

Pulmonary infection by RSV alters local immune response, promoting increased pathogenesis leading to enhanced lung disease.
Keywords: immune responses, cytokines, T cells
Abstract
In the present studies, we have established that RSV can elicit a more pathogenic environment dependent on improper DC-associated sensitization. Our initial studies demonstrated that RSV, but not influenza, infection during an allergen exposure into the airway induced a more severe allergen response. The RSV-induced exacerbation included an increased Th2 cytokine response and pathophysiology as monitored by AHR and mucus overproduction. DCs played a central role in the allergen-induced responses, as instilling RSV-infected BMDC into the airway could recapitulate a live virus challenge. With the use of CCR6−/− mice that have a primary defect in the recruitment of mDC subsets, reduced exacerbation of disease was observed when RSV was administered along with allergen. Furthermore, sensitization of mice with RSV-infected BMDC into the airway produced a more severe immune response to a live virus challenge. Subsequently, using RSV-infected BMDC from CCR7−/− mice (that do not migrate efficiently to LNs) to sensitize the exacerbated response demonstrated that the response was dependent on DC migration to the LN. Finally, the ability of RSV-infected DCs to elicit an exacerbated, allergen-induced pathogenic response could be maintained for as long as 3 weeks, suggesting that RSV-infected DCs themselves created an altered immune environment that impacts off-target mucosal responses that could have prolonged effects.
Introduction
The induction of immune responses in the host often determines the nature and the severity of the ensuing illness during infectious diseases and may even dictate future local immune responses. Studies have shown that patients with severe respiratory viral infections have an increased risk for the development of chronic pulmonary diseases [1–5]. A number of respiratory viruses have been implicated in the induction of pulmonary diseases, including infections with RSV, rhinovirus, influenza, parainfluenza, and adenovirus. How the interactions of specific viruses, underlying immune conditions, genetic predisposition, and local mucosal environments contribute to disease severity is not entirely clear. Recent studies in children [6] and adults [7] support this contention related to severe asthmatic exacerbations. Understanding the mechanisms that promote inappropriate immune responses to viral infections may lead to better therapy. RSV infections are especially detrimental and have been associated with early and persistent pulmonary disease, especially in premature infants.
The activation of the innate immune response, including antigen-presenting DCs, and recruitment of T cells that amplify and skew the immune response toward more intense pathology likely contribute to more severe disease and clinical crisis in asthmatic patients. Whereas various immune responses to viral infections may have common mechanisms of activation, RSV appears to promote responses that on their own can cause severe pulmonary problems [8–10]. Recent evidence suggests that RSV also has a significant role in elderly patient populations, as well as in patients with chronic obstructive pulmonary disease [11]. In addition, the specific mechanism(s) of immune regulation identified in RSV studies may be relevant to other viral infections that also must be recognized and properly cleared to prevent a more pathogenic disease progression.
A number of studies have shown that RSV infection of DCs significantly alters the ability of mDCs to express class II and costimulatory molecules, causes the preferential expression of IL-10, and impairs the induction of a Th1 response [12–16]. Correspondingly, mucosal mDCs have been shown to preferentially skew immune responses in the lung toward a more pathogenic Th2 phenotype and lead to severe asthma-like responses, including increase airway hyper-responsiveness and mucus overexpression [17–19]. The migration of DC subsets to the draining LNs indicates that lung-derived and LN resident DCs are involved in the presentation of antigen to CD4 and CD8 T cells [20]. The present work examines whether RSV alters the immune response to a simultaneous allergen sensitization and secondly, whether RSV-infected DCs played a role in this process. Our results show that RSV-infected DCs contribute to this altered immune environment and also that the DCs need to migrate back to the draining LNs for these perturbations to occur. We hypothesize that viral infections provide an inappropriate maturation stimulus that induces the DC presentation of environmental allergens, thus increasing the sensitization, and accelerates the chronic allergic responses.
MATERIALS AND METHODS
Animals and virus
Female BALB/c/J and C57BL/6 mice, 6–8 weeks of age, were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Animals with specific genetic deletions of CCR6 and CCR7 were graciously supplied by Professor Sergio Lira at Mount Sinai Medical School (New York, NY, USA). All mice were housed under specific pathogen-free conditions within the animal care facility at the University of Michigan (Ann Arbor, MI, USA). The RSV A strain was derived from a clinical isolate at the University of Michigan and was propagated and titered as Line 19 [21, 22]. The mouse-adapted influenza virus (Strain A/PR/8/34) was purchased from American Type Culture Collection (Manassas, VA, USA) and was used at a nonlethal dose following an initial experiment to determine an infectious dose that was tolerable by all mice (i.e., 100% survival).
Allergen sensitization
In our first set of experiments, the DC-instilled animals were simultaneously given a CRA (HollisterSteir, Spokane, WA, USA) sensitization into the trachea. Fourteen days later, a second administration of allergen (1 μg) was given into the nasal passages, followed by two intratrachea allergen challenges (4 μg/mouse) on Days 19 and 21 of the protocol. As our allergen is a skin-test allergen for clinical use, it has very low or no endotoxin and therefore, produces a strong Th2 response without induction of IFN or IL-17.
In a second protocol designed to determine whether RSV-infected DCs have a long-term alteration of the pulmonary immune environment, an intranasal administration of CRA was given 7 days (or 21 days) after the RSV-infected DC instillation. Subsequently, the animals were given intratrachea allergen challenges on Days 13 (or 27 days) and 15 (or 29 days) post-DC administration. Twenty-four hours after the final allergen challenge, AHR was assessed, and the lungs and LNs of mice were harvested for analysis.
Isolation and RSV infection of mouse DCs
BM cells were flushed from the femurs of mice. After RBC lysis, BM cells were cultured at 1 × 106 cells/ml in RMPI medium (RPMI 1640, 10% FCS, 1 μM Na pyruvate, 2 mM l-glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin) containing 10 ng/ml GM-CSF (R&D Systems, Minneapolis, MN, USA), and fresh medium was added after 3, 6, and 8 days. On Day 10, DCs were harvested. These cells represent a mDC population with an intermediate maturation status, as most cells were CD11c+ (90%) and also expressed CD11b (95%). DCs were washed and plated at 2 × 106/ml and then infected with RSV (∼0.8×106 PFU/ml) or medium alone overnight at 37°C.
CFSE staining of DCs
After RSV infection, 4 × 106 DCs/ml were stained with the green fluorescent dye CFSE (Molecular Probes, Eugene, OR, USA) at a final concentration of 2.5 μM for 5 min at room temperature. After extensive washing, RSV-infected or control CFSE+ DCs (0.5×106 cells/50 μl) were injected intratracheally on Day 0 after ketamin/xylazin anesthesia. Hereafter, 1, 2, or 3 days later, lung draining mediastinal LNs were isolated and dispersed using 0.2% collagenase (Type IV, Sigma-Aldrich, St. Louis, MO, USA) in RPMI 1640 with 10% FCS at 37°C for 45 min. Single-cell dispersions were used for flow cytometric analysis.
