

Abstract
The present study used conventional and quantitative microdialysis to assess glutamatergic and GABAergic neurotransmission in the hippocampal CA3 area of the rat following a moderate-dose ethanol treatment regimen. Male Wistar rats received 3.4 g/kg of ethanol or water for 6 days via gastric gavage. Microdialysis experiments commenced 2 days later. Basal and depolarization-induced glutamate overflow were significantly elevated in ethanol-treated animals. Basal and depolarization-induced gamma-aminobutyric acid (GABA) overflow were unaltered. Quantitative no-net-flux microdialysis was used to determine if changes in dialysate glutamate levels following ethanol administration are due to an increase in release or a decrease in uptake.To confirm the validity of this method for quantifying basal glutamate dynamics, extracellular concentrations of glutamate and the extraction fraction, which reflects changes in analyte clearance, were quantified in response to retro-dialysis of the glutamate uptake blocker trans-pyrrolidine-2,4-dicarboxylic acid (tPDC). tPDC significantly decreased the extraction fraction for glutamate, resulting in augmented extracellular glutamate concentrations. Repeated ethanol administration did not alter the glutamate extraction fraction. However, extracellular glutamate concentrations were significantly elevated, indicating that glutamate release is increased as a consequence of repeated ethanol administration. These data demonstrate that repeated bouts of moderate ethanol consumption alter basal glutamate dynamics in the CA3 region of the dorsal hippocampus. Basal glutamate release is augmented, whereas glutamate uptake is unchanged. Furthermore, they suggest that dysregulation of glutamate transmission in this region may contribute to the previously documented deficits in cognitive function associated with moderate dose ethanol use.
Keywords: Ethanol, GABA, glutamate, hippocampus, microdialysis, rat
INTRODUCTION
Chronic ethanol intake can lead to neuroadaptations that have long-term consequences on brain function (Rudolph et al. 1997; Vengeliene et al. 2008). For example, chronic ethanol exposure induces changes in N-methyl-D -aspartate (NMDA) receptors and voltage-sensitive calcium channels (Grant et al. 1990; Hoffman et al. 1990; Follesa & Ticku 1995; Walter & Messing 1999), which can result in altered neurotransmitter release. Such changes are implicated in the hyperexcitability associated with withdrawal from chronic ethanol use (De Witte 2004) and likely contribute to its long-term effects. Chronic ethanol use is also associated with enduring changes in learning and memory (Pereira et al. 1998; Lukoyanov, Madeira & Paula-Barbosa 1999; Farr et al. 2005). Increasing evidence suggests that drug-evoked alterations in cognition may be a critical factor leading to the habitual drug seeking and taking that characterizes addiction (Crews & Boettiger 2009; Robbins & Arnsten 2009).
Most studies documenting ethanol-evoked alterations in learning and memory in rodents have employed high doses of ethanol administered over a period of days (Majchrowicz 1975; Obernier et al. 2002; Faingold 2008) or repeated bouts of extended ethanol administration followed by periods of withdrawal (Stephens & Duka 2008). In these experiments, blood ethanol concentrations (BECs) greater than 250 mg/dl were achieved (Obernier et al. 2002; Faingold 2008). Although similar BECs are detected in heavy drinkers (Bedford, O'Farrell & Howell 2006; Jones 2008; Lamminpää & Hoppu 2009), binge drinkers often exhibit lower BECs (Jones 2008), and binge alcohol drinking is defined as alcohol consumption that yields BECs of 80 mg/dl within a ~2 hours drinking period (National Institute on Alcohol Abuse and Alcoholism 2004).
Seminal work by De Witte and colleagues showed that long-term exposure to ethanol increases dialysate glutamate levels in the hippocampus (Dahchour & De Witte 1999, 2003a,b; De Witte, 2004). Fundamental questions, however, exist as to whether changes in dialysate glutamate levels using conventional microdialysis techniques reflect changes in uptake or release. Furthermore, it is unclear whether hippocampal glutamate dynamis are altered following short-term exposure to a moderate-dose ethanol treatment regimen. The present study used conventional and quantitative microdialysis techniques to quantify gamma-aminobutyric acid A (GABA) and glutamate dynamics in the CA3 region of the dorsal hippocampus, a region implicated in spatial working memory, in control and ethanol-treated animals.
MATERIALS AND METHODS
Animals
Male Wistar rats, weighing 250–300 g (Charles River Laboratories, Germantown, MD, USA) were used. Animals were housed in facilities approved by the American Association for the Accreditation of Laboratory Animal Care. They were maintained at a constant temperature (22 ± 2°C) on a 12-hour light/dark cycle. Food and water were available ad libitum. All experiments were conducted in accordance with the guidelines of the Institutional Care and Use Committee of the National Institute on Drug Abuse, National Institutes of Health.
