

Abstract
Purpose of review
To summarize recent evidence that IGF1 mediates growth effects of multiple trophic factors and discuss clinical relevance.
Recent findings
Recent reviews and original reports indicate benefits of growth hormone (GH) and long-acting glucagon-like peptide 2 (GLP2) analogues in short bowel syndrome and Crohn’s disease. This review highlights evidence that biomarkers of sustained small intestinal growth or mucosal healing and evaluation of intestinal epithelial stem cell biomarkers may improve clinical measures of intestinal growth or response to trophic hormones. Compelling evidence that IGF1 mediates growth effects of GH and GLP2 on intestine or linear growth in preclinical models of resection or Crohn’s disease is presented, along with a concept that these hormones or IGF1 may enhance sustained growth if given early after bowel resection. Evidence that SOCS protein induction by GH or GLP2 in normal or inflamed intestine, may limit IGF1-induced growth, but protect against risk of dysplasia or fibrosis is reviewed. Whether IGF1 receptor mediates IGF1 action and potential roles of insulin receptors are addressed.
Summary
IGF1 has a central role in mediating trophic hormone action in small intestine. Better understanding of benefits and risks of IGF1, receptors that mediate IGF1 action, and factors that limit undesirable growth are needed.
Keywords: intestinal growth, enterotrophic therapy, short bowel syndrome, Crohn’s disease
Introduction
There is considerable interest in trophic factors that may promote sustained increases in mass and function of intestinal epithelium in patients at risk of short bowel syndrome (SBS) and intestinal failure, or promote mucosal healing in Crohn’s disease. This chapter will briefly review cellular mechanisms and measures used to assess intestinal epithelial growth, and a wealth of preclinical evidence demonstrating potent effects of insulin-like growth factor 1 (IGF1) on growth of small intestinal epithelium in multiple physiological and clinically relevant settings of intestinal loss or injury. Areas of emphasis will be new and emerging evidence that IGF1 preferentially targets intestinal epithelial stem cells (IESC) and that IGF1 is a critical mediator of the enterotrophic effects of growth hormone (GH), or glucagon-like peptide 2 (GLP2) and analogues, which are in clinical trial for SBS and Crohn’s disease. We will address the barriers to consideration of IGF1 as a clinically useful enterotrophic therapy and whether the insulin-like growth factor 1 receptor (IGF1R) is the primary mediator of the trophic actions of IGF1 on intestinal epithelial cells (IEC), or whether some effects may be mediated by a splice variant of the insulin receptor (IR).
Plasticity or adaptive changes in intestinal epithelial growth in health and disease
Continuous renewal of the small intestinal epithelium is essential to nutrient digestion and absorption and barrier function. Renewal is mediated by IESC located at the crypt base which divide to renew themselves and yield more rapidly dividing, transit amplifying progenitor cells that then give rise to the terminally-differentiated lineages on the villus (enterocytes, goblet cells and enteroendocrine cells) or crypt (Paneth cells and enteroendocrine cells). Within the small intestinal crypts there is a small but significant rate of spontaneous apoptosis. Under homeostatic conditions, apoptosis may limit excess production of new IEC and thereby maintain constant epithelial mass, and may also remove genetically damaged stem or progenitor cells to protect against dysplasia [1, 2].
Figure 1 [3] summarizes evidence, largely derived from animal models, for dramatic changes in growth and mass of small bowel epithelium in response to physiological or pathophysiological challenges. Fasting, nutrient restriction or lack of luminal nutrients, as occurs in total parenteral nutrition (TPN), lead to hypoplasia. Surgical resection of portions of small bowel leads to compensatory increases in mass of remaining small bowel epithelium per unit length, a phenomenon termed ‘adaptive growth’ or ‘intestinal adaptation’. While well documented in animal models, these responses have been much less well characterized in humans. Some studies suggest that TPN induces atrophy [4–6], but others do not [7]. These differences likely reflect duration of TPN. Similarly, a few studies have documented crypt and villus hyperplasia in humans after bowel resection [6, 8–10].
Figure 1. Schematic of altered growth of small intestinal epithelium in response to altered nutrient, resection, inflammation, or injury.