Flow cytometric analysis of LNs
After blocking nonspecific binding by FcR, cells were stained with anti-mouse CD11C, CD11b, MHC II, CD80, CD86 or CD40, 7-amino actinomycin D, and Annexin V (all from PharMingen, San Diego, CA, USA; and eBioscience, San Diego, CA, USA). Cells were analyzed on a Cytomics FC500 flow cytometer (Beckman-Coulter, Brea, CA, USA).
RSV infection and allergen sensitization (see Fig. 1)
Figure 1. RSV enhances and exacerbates CRA-induced disease pathology.
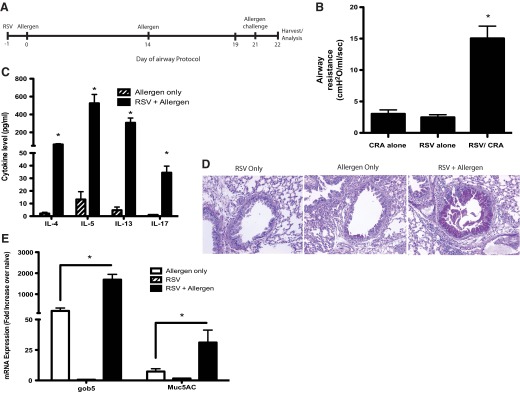
As indicated in the time line (A), when RSV is given into the airway of naïve mice 1 day prior to allergen, followed by a 21-day sensitization protocol, there is an increased clinical disease, as represented by methacholine-induced AHR after a final allergen challenge (B). Allergen-restimulated lung draining LNs demonstrated enhanced Th2 cytokines and IL-17 in animals given RSV along with allergen (C). The overproduction of mucus, as depicted histologically (D) and by mucus-associated gene expression profiles (E), was also increased significantly by the presence of a RSV infection at the time of initial allergen sensitization. Data represent mean ± se from five mice/group. *P < 0.05.
Mice were infected with RSV (∼1×105 PFU/mice) on Day −1 and the next day, were given a CRA (HollisterStier; 3 μg/mouse) sensitization into the trachea. Fourteen days later, a second administration of allergen (1.5 μg/mouse) was given intranasally, followed by two intratrachea allergen challenges (4 μg/mouse) on Days 19 and 21. Twenty-four hours after the final allergen challenge, AHR was assessed, and the lungs and LNs of mice were harvested for analysis.
DC transfer and RSV or CRA exposure
After extensive washing, RSV-infected or control DCs (2×105 cells/50 μl) were injected intratracheally on Day 0 after ketamin/xylazin anesthesia. Whereas we cannot rule out completely that the virus was not transferred, when we examined the lungs at 2–4 days post-DC transfer (a time that is normally peak virus propagation), we do not find any evidence by PCR of viral mRNA expression. Thus, the magnitude of any low-level infection must be very limited.
RSV infection
On Day 7, mice were anesthetized lightly and given RSV (1×105 PFU/mouse) by intratracheal infection, and after 8 additional days, the animals were analyzed for AHR, cytokine responses, and pathologic changes.
Measurement of airway responses
Airway reactivity in anesthetized mice was measured as described previously [23]. Briefly, mice were anesthetized with sodium pentobarbital, and the trachea was cannulated and ventilated using a pump ventilator in a mouse plethysmograph. After baseline measurements, mice were injected i.v. with 2.5 μg methacholine (Sigma-Aldrich), and the peak airway resistance was recorded.
LN restimulation assays
Single-cell suspensions of lung draining LNs were made by isolating draining lung LNs, pushing cells through a nylon mesh using a syringe, and then lysing RBCs. Cells were cultured at a concentration of 6 × 106 cells/ml in the presence of CRA (2 μg/ml) or medium and incubated at 37°C. After 2 days, supernatants were isolated and stored at −80°C until further analysis. A standardized sandwich ELISA (R&D Systems) or Bio-Plex (Bio-Rad Laboratories, Hercules, CA, USA) was performed to measure cytokine and chemokine levels in culture supernatants.
Histology
Lungs were maintained in formalin for a further 24 h before being processed into paraffin using standard histological techniques. Lung tissue sections were stained with H&E for analysis of inflammatory cell accumulation and alcian blue/PAS stain for assessment of mucus production.
Statistical analysis
Significance was determined using one-way ANOVA with 95% confidence intervals, followed by a Student Neuman Keuhl's post-test. With a P value of <0.05, differences were considered significant.
RESULTS
RSV infection enhances allergen sensitization and severe pathology associated with mDC accumulation and responses
A strong correlation in patient populations has been found between early pulmonary viral infection and subsequent development of allergic asthma [24–27]. Although the mechanism is unknown, it may be that the coincidental exposure of the viral infection along with an environmental allergen lead to the most severe disease. To address this latter concept, we examined whether instilling allergen along with viral infection would alter the development/sensitization of the allergen response. In our studies, the infection of mice with RSV (1×105 PFU/mouse) along with allergen sensitization into the airway clearly elicited a more severe allergic disease after an additional challenge with the allergen 21 days later (Fig. 1A). These data include enhanced AHR responses to methacholine (Fig. 1B), increased potentially pathogenic Th cytokines, IL-4, IL-5, IL-13, and IL-17 (Fig. 1C), more intense goblet cell development in the upper airways (Fig. 1D), and increased expression of mucus-associated genes gob5 and muc5ac (Fig. 1E). In contrast, when animals were infected with H1N1 influenza virus and simultaneously sensitized to allergen using a similar protocol as RSV, we observed a decrease in allergen-specific cytokine production and no change in the mucus gene expression or the histologic appearance (Supplemental Fig. 1). These data suggest that the RSV, but not influenza, infection leads to increased sensitization to allergen and caused an intensified local skewing response [28]. Thus, not all viral infections can promote an enhanced allergic airway response.
To determine the contribution of the mDC to the intensified response, BMDCs were infected with RSV in vitro (MOI, 1.0). This procedure addressed the effects of the RSV infection of airways, as DCs do not propagate RSV but did sensitize animals to the responses as described above. To address virus propagation from DCs that were transferred, we examined a time-course analysis (24–72 h postinfection) of RSV-infected DCs in vitro and found no virus propagation from DCs by plaque analysis at any time-point based on continuous decreasing levels of plaqued virus (0.04% of input at 24 h, 0.004% by 48 h, and none by 72 h; Supplemental Fig. 2A). However, when we examined RSV protein mRNA expression in lungs of mice at 2 days post-DC transfers, we found some evidence of RSV infection based on qPCR results, although at a barely detectable level compared with a live virus infection (Supplemental Fig. 2B). The data in Fig. 2 demonstrate that RSV-infected DC leads to an exacerbation of the allergic response with a significantly increased AHR response compared with controls (Fig. 2A). Additionally, IL-13, IL-17, and IFN were increased significantly, and mucus gene expression was also increased compared with controls (Fig. 2B and C). Interestingly, it was only in the presence of RSV-infected DCs that the allergen-rechallenged LN cells produced IL-17 (Fig. 2B). Moreover, simply delivering additional DCs into the airway at the time of allergen sensitization enhanced the Th2 cytokine sensitization; however, the activation of those DCs with RSV prior to instillation induced an even more intense and broader profile response (Fig. 2B). Thus, the DCs enhance the pathologic response, not only by promoting allergen sensitization but also by inducing potentially pathogenic IL-17. Characterization of the in vitro-infected DC demonstrated that RSV up-regulated CD80, CD86, and CD40 (Supplemental Fig. 2C) but did not induce apoptotic or necrotic cell death (Supplemental Fig. 2D).