Surgical procedures
Rats were anesthetized with Equithesin (NIH Pharmacy, Bethesda, MD, USA) [sodium pentobarbital, chloral hydrate, and magnesium sulphate; 9.72 mg/ml; 3 ml/kg, intraperitoneal (i.p.)] and placed in a Kopf stereotaxic apparatus (David Kopf, Tujunga, CA, USA) to enable implantation of a guide cannula (CMA/11, CMA Inc., North Chelmsford, MA, USA) into the CA3 region of the dorsal hippocampus using stereotaxic coordinates (antero–posterior: −3.3 mm from bregma, lateral: −2.6 mm and ventral: −3.0 mm from the skull surface) (Paxinos & Watson 1998). The cannula was secured to the skull using acrylic cement and small stainless steel screws. After surgery, the rats were housed individually and allowed to recover for 5 days before the commencement of repeated ethanol or water treatment regimen.
Upon completion of all experiments, animals were euthanized by i.p. injection of chloralhydrate (1 g/kg) for anatomical confirmation of probe placement. Dialysis probes were dipped briefly in cresyl violet and inserted into the guide cannula, allowing for visualization of their placement on serial coronal sections of the brain (20–30 μm). Only data from animals with microdialysis cannulae placements in CA3 were used for subsequent statistical analysis.
Ethanol treatment
Rats received ethanol (40% v/v, 10 ml/kg, 3.4 g/kg) or an equivalent volume of water (10 ml/kg) via gastric gavage (Collins, Corso & Neafsey 1996; Zou et al. 1996) once each day for 6 days. This treatment regimen resulted in BECs of 72–5 mg/dl 2 hours after gavage, and peak BECs (150 mg/dl) were obtained 4 hours post gavage.
Microdialysis procedures
Microdialysis experiments commenced 2 days after the final ethanol treatment. Approximately 12–16 hours prior to the start of experiments, a microdialysis probe (CMA/11, CMA; 1-mm probe) was connected to the microdialysis assembly and flushed with artificial cerebrospinal fluid (aCSF) containing (in mmol/l) NaCl, 145; KCl, 2.8; CaCl2, 1.2; MgCl2, 1.2; ascorbic acid, 0.25; d-glucose, 5.4, adjusted to pH 7.4 with 0.5 mol/l NaOH. The rat was gently restrained, and the probe was inserted into the guide cannula. The rat was then placed in the microdialysis chamber with food and water freely available, and the probe was perfused overnight with aCSF at a flow rate of 0.3 μl/min.
One hour prior to the initiation of the microdialysis experiments, the syringes were filled with fresh aCSF, and the flow rate was changed to 1.0 μl/min. Following a 1-hour equilibration period, six consecutive 5-minute dialysis samples were collected for determination of baseline GABA and glutamate concentrations. The aCSF was then changed to aCSF containing high KCl (in mmol/l: NaCl, 87.8; KCl, 60.0; CaCl2, 1.2; MgCl2, 1.2; ascorbic acid, 0.25; d-glucose, 5.4, adjusted to pH 7.4 with 0.5 mol/l NaOH), and, following a 30-minute equilibration period, six consecutive 5-minute samples were collected. The perfusate was then changed to fresh aCSF and six consecutive 5-minute samples were collected following a 30-minute equilibration period.
Immediately after collection, samples were frozen on dry ice and stored at −80°C. Quantification of amino acid neurotransmitters in dialysate samples was performed by capillary electrophoresis coupled to laser-induced fluorescence detection (CE-LIFD) as described in Chefer et al. (2009).
No-net-flux microdialysis experiments were conducted in separate groups of animals in order to assess basal glutamate dynamics in control and ethanol-treated animals. Five different concentrations of glutamate in aCSF (Cin of 0, 1, 2.5, 5 and 10 μM glutamate) were perfused through the probes in random order to determine the extracellular concentration and extraction fraction (Ed) for glutamate. After a 30-minute equilibration period, three 5-minute dialysis samples were collected at each Cin concentration. Glutamate in the perfusates (Cout) was quantified using CE-LIFD.
To confirm that the extraction fraction is altered in response to pharmacological manipulations that affect glutamate uptake, no-net-flux microdialysis was conducted to determine whether retro-dialysis of the glutamate transporter blocker pyrrolidine-2,4-dicarboxylic acid (tPDC; 0.5 mM; Tocris Bioscience, Ellis-ville, MO, USA) into the CA3 region decreases the extraction fraction. No-net-flux microdialysis was performed as described above in the presence and absence of tPDC in aCSF.
Statistical analysis
All values were expressed as means ± SEMs, and statistical analyses were performed using SPSS (SPSS Inc., Chicago, IL, USA). The effects of ethanol treatment were analyzed using a two-factor repeated-measures analysis of variance (ANOVA), with one between-subjects factor (water versus ethanol) and two within-subjects factor (baseline versus KCl-induced neurotransmitter values and time).
The area under the curve (AUC) value for the samples collected after KCl-aCSF perfusion was calculated for each animal, according to the standard trapezoid method. The formula used was AUC = [0.5(B + S1) d + 0.5(S1 + S2)d + 0.5 (S2 + S3)d + … + 0.5 (Sn−1 + Sn)d] – (Bdn), where B is the average of the samples collected during baseline, Sx are the values of each fraction collected during drug challenge, n is the total number of fractions collected during drug challenge and d is the duration of each fraction (in minutes). AUC values and values for the average basal neurotransmitter concentrations were analyzed using a one-way ANOVA with ethanol treatment as an independent factor. Absolute values for dialysate glutamate and GABA levels were analyzed using a repeated-measures ANOVA with two between-subjects factors (depolarization challenge: basal versus evoked levels and time) and one within-subjects factor (ethanol treatment: vehicle versus ethanol).