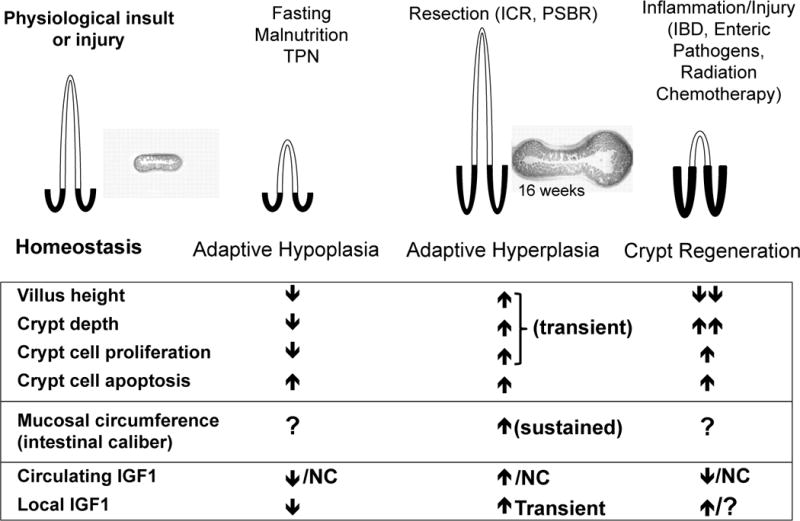
The schematic summarizes data from preclinical animal models about changes in crypt depth, villus height, crypt cell proliferation and apoptosis after TPN, resection, or chronic inflammation. Note that in preclinical models of resection, crypt and villus hyperplasia are transient and may not reflect long-term changes. Increased intestinal caliber (as illustrated at 16 weeks after ICR; from Dekaney 2007 [3]) may more accurately reflect long-term adaptive growth after resection. Relatively few clinical studies have evaluated crypt and villus morphology in these settings, but the limited data suggest similar, although possibly less pronounced changes (see text). Evaluation of intestinal caliber may be a useful measure, but is difficult to assess clinically. As depicted in animal models of TPN and nutrient restriction, decreases in circulating and local intestinal IGF1 correlate with adaptive hypoplasia. Resection-induced hyperplasia is associated with transient increases in local IGF1. Settings of inflammation and injury can lead to decreased circulating, but increased locally expressed IGF1. These changes have been documented in animal models. Fewer studies have assessed circulating or local IGF1 in humans, but the evidence that exists supports decreased circulating IGF1 [28**, 31] and elevated local IGF1 in active Crohn’s disease [32]. Thus roles and effects of circulating and local IGF1 may differ depending on the circumstance. PSBR = proximal short bowel resection NC = no change; ? = not examined.
Current views indicate that after massive small bowel resection or multiple small bowel resections, intestinal adaptation protects against the need for parenteral nutrition, SBS, and intestinal failure [11*]. This has led to a growing interest in identification of trophic factors that promote optimal adaptive growth after bowel loss [11*, 12].
Inflammation or injury of the small bowel epithelium as occurs in inflammatory bowel diseases (IBD), exposure to enteric pathogens and radiation or chemotherapy, can lead to crypt and villus loss. As illustrated in Figure 1, this triggers crypt hyperplasia and regenerative responses. If transient, such responses may promote beneficial mucosal healing and normalization of function and integrity. Most clinical studies of histopathology in Crohn’s disease focus on immunological features, but as recently described, a majority of patients with active Crohn’s disease, show villus blunting, crypt hyperplasia, or crypt disorganization [13].
Recent findings that mucosal healing is a major predictor of sustained clinical remission in patients with early stage Crohn’s disease [14*] have prompted significant interest in growth factors as endogenous mediators or potential therapies that may promote mucosal healing [15*]. However, it is important to consider whether commonly used measures of intestinal epithelial growth or responses to growth factors accurately reflect functionally beneficial or sustained adaptive growth or mucosal healing.
Measures of intestinal adaptation, mucosal healing or response to growth factors
Crypt depth, proliferation and villus height, are typical preclinical measures of intestinal adaptation or response to growth factors. However, recent findings in animal models of ileo-cecal resection (ICR) suggest that there may be only early and transient increases in jejunal crypt depth or villus height after ICR or in response to trophic therapies [3, 16*, 17**]. Long-term increases in crypt number, mucosal and epithelial circumference, and caliber of the remnant small intestine (Figure 1) may more accurately reflect sustained and functionally relevant adaptive growth or response to trophic therapies [3, 16*]. These are difficult to assess in clinical settings, but should at least be considered in preclinical evaluation of enterotrophic factors. Our recent studies have shown that these longer-term adaptive responses to resection depend on very early expansion of putative IESC and subsequent crypt fission [3, 16*]. Recently identified IESC biomarkers such as Lgr5, Bmi1 and Sox9 and availability of reporter mice expressing fluorescent proteins in IESC [18–20] provide new preclinical tools to assess, for the first time, direct actions of trophic factors on IESC. However useful antibodies to these biomarkers are either not available or not yet validated for broader application to evaluation of IESC in clinical settings. Emerging data on genes enriched in IESC [21] or IESC-specific genes affected by trophic therapies [22] should permit future identification of suitable biomarkers to assess the impact of trophic therapies on IESC in clinical situations. Crypt fission may represent a useful surrogate marker of sustained IESC expansion and adaptive growth or response to growth factors, at least in a setting of SBS [3, 16*, 23*]. However, in a setting of active Crohn’s disease, increased crypt depth, crypt fission or increases in IESC may not always reflect beneficial responses since excessive or chronic hyperproliferative or anti-apoptotic responses in IBD are associated with increased risk of dysplasia [23*, 24]. A recent Gates Foundation Grand Challenge to develop ‘Biomarkers of gut function and health’ illustrates the critical need for better biomarkers of bowel health and growth, or response to interventions designed to improve gut function.