Figure 2. RSV-infected DC instilled into the airways of mice can replace a live RSV infection for exacerbation of allergen-induced responses.

After an overnight infection, the washed DCs were transferred into the lungs of naïve mice by intratracheal administration, along with CRA. With the use of the same timeline as in Fig. 1, mice were challenged with CRA by intranasal (Day 14) and intratrachea delivery (Days 19 and 21), and 24 h after final allergen exposure, theresponses were assessed. The RSV-infected DCs induced a significant increase in AHR (A) compared with results with no DC + allergen or (continued) uninfected DC + allergen. When lung draining LN cells were restimulated in vitro with allergen at the end of the in vivo responses, changes in secreted cytokines were observed in the RSV-DC-treated mice compared with the other groups, as assessed in 48-h culture supernatants by Bio-Plex analysis (B). There was also a significant increase in mucus-associated genes in whole lung qPCR (C). Data represent mean ± se from five mice/group. *P < 0.05.
To further address whether the recruitment and accumulation of mDC subsets are necessary for the development of the RSV-induced enhanced allergic responses described above, CCR6−/− mice were used. Studies from several laboratories, including our own, have demonstrated that CCR6−/− animals have a defect in mDC accumulation, but not T cells, within the lungs of RSV-infected or allergen-sensitized animals [29–35]. The infection of WT and CCR6−/− mice with RSV, along with allergen sensitization, was repeated as described above in Fig. 1A. As published previously, we also observed decreases in CD11b/CD11c+ DC accumulation but no difference in T cell populations that accumulated in the sensitized and challenged mice (data not shown). Mice rechallenged at Day 21 demonstrated a striking reduction of RSV-induced exacerbation of AHR in the CCR6−/− mice compared with WT control mice (Fig. 3A). Also, the CCR6−/− animals histologically displayed a significant reduction in overall inflammation and reduction in mucus production, and this was supported by quantification of mucus gene expression (Fig. 3B). Finally, when cytokine responses were examined, there was a decrease in the expression of Th2 cytokines in the RSV-exacerbated responses (Fig. 3C). However, whereas IL-17 was up-regulated by the RSV exposure, it was not reduced in the CCR6−/− mice (data not shown).
Figure 3. Attenuation of RSV exacerbated allergen sensitization in CCR6−/− mice.

With the use of the sensitization protocol established in Fig. 1, WT or CCR6−/− [knockout (KO)] was sensitized to CRA, with or without RSV infection. Mice were challenged with CRA intranasally (Day 14) and intratrachelly (Days 19 and 21), and the responses were assessed. Twenty-four hours after final allergen exposure, mice were examined for methacholine-induced AHR (A). Differences in mucus hypersecretion in RSV-enhanced allergic disease were depicted by examining histology and mucus-associated genes muc5ac and gob5 by qPCR of lung mRNA (B). There was also a significant increase in Th2 genes, IL-4 and IL-13, in lung qPCR (C). Data represent mean ± se from five mice/group. *P < 0.05.
RSV infection predisposes animals to more severe pulmonary responses via DC activation and LN localization
Previous data have indicated that the severity of RSV-induced pathology can be associated with the presence of defined DC subsets recruited to the lung [14, 36–39]. Specifically, local mucosal (lung) mDCs were shown to be responsible for skewing the immune responses toward a modified Th cell response that becomes more pathogenic [15, 17–19, 40]. Thus, we examined whether RSV-infected DCs also skewed a RSV infection challenge, as observed above with allergen. To examine this aspect specifically, BMDCs (CD11c/CD11b+) were cultured overnight, with or without RSV (MOI of 1), in vitro to allow infection, subsequently washed free of excess virus, and instilled into the trachea of naïve mice (2×105 DC/mouse). After 7 days, the mice were infected with RSV, and the immune response was assayed to determine if the infected BMDCs altered the immune environment. The data in Fig. 4A demonstrate that those animals that received RSV-infected BMDCs had an enhanced IL-4, IL-5, and IL-13 Th2-induced immune response, whereas IL-17 and IFN-γ production were down-regulated. This was accompanied by increases in mucus overexpression and airway pathology (Fig. 4B and C). Our studies also observed a decrease in RSV-specific mRNA, as might be expected with a local sensitization (data not shown) [41]. Thus, this response suggests that exogenously added DCs alter immune responses to the virus and resulted in increased pathology via Th2 cytokine overproduction in agreement with other studies [42–45].
Figure 4. Airway sensitization of mice with RSV-infected DCs elicited a Th2-skewed immune response upon a RSV infection challenge.
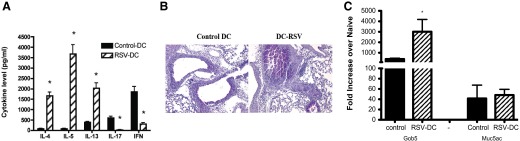
RSV-infected BMDC (2×105/mouse) were instilled into the airways of naïve mice and infected with live RSV (1×105 PFU/mouse), 7 days after BMDC instillation. Supernatants from RSV-restimulated lung draining LN cells were analyzed for cytokines by Bio-Plex (A). Lungs were taken 8 days after the additional RSV infection (15 days later, the DC-RSV instillation) and were stained with PAS (B) and assessed by qPCR for mucus genes (C). Data represent mean ± se from five mice/group. *P < 0.05.
We next determined whether RSV infection enhances migration of DCs to the LN. The data in Fig. 5A depict flow cytometry of the draining LNs from the instilled mice at different time-points, indicating that RSV-infected DCs traffic better to the LNs than do uninfected DCs. Thus, RSV does activate DCs and enhances their migration into the draining LNs of mice, potentially facilitating sensitization of the immune response.
Figure 5. BMDC from CCR6−/− mice do not migrate to draining LNs and fail to sensitize mice for an exacerbated RSV infection response.