Glutamate no-net-flux microdialysis data were analyzed analogous to that described for dopamine no-net-flux microdialysis (Chefer et al. 2005) Extracellular glutamate concentrations and extraction fractions were determined for each animal by plotting the net change in glutamate (Cin – Cout) against Cin followed by linear regression analysis. The point when no glutamate was gained or lost (Cin – Cout = 0) represents an unbiased estimate of extracellular concentrations. The slope of the linear regression line represents the extraction fraction (Ed), an indirect measure of glutamate uptake (Melendez et al. 2005). The slopes of the linear regression and estimates of the extracellular glutamate concentrations were analyzed with linear regression analysis and unpaired t-test.
A value of P ≤ 0.05 was considered to indicate a statistically significant difference between data sets.
RESULTS
Schematic representation of probe placements in the dorsal hippocampus is shown in Fig. 1 using histological drawing from the atlas of Paxinos & Watson (1998).
Figure 1.
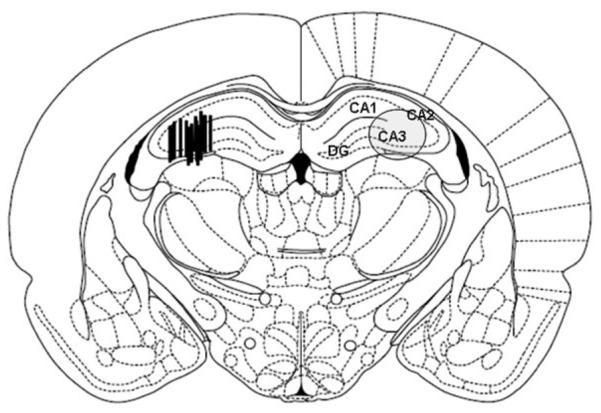
Location of microdialysis probes in the hippocampus. The actual probe placements are shown in the left brain section: black bars indicate the location of 1-mm microdialysis membrane. The overall area under investigation is shown with an oval in the right brain section. The image is adapted from Paxinos & Watson (1998), and AP coordinates (in mm) are given relative to bregma
There was no effect of ethanol treatment [F(1,11) = 0.02; P > 0.05; Fig. 2a,b] on dialysate GABA levels. Likewise, there was no ethanol depolarization challenge interaction [F(1,11) = 0.74; P > 0.05; Fig. 2a,c] for dialysate GABA levels. These results suggest that the ethanol treatment failed to significantly alter GABA neurotransmission in the CA3 area of hippocampus.
Figure 2.

Repeated intermittent administration of ethanol (3.4 g/kg, intragastrically via gavage, once daily × 6 days) does not change basal and KCl-induced GABA levels in dorsal hippocampus (CA3). (a) Time course of dialysate GABA levels in the CA3 area of hippocampus in water- (white circles) and ethanol-treated (black circles) animals before and after reversed dialysis of KCl (60 mM.; arrow and dotted line). Data are expressed as the means ± SEM. n = number of animals in each experimental group (6–7 animals per group). (b) Bar graphs of basal GABA levels in water- and ethanol-treated animals expressed as the means ± SEM. (c) Bar graphs of AUC values for the GABA response to KCl expressed as the means ± SEM. AUC, area under the curve; GABA, gamma-aminobutyric acid; SEM, standard error of the mean
In contrast, ethanol treatment increased basal and depolarization-evoked glutamate overflow in the CA3 region (Fig. 3a–c). Both basal and KCl-induced glutamate levels were increased, on average, by 400% in ethanol-treated animals compared with vehicle-treated controls: from 388 ± 97 nM to 1576 ± 770 nM and from 683 ± 120 nM to 2748 ± 869 nM for basal and stimulated overflow, respectively. Despite the observed increases, there was no significant main effect of ethanol treatment [F(1,11) = 2.29; P > 0.05] on glutamate overflow. However, there was a significant ethanol depolarization challenge interaction [F(1,11) = 7.59; P < 0.05]. Pair-wise analyses revealed a significant effect of ethanol treatment on KCl-induced glutamate levels [F(1,12) = 4.74; P < 0.05] and no significant effect of ethanol treatment on basal glutamate levels [F(1,11) = 1.56; P > 0.05]. Thus, ethanol treatment significantly augmented depolarization-induced glutamate release.
Figure 3.

