JP, Kruglyak L. Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross. Cold Spring Harb Symp Quant Biol. 2009;74:145–153. doi: 10.1101/sqb.2009.74.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson J, Hsieh L, Sung H, Wang T, Huang C, Lu H, Lu M, Wu S, Li W. Natural selection on cis and trans regulation in yeasts. Genome Res. 2010;20:826–836. doi: 10.1101/gr.101576.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi M, Jelier R, Lehner B. Integrated genome-scale prediction of detrimental mutations in transcription networks. PLoS Genet. 2011;7:e1002077. doi: 10.1371/journal.pgen.1002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. Improved pairwise alignment of genomic DNA [PhD thesis] [University Park (PA)]: The Pennsylvania State University. [Google Scholar]
- He B, Holloway A, Maerkl SJ, Kreitman M. Does positive selection drive transcription factor binding site turnover? A test with Drosophila cis-regulatory modules. PLoS Genet. 2011;7:e1002053. doi: 10.1371/journal.pgen.1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, et al. (26 co-authors) Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Styles CA, Fink GR. Saccharomyces cerevisiae S288C has a mutation in FLO8, a gene required for filamentous growth. Genetics. 1996;144:967–978. doi: 10.1093/genetics/144.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mizuno A, Tabei H, Iwahuti M. Characterization of low-acetic-acid-producing yeast isolated from 2-deoxyglucose-resistant mutants and its application to high-gravity brewing. J Biosci Bioeng. 2006;101:31–37. doi: 10.1263/jbb.101.31. [DOI] [PubMed] [Google Scholar]
- Moses AM, Pollard DA, Nix DA, Iyer VN, Li XY, Biggin MD, Eisen MB. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou C, Pitel F, Gourichon D, et al. (12 co-authors) Cryptic patterning of avian skin confers a developmental facility for loss of neck feathering. PLoS Biol. 2011;9:e1001028. doi: 10.1371/journal.pbio.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustonen V, Lässig M. Evolutionary population genetics of promoters: predicting binding sites and functional phylogenies. Proc Natl Acad Sci U S A. 2005;102:15936–15941. doi: 10.1073/pnas.0505537102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, et al. (37 co-authors) An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Meadows J, Truvé K, et al. (24 co-authors) A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. 2011;7:e1001332. doi: 10.1371/journal.pgen.1001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme B, Gompel N, Rokas A, Kassner V, Williams T, Yeh S, True JR, Carroll SB. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature. 2006;440:1050–1053. doi: 10.1038/nature04597. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suites of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. Vienna (Austria): R Foundation for Statistical Computing; 2011. R: a language and environment for statistical computing. R Version 2.12.2 (2011-02-25). ISBN 3-900051-07-0. Available from: http://www.R-project.org/ [Google Scholar]
- Rockman MV, Kruglyak L. Genetics of global gene expression. Nat Rev Genet. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- Ronald J, Akey J. The evolution of gene expression QTL in Saccharomyces cerevisiae. PLoS One. 2007;2:e678. doi: 10.1371/journal.pone.0000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D, Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, et al. (16 co-authors) Evolutionarily conserved elements in vertebrate, insect, worm and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Ronald J, Akey JM. Inherited variation in gene expression. Annu Rev Genomics Hum Genet. 2009;10:313–332. doi: 10.1146/annurev-genom-082908-150121. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Cox D, Frazer K. Allele-specific KRT1 expression is a complex trait. PLoS Genet. 2006;2:e93. doi: 10.1371/journal.pgen.0020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Regev A. Fungal regulatory evolution: cis and trans in the balance. FEBS Lett. 2009;583:3959–3965. doi: 10.1016/j.febslet.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, et al. (62 co-authors) The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008;18:1084–1091. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy A, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–662. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat Genet. 2006;38:830–834. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- Vernot B, Stergachis AB, Maurano MT, Vierstra J, Neph S, Thurman RE, Stamatoyannopoulos JA, Akey JM. Personal and population genomics of human regulatory variation. Genome Res. 2012;22:1689–1697. doi: 10.1101/gr.134890.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfiffner J, French C, Socha A, Thompson DA, Regev A. Gene duplication and the evolution of ribosomal protein gene regulation in yeast. Proc Natl Acad Sci U S A. 2010;107:5505–5510. doi: 10.1073/pnas.0911905107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, True JR, Carroll SB. Reciprocal functions of the Drosophila yellow and ebony proteins in the development and evolution of pigment patterns. Development. 2002;129:1849–1858. doi: 10.1242/dev.129.8.1849. [DOI] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Yu J, Pressoir G, Briggs W, et al. (12 co-authors) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Dietrich FS. Mapping of transcription start sites in Saccharomyces cerevisiae using 5' SAGE. Nucleic Acids Res. 2005;33:2838–2851. doi: 10.1093/nar/gki583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Zhao H, Mancera E, Steinmetz LM, Snyder M. Genetic analysis of variation in transcription factor binding in yeast. Nature. 2010;464:1187–1191. doi: 10.1038/nature08934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.