BMDCs were cultured overnight, with or without RSV, and stained with CFSE. After an injection of DCs into mice by intratrachea injection, lung draining LNs were isolated at different time-points, and single-cell dispersions were used for flow cytometry analysis (A). Left panel is flow cytometry plot at 24 h time-point. R1 represents CD11c+ CFSE-positive cells. Similar results were observed at 48-h and 72-h later time-points (right panel). WT and CCR7−/− BMDCs were infected with RSV and were stained with CFSE. Lung draining LNs were isolated after 24 h and 48 h and performed flow cytometric analysis (B). Supernatants from RSV-restimulated LN cells were analyzed for cytokines by Bio-Plex (C). Data represent mean ± se from five mice/group. *P < 0.05.
Our next series of studies was set up to determine whether the trafficking of the RSV-infected DCs to the LN was required for the altered responses, as previous studies have demonstrated that lung as well as LN resident DCs are involved in RSV-specific CD4 and CD8 T cell activation [20]. To address this question, studies used a similar system as described above with BMDCs from WT or CCR7−/− mice. To verify that the CCR7−/− DCs were impaired in their ability to traffic to draining LNs, as reported [46], the nodes were harvested at 24 h and 48 h postinstillation of CFSE-labeled, RSV-infected DCs. The data in Fig. 5B confirmed that DCs from CCR7−/− animals do not traffic efficiently to the LNs after RSV infection. Seven days after the instillation of RSV-infected DC into the lungs of naïve mice, the animals were infected with RSV. Following 8 days of infection, the draining LNs were harvested, and the isolated cells rechallenged in vitro with a RSV infection. The resulting response indicated that when CCR7−/− DCs were used for airway sensitization, there was a significant reduction of Th2 cytokines with no change in IL-17 expression (Fig. 5C). Thus, the exacerbated immune response appears to be, at least partially, dependent on LN accumulation of the instilled, RSV-infected DCs and also enhances the pathogenic response to subsequent challenges. These responses should help identify the locational requirements for altering the immune responses during respiratory viral infections that may enhance allergen responses.
RSV-infected DC enhancement of allergic responses does not need to be simultaneous
We next examined whether the RSV-infected DCs have a long-term effect on allergen sensitization. Isolated DCs were infected with RSV and instilled into the airway as above. After 7 days of sensitization, the animals were given an allergen sensitization into the airway via intranasal delivery. Following an additional 6 days, the animals were again challenged twice with intratracheal allergen, 48 h apart. This suboptimal allergen-sensitization process led to increases in AHR responses (Fig. 6A) and increases in Th2 cytokines IL-4, IL-5, and IL-13 (Fig. 6B), with no allergen-specific increase in IL-17 detected (data not shown). Histologic studies of PAS-stained lungs showed that although the mice receiving DCs alone displayed significant inflammatory cell infiltration, a significantly more mucogenic response was observed in animals given RSV-infected DCs prior to the allergen (Fig. 6C). Furthermore, when we waited for 21 days after RSV-infected DC instillation, prior to the allergen sensitization, the Th2 cytokine response continued to be enhanced by the RSV-DC transfer, with increased pathology characterized by mucus overproduction (Supplemental Fig. 3). Thus, these data indicate that RSV-infected DCs accelerated the allergen-sensitization process and caused Th2 cytokine skewing, leading to more severe disease, even when given up to 21 days prior to allergen exposure.
Figure 6. RSV-infected BMDC-induced allergen exacerbation is long-lived and does not need to be simultaneous.

The BMDCs were infected with RSV (MOI=1.0) and instilled into naïve Balb/c mice. After 1 week, the mice were exposed to CRA (1 μg/mouse) by intranasal instillation with repeated instillations, 6 and 8 days later (Days 13 and 15 of the protocol). Twenty-four hours after final allergen exposure (Day 16), the animals were assessed for AHR (A) by methacholine challenge. Cont, Control. Draining LNs were prepared to single-cell suspension and restimulated with allergen (2 μg/ml) and 48-h culture supernatant assessed for cytokines by Bio-Plex analysis (B). Formalin-preserved lung tissue (left lobe) was assessed for mucus and goblet cell hypertrophy by PAS staining of histology sections (C). Data represent mean ± se from five mice/group. *P < 0.05.
DISCUSSION
In these studies, we have begun to address several important mechanistic questions, including: are there detrimental effects of an antiviral immune response within the pulmonary environment that lead to a more pathogenic response to noninfectious (allergen) stimuli? Can innate immune cells (DCs) dictate the nature of a subsequent immune response? Although the present studies have only begun to address these issues, the data do correlate with clinical observations, suggesting that early viral infections can lead to long-term effects that alter subsequent immune responses [26, 27, 47, 48]. The role of RSV for development of childhood pulmonary disease has been epidemiologically controversial [9, 25, 47, 49–54], and several reports have begun to address the role of RSV in model allergen systems [55–61]. In fact, a recent publication has indicated that RSV can break an induced, antigen-specific tolerance response by altering regulatory T cell function [62]. Although RSV itself appears to promote a dysregulated innate immune response via specific NS proteins (NS1 and NS2) [63–66], it may be that the virus infection is a stimulus for enhancing sensitization and leads to the altered immune responses. This was manifested in our studies using the DC sensitization model, where the active infection was replaced with RSV-infected DCs that propagate undetectable levels of active virions. In contrast, influenza infection did not induce a similar state of exacerbated disease and in fact, inhibited the skewing of allergen-specific responses. This latter change may be associated with differences in key mediators that are induced by the two viruses, such as type I IFN, which is highly induce by influenza [67] but inhibited by RSV NS1 and NS2 proteins [63, 65, 68]. With the use of DCs from CCR7−/− mice, studies established that migration into the LN was one necessary step for the altered immune response to a subsequent RSV infection. In patient populations, especially immune “inexperienced” infants, early viral infection may aid in the improper sensitization of individuals to environmental allergens. This effect, along with a genetic predisposition to an altered pulmonary environment, could lead to an enhanced pathologic response within the lung. The results, showing that the infected DC-induced immune deviation lasted 1 week and as long as 21 days, suggest that these cells are primarily involved in the process, either directly via APC function or indirectly by mediator release, such as IL-10 [69, 70]. Although the use of BMDCs is not entirely physiologic and the relevance to lung DC unclear, the studies help to identify a potential cellular mechanism of how RSV can alter an immune environment. Future studies will be needed to resolve these issues and to test exactly how long this effect persists.