Repeated intermittent administration of ethanol (3.4 g/kg, intragastrically via gavage, once daily × 6 days) increases dialysate glutamate levels in dorsal hippocampus (CA3). (a) Time course of dialysate glutamate levels in the CA3 area of hippocampus in water- (white circles) and ethanol-treated (black circles) animals before and after reversed dialysis of KCl (60 mM.; arrow and dotted line). Data are expressed as the means ± SEM. n = number of animals in each experimental group (6–7 animals per group). (b) Bar graphs of basal GABA levels in water- and ethanol-treated animals expressed as the means ± SEM. (c) Bar graphs of AUC values for the GABA response to KCl expressed as the means ± SEM. *Significant difference in KCl-induced glutamate responses between water- and ethanol-treated animals (P ≤ 0.05). AUC, area under the curve; GABA, gamma-aminobutyric acid; SEM, standard error of the mean
The lack of significance for basal glutamate could be due to the reported variability of basal dialysate glutamate levels measured by conventional microdialysis (see Timmerman & Westerink 1997 for review). To quantify the basal extracellular glutamate levels in the CA3 and to test if glutamate uptake was changed in ethanol-treated animals, a no-net-flux experiment was performed in vehicle- and ethanol-treated animals at the same abstinence time point (2 days).
The use of no-net flux microdialysis for measuring changes in glutamate uptake was validated in experiments in which two separate groups of animals were perfused with aCSF containing different concentrations of glutamate in the absence or presence of the glutamate transporter blocker tPDC (0.5 mM). Figure 4, A illustrates the gain or loss of glutamate (Cin – Cout) as a function of concentration added to the perfusate (Cin). The slope of the regression lines is equal to the extraction fraction, and the x-intercept provides an unbiased estimate of extracellular transmitter concentrations. The negative value of the y-intercept corresponds to dialysate concentrations obtained in a conventional microdialysis experiment. Linear regression analysis revealed a significant difference in slope between the two groups [0.53 ± 0.04 versus 0.39 ± 0.04 for control and tPDC, respectively; F(1,6) = 6.25; P = 0.046; Fig. 4a,b], indicating a decrease in glutamate uptake in tPDC-perfused animals. Because of the significant slope difference, linear regression analysis could not be performed to test statistical differences between the x-intercepts. However, an unpaired t-test showed that the value of the x-intercept differed significantly between groups (668 ± 103 versus 3148 ± 883 for control and tPDC group, respectively; unpaired t-test: P = 0.01, t = 3.08, degrees of freedom = 9; Fig. 4a,b). These results suggest that tPDC increases extracellular glutamate concentration by decreasing glutamate uptake.
Figure 4.

Validation of the no-net-flux method for measuring changes in glutamate uptake. (a) Plot of the mean ± SEM gain or loss of dialysate glutamate concentrations to and from the brain as a function of the perfusate glutamate concentration and the average linear regression fit of the data: data from animals perfused with regular aCSF (open circles), data from animals perfused with 0.5 mM tPDC in the aCSF (filled circles). The slope of the regression line represents the extraction fraction (Ed), an indirect estimate of glutamate uptake. The zero point on the y-axis at which no glutamate is gained or lost from the perfusate represents an unbiased estimate of the apparent extracellular concentration of glutamate. (b) Bar graphs of extracellular glutamate and Ed values expressed as the mean ± SEM. Significant difference between aCSF and tPDC group in *Ed and **extracellular glutamate values. aCSF, artificial cerebrospinal fluid; Glut, glutamate; SEM, standard error of the mean; tPDC, pyrrolidine-2,4-dicarboxylic acid
Figure 5 illustrates the results of the no-net-flux microdialysis experiment in vehicle- and ethanol-treated animals. There was no significant difference between pre-treatment groups in the slope of the linear regression [0.52 ± 0.01 versus 0.54 ± 0.02 for vehicle- and ethanol-treated animals, respectively; F(1,6) = 1.38, P = 0.29; Fig. 5a,b], indicating no change in hippocampal glutamate uptake following chronic ethanol administration. However, a significant difference in the value of the x-intercept [435 ± 77 versus 1524 ± 713 for control and ethanol-treated group, respectively; F(1,6) = 52.85, P < 0.001; Fig. 5a,b] was seen, indicating that basal extracellular glutamate levels are increased following repeated, moderate-dose ethanol administration. Extracellular concentrations reflect release and uptake. Therefore, the lack of change in extraction fraction coupled to the increase in extracellular glutamate levels indicates an increase in basal glutamate release.
Figure 5.

Repeated intermittent administration of ethanol increases glutamate release but does not change its uptake in dorsal hippocampus (CA3). (a) Plot of the mean ± SEM gain or loss of dialysate glutamate concentrations to and from the brain as a function of the perfusate glutamate concentration and the average linear regression fit of the data from animals treated with vehicle (water; 10 ml/kg; open circles) or ethanol (40% v/v, 10 ml/kg; 3.4 g/kg; filled circles) for 6 days. The slope of the regression line represents the extraction fraction (Ed), an indirect estimate of glutamate uptake. The zero point on the y-axis at which no glutamate is gained or lost from the perfusate represents an unbiased estimate of the apparent extracellular concentration of glutamate. (b) Bar graphs of extracellular glutamate and Ed values expressed as the mean ± SEM *Significant difference between vehicle- and ethanol-treated group in extracellular glutamate values. Glu, glutamate; SEM, standard error of the mean
In summary, microdialysis experiments demonstrated that ethanol treatment resulted in an increase in glutamatergic neurotransmission in the CA3 area of the hippocampus attributed to an augmentation of basal glutamate release.