One of the primary implications of our data is the relative lack of specificity of the altered pulmonary immune response that extends its influence on a noninfectious allergen response. This raises the possibility that specific viral infections preferentially alter the immune environment as a result of a modified immune response. As mentioned above, this may be a result of specific viral proteins, such as the NS proteins of RSV that can inhibit key innate cytokines, and/or the entry of RSV via a nonendosomic route [71]. This latter concept is intriguing, as a primary mechanism for type I IFN production during RSV infection in epithelial cells is via the cytoplasmic sensor RIG-I that recognizes dsRNA [72, 73]. Recent studies have shown that the NS2 protein of RSV antagonizes IFN-β production induced by RIG-I [74]. Other studies have clearly demonstrated a skewed pathogenic environment in the absence of STAT1, an essential downstream signaling molecule for type I IFN expression [75–78]. Thus, DC activation and mediator release are affected directly by RSV infection. Subsequently, TLRs found in the endosome are also activated and contribute to the innate immune response during RSV infection [79–81]. Other studies have suggested that RSV G protein can inhibit TLR-induced activation [82]. Interestingly, when the RIG-I pathway and MyD88-dependent TLR pathways were blocked in mitochondrial antiviral-signaling protein/MyD88 double-knockout mice, no alteration in cytotoxic T cell development and viral clearance was observed [83]. Likewise, the use of any one of several knockout mice deficient in TLR or MyD88 signaling did not alter viral clearance; however, significantly altered immune responses and increased pathogenesis during RSV infection were observed [79, 80, 84–86]. Thus, there may be multiple mechanisms for immune response alteration during RSV infection that can influence the direction and/or quality of the immune responses and therefore, the pathologic outcome.
The cytokine profile induced with a primary RSV-infected DC sensitization into the airway was a skewed Th2 response with decreases in IFN and IL-17 upon a RSV infection. A different profile was recognized when mice were infected with RSV coincident with allergen sensitization with an allergen-specific increase in Th2 and Th17 cytokines. Although one could explain these responses by the ability of RSV to cause enhanced DC migration to the LNs (as demonstrated with the CCR7−/− DC studies), the ability to achieve enhanced allergen-induced Th2 responses, 1–3 weeks after RSV-DC instillation into the airway, is more difficult to explain. Previous studies have clearly suggested that pulmonary mDC populations have a preferential ability to skew the local immune responses toward a Th2-type immune environment [17–19, 87]. However, as described in a recent study by Guerrero-Plata et al. [88], pDCs also appear to be altered upon RSV infection. This latter effect may be important, as pDCs have a pivotal role in establishing an appropriate anti-RSV response and are necessary for maintaining a toleragenic immune environment against locally administered antigens/allergens [36, 38, 39]. Recent evidence has suggested that the NS proteins from RSV also suppress the maturation of DCs, including costimulatory proteins CD80, CD83, and CD86, and are associated with previously described reductions in type I IFN production [16]. Thus, the altered immune environment that was characterized by elevated Th cytokine production may have been caused by altered DC maturation. Although our studies have focused on using a BMDC that best resembles a monocyte-derived DC subset (CD11b+/CD11c+), there are other important subsets in the lung, especially the CD103+ subset that appears to be relevant in RSV infection [20]. The CD103+ subset is migratory and has been suggested to better promote an antiviral Th1-type cytotoxic T cell response as a result of its ability to cross-present antigens [20, 89–93]. In contrast, a more recent study indicated that pulmonary CD103+ DCs were better able than CD11b+ DCs at promoting Th2 cytokine responses when an inert antigen (OVA) was used [94]. Thus, offsetting the balance of the subset ratios by adding a CD11b+/CD11c+ DCs may represent a highly inflamed environment that promotes the recruitment of more “inflammatory”-type DCs that migrate into the lung and initiate pathologic responses. In our studies, when we examined the CFSE trafficking of infected DCs into the LN, we had no evidence that the CD103+ cells were involved in the process, but we cannot rule out their contribution to the process. However, the use of CCR6−/− mice, which have altered inflammatory CD11b/CD11c DC accumulation after RSV and allergen exposures [29, 34, 35], further supports the concept that altering DC subset ratios can affect the outcome of the pulmonary response. Interestingly, our studies observed no alteration in IL-17 production in CCR6−/− or CCR7−/− studies in the presence of RSV, but we have not pursued this further at this time.
The data presented in these studies suggest that alteration of the pulmonary immune environment by RSV infection may have an underlying effect on the progression of disease pathology. Although several inter-related mechanisms may be induced, these studies suggest that RSV infections may predispose the host to an altered and modified disease phenotype through activation of DC subsets that may allow immune responses to allergens to be altered. Future studies will explore the nature of the RSV-mediated DC alteration.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants HL036302 and AI073876.
SEE CORRESPONDING EDITORIAL ON PAGE 1
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- AHR
- airway hyper-reactivity
- BMDC
- bone marrow-derived DC
- CRA
- cockroach allergen
- mDC
- myeloid DC
- NS
- influenza nonstructural
- PAS
- periodic acid Schiff
- pDC
- plasmacytoid DC
- qPCR
- quantitative PCR
- RIG-I
- retinoic acid-inducible gene 1
- RSV
- respiratory syncytial virus
AUTHORSHIP
S.J. contributed to design, writing, and execution. J.S. contributed to the design and execution. L.E.K. contributed to the execution of some of the experiments. N.W.L. contributed to the writing, execution, and design of the studies.
REFERENCES
- 1. Busse W. W., Gern J. E., Dick E. C. (1997) The role of respiratory viruses in asthma. Ciba Found. Symp. 206, 208–213 [DOI] [PubMed] [Google Scholar]
- 2. Stempel D. A., Boucher R. C. (1981) Respiratory infection and airway reactivity. Med. Clin. North Am. 65, 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hogg J. C. (1999) Childhood viral infection and the pathogenesis of asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 160, S26–S28 [DOI] [PubMed] [Google Scholar]
- 4. Holtzman M. J., Morton J. D., Shornick L. P., Tyner J. W., O'Sullivan M. P., Antao A., Lo M., Castro M., Walter M. J. (2002) Immunity, inflammation, and remodeling in the airway epithelial barrier: epithelial-viral-allergic paradigm. Physiol. Rev. 82, 19–46 [DOI] [PubMed] [Google Scholar]
- 5. Nicholson K. G., Kent J., Ireland D. C. (1993) Respiratory viruses and exacerbations of asthma in adults. BMJ 307, 982–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao J., Takamura M., Yamaoka A., Odajima Y., Iikura Y. (2002) Altered eosinophil levels as a result of viral infection in asthma exacerbation in childhood. Pediatr. Allergy Immunol. 13, 47–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simpson J. L., Moric I., Wark P. A., Johnston S. L., Gibson P. G. (2003) Use of induced sputum for the diagnosis of influenza and infections in asthma: a comparison of diagnostic techniques. J. Clin. Virol. 26, 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoffman S. J., Laham F. R., Polack F. P. (2004) Mechanisms of illness during respiratory syncytial virus infection: the lungs, the virus and the immune response. Microbes Infect. 6, 767–772 [DOI] [PubMed] [Google Scholar]
- 9. Everard M. L. (2006) The role of the respiratory syncytial virus in airway syndromes in childhood. Curr. Allergy Asthma Rep. 6, 97–102 [DOI] [PubMed] [Google Scholar]
- 10. Falsey A. R. (2007) Respiratory syncytial virus infection in adults. Semin. Respir. Crit. Care Med. 28, 171–181 [DOI] [PubMed] [Google Scholar]
- 11. Thompson W. W., Shay D. K., Weintraub E., Brammer L., Cox N., Anderson L. J., Fukuda K. (2003) Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 [DOI] [PubMed] [Google Scholar]
- 12. Tripp R. A., Moore D., Anderson L. J. (2000) TH(1)- and TH(2)-type cytokine expression by activated T lymphocytes from the lung and spleen during the inflammatory response to respiratory syncytial virus. Cytokine 12, 801–807 [DOI] [PubMed] [Google Scholar]
- 13. Bartz H., Buning-Pfaue F., Turkel O., Schauer U. (2002) Respiratory syncytial virus induces prostaglandin E2, IL-10 and. IL-11 generation in antigen presenting cells. Clin. Exp. Immunol. 129, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartz H., Turkel O., Hoffjan S., Rothoeft T., Gonschorek A., Schauer U. (2003) Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-γ in naive T cells. Immunology 109, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kondo Y., Matsuse H., Machida I., Kawano T., Saeki S., Tomari S., Obase Y., Fukushima C., Kohno S. (2004) Regulation of mite allergen-pulsed murine dendritic cells by respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 169, 494–498 [DOI] [PubMed] [Google Scholar]
- 16. De Graaff P. M., de Jong E. C., van Capel T. M., van Dijk M. E., Roholl P. J., Boes J., Luytjes W., Kimpen J. L., van Bleek G. M. (2005) Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate CD4 T cells. J. Immunol. 175, 5904–5911 [DOI] [PubMed] [Google Scholar]
- 17. Julia V., Hessel E. M., Malherbe L., Glaichenhaus N., O'Garra A., Coffman R. L. (2002) A restricted subset of dendritic cells captures airborne antigens and remains able to activate specific T cells long after antigen exposure. Immunity 16, 271–283 [DOI] [PubMed] [Google Scholar]
- 18. Lambrecht B. N., Salomon B., Klatzmann D., Pauwels R. A. (1998) Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J. Immunol. 160, 4090–4097 [PubMed] [Google Scholar]
- 19. Lambrecht B. N., De Veerman M., Coyle A. J., Gutierrez-Ramos J. C., Thielemans K., Pauwels R. A. (2000) Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J. Clin. Invest. 106, 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lukens M. V., Kruijsen D., Coenjaerts F. E., Kimpen J. L., van Bleek G. M. (2009) Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J. Virol. 83, 7235–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller A. L., Bowlin T. L., Lukacs N. W. (2004) Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J. Infect. Dis. 189, 1419–1430 [DOI] [PubMed] [Google Scholar]
- 22. Miller A. L., Strieter R. M., Gruber A. D., Ho S. B., Lukacs N. W. (2003) CXCR2 regulates respiratory syncytial virus-induced airway hyperreactivity and mucus overproduction. J. Immunol. 170, 3348–3356 [DOI] [PubMed] [Google Scholar]
- 23. Tekkanat K. K., Maassab H. F., Cho D. S., Lai J. J., John A., Berlin A., Kaplan M. H., Lukacs N. W. (2001) IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 166, 3542–3548 [DOI] [PubMed] [Google Scholar]
- 24. Kim E. Y., Battaile J. T., Patel A. C., You Y., Agapov E., Grayson M. H., Benoit L. A., Byers D. E., Alevy Y., Tucker J., Swanson S., Tidwell R., Tyner J. W., Morton J. D., Castro M., Polineni D., Patterson G. A., Schwendener R. A., Allard J. D., Peltz G., Holtzman M. J. (2008) Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 14, 633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lemanske R. F., Jr., (2002) The childhood origins of asthma (COAST) study. Pediatr. Allergy Immunol. 13 (Suppl. 15), 38–43 [DOI] [PubMed] [Google Scholar]
- 26. Martinez F. D. (2009) The origins of asthma and chronic obstructive pulmonary disease in early life. Proc. Am. Thorac. Soc. 6, 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walton R. P., Johnston S. L. (2008) Role of respiratory viral infections in the development of atopic conditions. Curr. Opin. Allergy Clin. Immunol. 8, 150–153 [DOI] [PubMed] [Google Scholar]
- 28. Al-Garawi A., Fattouh R., Botelho F., Walker T. D., Goncharova S., Moore C. L., Mori M., Erjefalt J. S., Chu D. K., Humbles A. A., Kolbeck R., Stampfli M. R., O'Byrne P. M., Coyle A. J., Jordana M. (2011) Influenza A facilitates sensitization to house dust mite in infant mice leading to an asthma phenotype in adulthood. Mucosal Immunol. 4, 682–694 [DOI] [PubMed] [Google Scholar]
- 29. Kallal L. E., Schaller M. A., Lindell D. M., Lira S. A., Lukacs N. W. (2010) CCL20/CCR6 blockade enhances immunity to RSV by impairing recruitment of DC. Eur. J. Immunol. 40, 1042–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wen H., Hogaboam C. M., Lukacs N. W., Cook D. N., Lira S. A., Kunkel S. L. (2007) The chemokine receptor CCR6 is an important component of the innate immune response. Eur. J. Immunol. 37, 2487–2498 [DOI] [PubMed] [Google Scholar]
- 31. Schutyser E., Struyf S., Van Damme J. (2003) The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 14, 409–426 [DOI] [PubMed] [Google Scholar]
- 32. Vanbervliet B., Homey B., Durand I., Massacrier C., Ait-Yahia S., de Bouteiller O., Vicari A., Caux C. (2002) Sequential involvement of CCR2 and CCR6 ligands for immature dendritic cell recruitment: possible role at inflamed epithelial surfaces. Eur. J. Immunol. 32, 231–242 [DOI] [PubMed] [Google Scholar]
- 33. Lundy S. K., Lira S. A., Smit J. J., Cook D. N., Berlin A. A., Lukacs N. W. (2005) Attenuation of allergen-induced responses in CCR6−/− mice is dependent upon altered pulmonary T lymphocyte activation. J. Immunol. 174, 2054–2060 [DOI] [PubMed] [Google Scholar]
- 34. Lukacs N. W., Prosser D. M., Wiekowski M., Lira S. A., Cook D. N. (2001) Requirement for the chemokine receptor CCR6 in allergic pulmonary inflammation. J. Exp. Med. 194, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook D. N., Prosser D. M., Forster R., Zhang J., Kuklin N. A., Abbondanzo S. J., Niu X. D., Chen S. C., Manfra D. J., Wiekowski M. T., Sullivan L. M., Smith S. R., Greenberg H. B., Narula S. K., Lipp M., Lira S. A. (2000) CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity 12, 495–503 [DOI] [PubMed] [Google Scholar]