DISCUSSION
The present microdialysis study characterized the effects of repeated moderate-dose ethanol binges on hippocampal glutamate and GABA transmission. The results indicate that 6 days of ethanol (3.4 mg/kg/day, intragastrically) administration via gavage increases dialysate glutamate levels in the CA3 area of the hippocampus, but does not significantly alter dialysate GABA levels. No-net-flux microdialysis experiments further revealed that the augmentation in dialysate glutamate levels reflects an increase in the extracellular concentration of this neurotransmitter with no apparent change in uptake, suggestive of elevated glutamate release.
Ethanol treatment increased basal and depolarization-evoked glutamate overflow in the CA3 region by approximately 400%. Only the change in depolarization-evoked glutamate overflow was statistically significant. The lack of significance for basal glutamate overflow appears incongruous with the observed ethanol treatment-associated increase in extracellular glutamate concentrations. However, it is not surprising in light of the reported variability in basal glutamate levels measured by microdialysis (see Timmerman & Westerink 1997 for review). Indeed, basal extracellular glutamate is derived from multiple sources and regulated by multiple mechanisms (Fillenz 1995; Timmerman & Westerink 1997; Palmada & Centelles 1998). In contrast, potassium-induced glutamate release has been shown to be mostly of vesicular origin (Paulsen & Fonnum 1989; Forray, Bustos & Gysling 1999) and is therefore regulated by a limited number of mechanisms.
While the ability of ethanol to interact with glutamatergic pathways and NMDA receptors is well established, relatively little is known about its ability to modulate endogenous release of glutamate in vivo. Depending on the dose administered, acute ethanol exposure has been reported to either increase, decrease or have no effect on glutamate levels in the hippocampus (Moghaddam & Bolinao 1994; Shimizu et al. 1998). However, withdrawal from long-term ethanol vapour exposure has consistently been shown to elevate dorsal hippocampal microdialysate glutamate levels (Dahchour & De Witte 1999, 2003a). This effect was transient and augmented with subsequent withdrawal periods (Dahchour & De Witte 2003a; De Witte 2004). Similar increases in glutamate levels were also described in ventral hippocampus following 3 weeks of ethanol administration (2 or 3 g/kg, three times per day for two consecutive days, followed by 5 days of abstinence, `binge drinking' rat model) (Ward et al. 2009). Interestingly, all of these studies showed no significant changes in GABA neurotransmission. To date, there are no data in the literature regarding hippocampal glutamate or GABA levels following repeated ethanol binges of shorter duration and more moderate doses.
It is well known that chronic repeated exposure to ethanol increases NMDA receptor binding sites (Grant et al. 1990; Gulya et al. 1991; Snell, Tabakoff & Hoffman 1993) and NMDA receptor-mediated function (Trevisan et al. 1994). This neuroadaptation is probably caused by chronic blockade of NMDA receptors by ethanol (Lovinger, White & Weight 1989; Whittington, Lambert & Little 1996). Furthermore, it has been suggested that NMDA receptors are involved in a positive feedback mechanism during ethanol withdrawal, leading to a facilitation of glutamate release. Indeed, Rossetti, Carboni & Fadda (1999) demonstrated that ethanol withdrawal facilitated glutamate release in striatum. It is important, however, to note that the animals used in the present study were not in withdrawal when dialysis studies were performed. Thus, we previously showed that neither seizures nor other quantifiable somatic withdrawal signs were evident 24 or 48 hours after treatment cessation (Bakalkin et al. 2008).
Although, the exact mechanism through which the NMDA receptor mediates increases in glutamate is not clear, several NMDA receptor-mediated pathways may be involved (see Fadda & Rossetti 1998 for review). Recent electron microscopy studies indicate that the NMDA-1 receptor subunit is present on dendrites of CA3 pyramidal cells and mossy fiber axons of the primate hippocampus (Siegel et al. 1994). This histochemical evidence for a pre-synaptic localization of NMDA receptors on excitatory neurons is suggestive of NMDA-induced glutamate release in this area of hippocampus.
The present results indicating that repeated ethanol (3.4 g/kg/day for 6 days) did not significantly alter basal or evoked dialysate GABA levels in the CA3 region were unexpected. Behavioral, neurochemical and electro-physiological studies have provided compelling evidence that ethanol can enhance GABAergic synaptic activity via both pre- and post-synaptic mechanisms (see Criswell & Breese 2005 for review). Moreover, ethanol-mediated changes in GABAergic neurotransmission can alter the activity of pyramidal neurons in CA3, thereby affecting glutamate outflow. The lack of difference in GABA levels between ethanol-treated animals and controls in the present findings suggests that if GABAergic mechanisms contribute to the ethanol-induced enhancement of glutamate release, then they are post-synaptic rather than pre-synaptic.
Recent studies have suggested a role for GABAA receptors in the acute and chronic actions of ethanol. Typically, GABAA receptors in the brain adapt to chronic ethanol exposure via changes in receptor function and subunit expression. Acute ethanol enhances GABAA receptor-mediated synaptic transmission in the hippocampus and inhibitory synaptic transmission in the brain (Proctor et al. 2006; Yan et al. 2009). Conversely, chronic ethanol consumption results in tolerance to many GABAergic-related effects of ethanol and increased central nervous system excitability. The mechanisms responsible for adaptation of GABAA receptors to chronic ethanol exposure are complex and involve ethanol-induced changes in cell surface expression, synaptic localization, receptor phosphorylation, neurosteroids and/or changes in GABAA receptor subunit composition (see Kumar, Fleming & Morrow. 2004 for review). These post-synaptic changes and corresponding decreases in GABAergic tone may be one mechanism contributing to the augmentation in glutamatergic neurotransmission seen in the present study.
Hypofunction of GABAA receptors and enhanced function of NMDA receptors are suggested to be responsible for the increase in glutamate levels and the behavioral susceptibility during ethanol withdrawal (De Witte 2004). However, the same mechanisms may also contribute to increased glutamate release at later stages of abstinence. Thus, a single intoxicating dose of ethanol (5 mg/kg, gavage) produces changes in GABAA receptors function that are fully reversed only 2 weeks after ethanol administration (Liang et al. 2007).
Interestingly, increased GABA transporter density is observed in the hippocampus following 2 weeks of ethanol administration in a liquid diet (Devaud 2001). Therefore, changes in basal GABA uptake following moderate dose ethanol treatment can not be ruled out. Future studies, using quantitative microdialysis, are needed to address this issue.
The moderate-dose repeated ethanol treatment employed in the present study resulted in no quantifiable withdrawal signs, while markedly impaired spatial learning and memory 3–6 days after cessation of ethanol exposure (Bakalkin et al. 2008). Spatial learning and memory were recovered by the 11th–14th days, which is consistent with cognitive recovery of human alcohol abusers in the course of abstinence. The results of the present study indicate that the employed treatment leads to a significant augmentation of glutamate release in CA3 area of hippocampus. Aberrant glutamate neurotransmission is implicated in hippocampal-related learning/memory impairments (see Vizi & Kiss 1998; McEwen 2001 for review). Therefore, the observed increase in glutamate release may be one mechanism that contributes to alcohol-related deficits in learning and memory following repeated ethanol binges.
Here, we provide evidence that that even short-term exposure to a moderate-dose ethanol treatment regimen can lead to increases in dialysate glutamate levels in CA3 region of the hippocampus, which reflect an increase in glutamate release. As a consequence, the intricate balances between excitatory and inhibitory mechanisms that are normally in place to regulate hippocampal function are impaired. Further studies examining the neural substrates by which moderate-dose ethanol administration disrupts hippocampal homeostasis and produces cognitive impairment are warranted.
Acknowledgements
This work was supported by the National Institute on Drug Abuse/Intramural Research Program (NIDA/IRP) and grants from the Swedish Council for Working Life and Social Research (FAS), AFA Forsäkring, Swedish Science Research Council, Alcohol Research Council of the Swedish Retailing Monopoly, and funds from Uppsala University and Karolinska Institutet.
Footnotes
Authors Contribution
TS, GB and VC were responsible for the study concept and design. VC oversaw all aspects of microdialysis experiments as well as data collection and analysis. GW performed surgeries and treatments. VC and JM collected and analyzed the data. VC drafted the manuscript and TS and GB provided critical revisions of the manuscript. All authors critically reviewed the content of the manuscript and approved the final version for submission.