- 36. Smit J. J., Lindell D. M., Boon L., Kool M., Lambrecht B. N., Lukacs N. W. (2008) The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS One 3, e1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez P. A., Prado C. E., Leiva E. D., Carreno L. J., Bueno S. M., Riedel C. A., Kalergis A. M. (2008) Respiratory syncytial virus impairs T cell activation by preventing synapse assembly with dendritic cells. Proc. Natl. Acad. Sci. USA 105, 14999–15004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Peters N., Schwarze J. (2006) Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J. Immunol. 177, 6263–6270 [DOI] [PubMed] [Google Scholar]
- 39. Smit J. J., Rudd B. D., Lukacs N. W. (2006) Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 203, 1153–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stampfli M. R., Wiley R. E., Neigh G. S., Gajewska B. U., Lei X. F., Snider D. P., Xing Z., Jordana M. (1998) GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J. Clin. Invest. 102, 1704–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McDermott D. S., Weiss K. A., Knudson C. J., Varga S. M. (2011) Central role of dendritic cells in shaping the adaptive immune response during respiratory syncytial virus infection. Future Virol. 6, 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Openshaw P. J. (1995) Immunity and immunopathology to respiratory syncytial virus. The mouse model. Am. J. Respir. Crit. Care Med. 152, S59–S62 [DOI] [PubMed] [Google Scholar]
- 43. Kim H. W., Leikin S. L., Arrobio J., Brandt C. D., Chanock R. M., Parrott R. H. (1976) Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr. Res. 10, 75–78 [DOI] [PubMed] [Google Scholar]
- 44. Graham B. S., Bunton L. A., Wright P. F., Karzon D. T. (1991) Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Invest. 88, 1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Waris M. E., Tsou C., Erdman D. D., Zaki S. R., Anderson L. J. (1996) Respiratory synctial virus infection in BALB/c mice previously immunized with formalin-inactivated virus induces enhanced pulmonary inflammatory response with a predominant Th2-like cytokine pattern. J. Virol. 70, 2852–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sanchez-Sanchez N., Riol-Blanco L., Rodriguez-Fernandez J. L. (2006) The multiple personalities of the chemokine receptor CCR7 in dendritic cells. J. Immunol. 176, 5153–5159 [DOI] [PubMed] [Google Scholar]
- 47. Peebles R. S., Jr., (2004) Viral infections, atopy, and asthma: is there a causal relationship? J. Allergy Clin. Immunol. 113, S15–S18 [DOI] [PubMed] [Google Scholar]
- 48. Friedlander S. L., Jackson D. J., Gangnon R. E., Evans M. D., Li Z., Roberg K. A., Anderson E. L., Carlson-Dakes K. T., Adler K. J., Gilbertson-White S., Pappas T. E., Dasilva D. F., Tisler C. J., Pleiss L. E., Mikus L. D., Rosenthal L. A., Shult P. A., Kirk C. J., Reisdorf E., Hoffjan S., Gern J. E., Lemanske R. F., Jr., (2005) Viral infections, cytokine dysregulation and the origins of childhood asthma and allergic diseases. Pediatr. Infect. Dis. J. 24, S170–S176 [DOI] [PubMed] [Google Scholar]
- 49. Sigurs N. (2002) Clinical perspectives on the association between respiratory syncytial virus and reactive airway disease. Respir. Res. 3 (Suppl. 1), S8–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sigurs N. (2002) A cohort of children hospitalised with acute RSV bronchiolitis: impact on later respiratory disease. Paediatr. Respir. Rev. 3, 177–183 [DOI] [PubMed] [Google Scholar]
- 51. Piedimonte G. (2002) Origins of reactive airways disease in early life: do viral infections play a role? Acta Paediatr. Suppl. 91, 6–11 [DOI] [PubMed] [Google Scholar]
- 52. Message S. D., Johnston S. L. (2002) Viruses in asthma. Br. Med. Bull. 61, 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gentile D. A., Skoner D. P. (2002) Effect of respiratory syncytial virus infection during early infancy on the ontogeny of cytokine immune responses. Allergy Asthma Proc. 23, 399–405 [PubMed] [Google Scholar]
- 54. Graham B. S., Johnson T. R., Peebles R. S. (2000) Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology 48, 237–247 [DOI] [PubMed] [Google Scholar]
- 55. Siegle J. S., Hansbro N., Herbert C., Rosenberg H. F., Domachowske J. B., Asquith K. L., Foster P. S., Kumar R. K. (2010) Early-life viral infection and allergen exposure interact to induce an asthmatic phenotype in mice. Respir. Res. 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. You D., Becnel D., Wang K., Ripple M., Daly M., Cormier S. A. (2006) Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir. Res. 7, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barends M., Van Oosten M., De Rond C. G., Dormans J. A., Osterhaus A. D., Neijens H. J., Kimman T. G. (2004) Timing of infection and prior immunization with respiratory syncytial virus (RSV) in RSV-enhanced allergic inflammation. J. Infect. Dis. 189, 1866–1872 [DOI] [PubMed] [Google Scholar]
- 58. Peebles R. S., Jr., Hashimoto K., Collins R. D., Jarzecka K., Furlong J., Mitchell D. B., Sheller J. R., Graham B. S. (2001) Immune interaction between respiratory syncytial virus infection and allergen sensitization critically depends on timing of challenges. J. Infect. Dis. 184, 1374–1379 [DOI] [PubMed] [Google Scholar]
- 59. Peebles R. S., Jr., Sheller J. R., Johnson J. E., Mitchell D. B., Graham B. S. (1999) Respiratory syncytial virus infection prolongs methacholine-induced airway hyperresponsiveness in ovalbumin-sensitized mice. J. Med. Virol. 57, 186–192 [DOI] [PubMed] [Google Scholar]
- 60. Dakhama A., Bramley A. M., Chan N. G., McKay K. O., Schellenberg R. R., Hegele R. G. (1999) Effect of respiratory syncytial virus on subsequent allergic sensitization to ovalbumin in guinea-pigs. Eur. Respir. J. 13, 976–982 [DOI] [PubMed] [Google Scholar]
- 61. Schwarze J., Hamelmann E., Bradley K. L., Takeda K., Gelfand E. W. (1997) Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J. Clin. Invest. 100, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krishnamoorthy N., Khare A., Oriss T. B., Raundhal M., Morse C., Yarlagadda M., Wenzel S. E., Moore M. L., Peebles R. S., Jr., Ray A., Ray P. (2012) Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat. Med. 18, 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Munir S., Le Nouen C., Luongo C., Buchholz U. J., Collins P. L., Bukreyev A. (2008) Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J. Virol. 82, 8780–8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bitko V., Shulyayeva O., Mazumder B., Musiyenko A., Ramaswamy M., Look D. C., Barik S. (2007) Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-κB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 81, 1786–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spann K. M., Tran K. C., Collins P. L. (2005) Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J. Virol. 79, 5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Spann K. M., Tran K. C., Chi B., Rabin R. L., Collins P. L. (2004) Suppression of the induction of α, β, and λ interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J. Virol. 78, 4363–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shahangian A., Chow E. K., Tian X., Kang J. R., Ghaffari A., Liu S. Y., Belperio J. A., Cheng G., Deng J. C. (2009) Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Munir S., Hillyer P., Le Nouen C., Buchholz U. J., Rabin R. L., Collins P. L., Bukreyev A. (2011) Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. 7, e1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bont L., Heijnen C. J., Kavelaars A., van Aalderen W. M., Brus F., Draaisma J. T., Geelen S. M., Kimpen J. L. (2000) Monocyte IL-10 production during respiratory syncytial virus bronchiolitis is associated with recurrent wheezing in a one-year follow-up study. Am. J. Respir. Crit. Care Med. 161, 1518–1523 [DOI] [PubMed] [Google Scholar]