References
- Bakalkin G, Bazov I, Yakovleva T, Kuntic V, Sheedy D, Gartrick T, Harper C, Ögren S, Kuzmin A. Alcoholism-associated molecular adaptations in endogenous opioids in brain neurocognitive circuits: human and animal correlates. European Neuropsychopharmacology. 2008;18(Suppl. 4):S194. [Google Scholar]
- Bedford D, O'Farrell A, Howell F. Blood alcohol levels in persons who died from accidents and suicide. Ir Med J. 2006;99:80–83. [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, Shippenberg TS. Endogenous kappa-opioid receptor systems regulate mesoaccumbal dopamine dynamics and vulnerability to cocaine. J Neurosci. 2005;25:5029–5037. doi: 10.1523/JNEUROSCI.0854-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Denoroy L, Zapata A, Shippenberg TS. Mu opioid receptor modulation of somatodendritic dopamine overflow: GABAergic and glutamatergic mechanisms. Eur J Neurosci. 2009;30:272–278. doi: 10.1111/j.1460-9568.2009.06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Corso TD, Neafsey EJ. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic `binge' intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcohol Clin Exp Res. 1996;20:284–292. doi: 10.1111/j.1530-0277.1996.tb01641.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Boettiger CA. Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav. 2009;93:237–247. doi: 10.1016/j.pbb.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell HE, Breese GR. A conceptualization of integrated actions of ethanol contributing to its GABAmimetic profile: a commentary. Neuropsychopharmacology. 2005;30:1407–1425. doi: 10.1038/sj.npp.1300750. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P. Effect of repeated ethanol withdrawal on glutamate microdialysate in the hippocampus. Alcohol Clin Exp Res. 1999;23:1698–1703. doi: 10.1111/j.1530-0277.1999.tb04063.x. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P. Excitatory and inhibitory amino acid changes during repeated episodes of ethanol withdrawal: an in vivo microdialysis study. Eur J Pharmacol. 2003a;459:171–178. doi: 10.1016/s0014-2999(02)02851-0. [DOI] [PubMed] [Google Scholar]
- Dahchour A, De Witte P. Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res. 2003b;27:465–470. doi: 10.1097/01.ALC.0000056617.68874.18. [DOI] [PubMed] [Google Scholar]
- De Witte P. Imbalance between neuroexcitatory and neuroinhibitory amino acids causes craving for ethanol. Addict Behav. 2004;29:1325–1339. doi: 10.1016/j.addbeh.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Devaud LL. Ethanol dependence has limited effects on GABA or glutamate transporters in rat brain. Alcohol Clin Exp Res. 2001;25:606–611. [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;55:1–47. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Faingold CL. The Majchrowicz binge alcohol protocol: an intubation technique to study alcohol dependence in rats. Curr Protoc Neurosci. 2008;28:1–9. doi: 10.1002/0471142301.ns0928s44. Chapter 9:Unit 9. [DOI] [PubMed] [Google Scholar]
- Farr SA, Scherrer JF, Banks WA, Flood JF, Morley JE. Chronic ethanol consumption impairs learning and memory after cessation of ethanol. Alcohol Clin Exp Res. 2005;29:971–982. doi: 10.1097/01.alc.0000171038.03371.56. [DOI] [PubMed] [Google Scholar]
- Fillenz M. Physiological release of excitatory amino acids. Behav Brain Res. 1995;71:51–67. doi: 10.1016/0166-4328(95)00045-3. [DOI] [PubMed] [Google Scholar]
- Follesa P, Ticku MK. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Brain Res Mol Brain Res. 1995;29:99–106. doi: 10.1016/0169-328x(94)00235-7. [DOI] [PubMed] [Google Scholar]
- Forray MI, Bustos G, Gysling K. Noradrenaline inhibits glutamate release in the rat bed nucleus of the stria terminalis: in vivo microdialysis studies. J Neurosci Res. 1999;55:311–320. doi: 10.1002/(SICI)1097-4547(19990201)55:3<311::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Grant KA, Valverius P, Hudspith M, Tabakoff B. Ethanol withdrawal seizures and the NMDA receptor complex. Eur J Pharmacol. 1990;176:289–296. doi: 10.1016/0014-2999(90)90022-x. [DOI] [PubMed] [Google Scholar]
- Gulya K, Grant KA, Valverius P, Hoffman PL, Tabakoff B. Brain regional specificity and time course changes in the NMDA-receptor-ionophore complex during ethanol withdrawal. Brain Res. 1991;547:129–134. [PubMed] [Google Scholar]
- Hoffman PL, Rabe CS, Grant KA, Valverius P, Hudspith M, Tabakoff B. Ethanol and the NMDA receptor. Alcohol. 1990;7:229–231. doi: 10.1016/0741-8329(90)90010-a. [DOI] [PubMed] [Google Scholar]
- Jones AW. Hospital alcohol tests not completely easy to use for legal purposes. Conversion of ethanol levels in plasma or serum to permillage level in blood. Lakartidningen. 2008;105:367–368. [PubMed] [Google Scholar]
- Kumar S, Fleming RL, Morrow AL. Ethanol regulation of gamma-aminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol Ther. 2004;101:211–226. doi: 10.1016/j.pharmthera.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Lamminpää A, Hoppu K. First-order alcohol elimination in severe alcohol intoxication in an adolescent: a case report. Am J Emerg Med. 2009;27:128.e5. doi: 10.1016/j.ajem.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Liang J, Suryanarayanan A, Abriam A, Snyder B, Olsen RW, Spigelman I. Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J Neurosci. 2007;27:12367–12377. doi: 10.1523/JNEUROSCI.2786-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Lukoyanov NV, Madeira MD, Paula-Barbosa MM. Behavioral and neuroanatomical consequences of chronic ethanol intake and withdrawal. Physiol Behav. 1999;66:337–346. doi: 10.1016/s0031-9384(98)00301-1. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia. 1975;43:245–254. doi: 10.1007/BF00429258. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann NY Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Hicks MP, Cagle SS, Kalivas PW. Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res. 2005;29:326–333. doi: 10.1097/01.alc.0000156086.65665.4d. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Bolinao ML. Biphasic effect of ethanol on extracellular accumulation of glutamate in the hippocampus and the nucleus accumbens. Neurosci Lett. 1994;178:99–102. doi: 10.1016/0304-3940(94)90299-2. [DOI] [PubMed] [Google Scholar]
- National Institute on Alcohol Abuse and Alcoholism Newsletter Feb 4, 2004.
- Obernier JA, White AM, Swartzwelder HS, Crews FT. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav. 2002;72:521–532. doi: 10.1016/s0091-3057(02)00715-3. [DOI] [PubMed] [Google Scholar]
- Palmada M, Centelles JJ. Excitatory amino acid neurotransmission. Pathways for metabolism, storage and reuptake of glutamate in brain. Front Biosci. 1998;20:d701–d718. doi: 10.2741/a314. [DOI] [PubMed] [Google Scholar]
- Paulsen RE, Fonnum F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J Neurochem. 1989;52:1823–1829. doi: 10.1111/j.1471-4159.1989.tb07263.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1998. [Google Scholar]
- Pereira SR, Menezes GA, Franco GC, Costa AE, Ribeiro AM. Chronic ethanol consumption impairs spatial remote memory in rats but does not affect cortical cholinergic parameters. Pharmacol Biochem Behav. 1998;60:305–311. doi: 10.1016/s0091-3057(97)00472-3. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Diao L, Freund RK, Browning MD, Wu PH. Synaptic GABAergic and glutamatergic mechanisms underlying alcohol sensitivity in mouse hippocampal neurons. J Physiol. 2006;575(Pt 1):145–159. doi: 10.1113/jphysiol.2006.112730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Arnsten AF. The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu Rev Neurosci. 2009;32:267–287. doi: 10.1146/annurev.neuro.051508.135535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S, Fadda F. Glutamate-induced increase of extracellular glutamate through N-methyl-D-aspartate receptors in ethanol withdrawal. Neuroscience. 1999;93:1135–1140. doi: 10.1016/s0306-4522(99)00250-x. [DOI] [PubMed] [Google Scholar]
- Rudolph JG, Walker DW, Iimuro Y, Thurman RG, Crews FT. NMDA receptor binding in adult rat brain after several chronic ethanol treatment protocols. Alcohol Clin Exp Res. 1997;21:1508–1519. [PubMed] [Google Scholar]
- Shimizu K, Matsubara K, Uezono T, Kimura K, Shiono H. Reduced dorsal hippocampal glutamate release significantly correlates with the spatial memory deficits produced by benzodiazepines and ethanol. Neuroscience. 1998;83:701–706. doi: 10.1016/s0306-4522(97)00339-4. [DOI] [PubMed] [Google Scholar]
- Siegel SJ, Brose N, Janssen WG, Gasic GP, Jahn R, Heineman SF. Regional, cellular and ultrastructural distribution of N-methyl-d-aspartate receptor subunit 1 in monkey hippocampus. Proc Natl Acad Sci U S A. 1994;91:564–568. doi: 10.1073/pnas.91.2.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell LD, Tabakoff B, Hoffman PL. Radioligand binding to the N-methyl-D-aspartate receptor/ionophore complex: alterations by ethanol in vitro and chronic in vivo ingestion. Brain Res. 1993;602:91–98. doi: 10.1016/0006-8993(93)90246-j. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Duka T. Review. Cognitive and emotional consequences of binge drinking: role of amygdala and prefrontal cortex. Philos Trans R Soc Lond B Biol Sci. 2008;363:3169–3179. doi: 10.1098/rstb.2008.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, Nestler EJ. Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem. 1994;62:1635–1638. doi: 10.1046/j.1471-4159.1994.62041635.x. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Molander A, Spanagel R. Neuropharmacology of alcohol addiction. Br J Pharmacol. 2008;154:299–315. doi: 10.1038/bjp.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizi ES, Kiss JP. Neurochemistry and pharmacology of the major hippocampal transmitter systems: synaptic and non-synaptic interactions. Hippocampus. 1998;8:566–607. doi: 10.1002/(SICI)1098-1063(1998)8:6<566::AID-HIPO2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Messing RO. Regulation of neuronal voltage-gated calcium channels by ethanol. Neurochem Int. 1999;35:95–101. doi: 10.1016/s0197-0186(99)00050-9. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Colivicchi MA, Allen R, Schol F, Lallemand F, de Witte P, Ballini C, Corte LD, Dexter D. Neuro-inflammation induced in the hippocampus of `binge drinking' rats may be mediated by elevated extracellular glutamate content. J Neurochem. 2009;111:1119–1128. doi: 10.1111/j.1471-4159.2009.06389.x. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Lambert JD, Little HJ. Increased NMDA receptor and calcium channel activity underlying ethanol withdrawal hyperexcitability. Alcohol Alcohol. 1996;30:105–114. [PubMed] [Google Scholar]
- Yan H, Li Q, Fleming R, Madison RD, Wilson WA, Swartzwelder HS. Developmental sensitivity of hippocampal inter-neurons to ethanol: involvement of the hyperpolarization-activated current. J Neurophysiol. 2009;101:67–83. doi: 10.1152/jn.90557.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Martinez DB, Neafsey EJ, Collins MA. Binge ethanol-induced brain damage in rats: effect of inhibitors of nitric oxide synthase. Alcohol Clin Exp Res. 1996;20:1406–1411. doi: 10.1111/j.1530-0277.1996.tb01141.x. [DOI] [PubMed] [Google Scholar]