- 70. Panuska J. R., Merolla R., Rebert N. A., Hoffmann S. P., Tsivitse P., Cirino N. M., Silverman R. H., Rankin J. A. (1995) Respiratory syncytial virus induces interleukin-10 by human alveolar macrophages. Suppression of early cytokine production and implications for incomplete immunity. J. Clin. Invest. 96, 2445–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Deretic V. (2009) Strange bedfellows expose ancient secrets of autophagy in immunity. Immunity 30, 479–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bitko V., Musiyenko A., Bayfield M. A., Maraia R. J., Barik S. (2008) Cellular La protein shields nonsegmented negative-strand RNA viral leader RNA from RIG-I and enhances virus growth by diverse mechanisms. J. Virol. 82, 7977–7987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu P., Jamaluddin M., Li K., Garofalo R. P., Casola A., Brasier A. R. (2007) Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 81, 1401–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ling Z., Tran K. C., Teng M. N. (2009) Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of β interferon transcription by interacting with RIG-I. J. Virol. 83, 3734–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Durbin J. E., Johnson T. R., Durbin R. K., Mertz S. E., Morotti R. A., Peebles R. S., Graham B. S. (2002) The role of IFN in respiratory syncytial virus pathogenesis. J. Immunol. 168, 2944–2952 [DOI] [PubMed] [Google Scholar]
- 76. Hashimoto K., Durbin J. E., Zhou W., Collins R. D., Ho S. B., Kolls J. K., Dubin P. J., Sheller J. R., Goleniewska K., O'Neal J. F., Olson S. J., Mitchell D., Graham B. S., Peebles R. S., Jr., (2005) Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J. Allergy Clin. Immunol. 116, 550–557 [DOI] [PubMed] [Google Scholar]
- 77. Johnson T. R., Mertz S. E., Gitiban N., Hammond S., Legallo R., Durbin R. K., Durbin J. E. (2005) Role for innate IFNs in determining respiratory syncytial virus immunopathology. J. Immunol. 174, 7234–7241 [DOI] [PubMed] [Google Scholar]
- 78. Ramaswamy M., Shi L., Monick M. M., Hunninghake G. W., Look D. C. (2004) Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 30, 893–900 [DOI] [PubMed] [Google Scholar]
- 79. Rudd B. D., Schaller M. A., Smit J. J., Kunkel S. L., Neupane R., Kelley L., Berlin A. A., Lukacs N. W. (2007) MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J. Immunol. 178, 5820–5827 [DOI] [PubMed] [Google Scholar]
- 80. Rudd B. D., Smit J. J., Flavell R. A., Alexopoulou L., Schaller M. A., Gruber A., Berlin A. A., Lukacs N. W. (2006) Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 176, 1937–1942 [DOI] [PubMed] [Google Scholar]
- 81. Schlender J., Hornung V., Finke S., Gunthner-Biller M., Marozin S., Brzozka K., Moghim S., Endres S., Hartmann G., Conzelmann K. K. (2005) Inhibition of Toll-like receptor 7- and 9-mediated α/β interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol. 79, 5507–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shingai M., Azuma M., Ebihara T., Sasai M., Funami K., Ayata M., Ogura H., Tsutsumi H., Matsumoto M., Seya T. (2008) Soluble G protein of respiratory syncytial virus inhibits Toll-like receptor 3/4-mediated IFN-β induction. Int. Immunol. 20, 1169–1180 [DOI] [PubMed] [Google Scholar]
- 83. Bhoj V. G., Sun Q., Bhoj E. J., Somers C., Chen X., Torres J. P., Mejias A., Gomez A. M., Jafri H., Ramilo O., Chen Z. J. (2008) MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 105, 14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Murawski M. R., Bowen G. N., Cerny A. M., Anderson L. J., Haynes L. M., Tripp R. A., Kurt-Jones E. A., Finberg R. W. (2009) Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 83, 1492–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lukacs N. W., Smit J. J., Schaller M. A., Lindell D. M. (2008) Regulation of immunity to respiratory syncytial virus by dendritic cells, Toll-like receptors, and notch. Viral Immunol. 21, 115–122 [DOI] [PubMed] [Google Scholar]
- 86. Kurt-Jones E. A., Popova L., Kwinn L., Haynes L. M., Jones L. P., Tripp R. A., Walsh E. E., Freeman M. W., Golenbock D. T., Anderson L. J., Finberg R. W. (2000) Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1, 398–401 [DOI] [PubMed] [Google Scholar]
- 87. Jones H. P., Hodge L. M., Fujihashi K., Kiyono H., McGhee J. R., Simecka J. W. (2001) The pulmonary environment promotes Th2 cell responses after nasal-pulmonary immunization with antigen alone, but Th1 responses are induced during instances of intense immune stimulation. J. Immunol. 167, 4518–4526 [DOI] [PubMed] [Google Scholar]
- 88. Guerrero-Plata A., Kolli D., Hong C., Casola A., Garofalo R. P. (2009) Subversion of pulmonary dendritic cell function by paramyxovirus infections. J. Immunol. 182, 3072–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hintzen G., Ohl L., del Rio M. L., Rodriguez-Barbosa J. I., Pabst O., Kocks J. R., Krege J., Hardtke S., Forster R. (2006) Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J. Immunol. 177, 7346–7354 [DOI] [PubMed] [Google Scholar]
- 90. Beaty S. R., Rose C. E., Jr., Sung S. S. (2007) Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178, 1882–1895 [DOI] [PubMed] [Google Scholar]
- 91. del Rio M. L., Rodriguez-Barbosa J. I., Kremmer E., Forster R. (2007) CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178, 6861–6866 [DOI] [PubMed] [Google Scholar]
- 92. Jakubzick C., Tacke F., Ginhoux F., Wagers A. J., van Rooijen N., Mack M., Merad M., Randolph G. J. (2008) Blood monocyte subsets differentially give rise to CD103+ and CD103− pulmonary dendritic cell populations. J. Immunol. 180, 3019–3027 [DOI] [PubMed] [Google Scholar]
- 93. Dunne P. J., Moran B., Cummins R. C., Mills K. H. (2009) CD11c+CD8α+ dendritic cells promote protective immunity to respiratory infection with Bordetella pertussis. J. Immunol. 183, 400–410 [DOI] [PubMed] [Google Scholar]
- 94. Nakano H., Free M. E., Whitehead G. S., Maruoka S., Wilson R. H., Nakano K., Cook D. N. (2012) Pulmonary CD103(+) dendritic cells prime Th2 responses to inhaled allergens. Mucosal Immunol. 5, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]