Circulating and locally expressed intestinal IGF1
IGF1 can act on the intestinal epithelium via endocrine effects of circulating IGF1 derived primarily from hepatocytes, and paracrine effects of locally expressed IGF1 synthesized by intestinal mesenchyme. Circulating IGF1 is positively regulated by growth hormone (GH), caloric and protein intake, and insulin. Local intestinal IGF1 is less subject to GH regulation, except in extremes of GH deficiency or excess, but is positively regulated by luminal nutrients [25–27] and is transiently up-regulated in response to ICR [16*, 27]. In active IBD, chronic elevation of pro-inflammatory cytokines is associated with reduced levels of circulating IGF1, which reflects at least in part GH resistance at the level of hepatocytes, as well as impact of malabsorption or malnutrition [28**], but increased local IGF1 expression was found (reviewed in [27]). Thus active intestinal inflammation leads to a disconnect between circulating and locally expressed IGF1. A key unanswered question is the extent to which circulating or locally expressed IGF1 contribute to or promote normal growth, adaptive growth or repair and regeneration of intestinal epithelium. Mouse models lacking local intestinal IGF1 expression due to disruption of both endogenous IGF1 alleles, but with normal circulating IGF1 due to a liver specific IGF1 transgene, provide potentially useful new model systems to dissect the roles of circulating IGF1 versus locally expressed intestinal IGF1 [29, 30].
IGF1 potently promotes growth of small intestinal epithelium and may preferentially target IESC
As summarized in Figure 1, data derived largely from animal models demonstrate that altered levels of endogenous circulating and/or local IGF1 accompany changes in growth of intestinal epithelium. The few existing studies suggest similar changes in humans [28**, 31, 32]. Prior reviews summarize evidence that IGF1 can promote growth of small intestinal epithelium [11*, 12, 27]. Briefly, systemically administered or transgene-derived IGF1 potently increase the mass of small bowel epithelium in normal adult animals and preclinical models of TPN, small bowel resection or ICR [33, 34]; these effects of IGF1 are associated with increased proliferation and decreased apoptosis of crypt epithelial cells. The local up-regulation of IGF1 in the intestine of multiple animal models of IBD and in patients with Crohn’s disease [27] may promote mucosal healing during inflammation-induced injury, although this has not yet been formally demonstrated. A recent study in mice and humans demonstrated that during sepsis, reduced levels of circulating IGF1 correlate with increased bacterial translocation from the gut. In the mouse model, anti-apoptotic effects of exogenous IGF1 were associated with dramatically reduced bacterial translocation [35]. Together these studies provide compelling evidence for potent, trophic, proliferative and anti-apoptotic effects of IGF1 on small intestinal epithelium in many clinically relevant situations.
Available and emerging evidence indicates that IGF1 may preferentially target IESC. Findings in models of radiation-induced apoptosis indicate preferential anti-apoptotic effects of IGF1 overexpression or IGF1 therapy on putative IESC [36, 37]. Recent studies using Sox9-EGFP mice to directly visualize IESC [19] indicate that IGF1 given after radiation promotes IESC expansion and crypt regeneration [22] [van Landeghem L, Lund PK, unpublished]. This is highly relevant to a growing interest in trophic factors that might promote crypt regeneration and help protect against radiation-induced gastrointestinal syndrome after accidental exposure to radiation. Studies in a murine ICR model also indicate that early and transient up-regulation of local IGF1 correlates with expansion of putative IESC and subsequent crypt fission, responses that are critical to sustained adaptive increases in crypt number and mucosal mass [16*].
IGF1 is a common mediator of the actions of growth hormone (GH) and glucagon-like peptide 2 (GLP2) or longer acting GLP2 analogues
Excellent reviews have summarized evidence from small clinical trials indicating that GH, and a long-acting form of GLP2 (teduglutide, [NPS Allelix, Mississauga, Ontario, Canada]), have beneficial effects in patients with SBS receiving supplemental parenteral nutrition [11*, 12, 38], or Crohn’s disease [15*, 39*]. To date the majority of evidence for benefits is based on reduced need for parenteral nutrition in SBS and improved Crohn’s disease activity index (CDAI) [11*, 12, 15*, 38–40]. Recent original reports in SBS patients indicate that GH [41**] or teduglutide [42, 43**] increased plasma citrulline, a surrogate marker of epithelial mass. Increases in villus height in SBS patients given teduglutide provide more direct evidence for growth effects on intestinal epithelium [43**]. However, there is little evidence that GH or GLP2/teduglutide directly act on IEC to promote growth or repair. Indeed the bulk of the evidence suggests that IGF1 is a critical mediator of the enterotrophic effects of GH or GLP2.
IGF1 as a mediator of GH action
Older studies in transgenic mice that overexpress GH or IGF1, and studies in TPN or resection models given systemic GH or IGF1, indicate that IGF1 more consistently and more potently increases epithelial mass, crypt proliferation, and small bowel length than GH, and that IGF1 but not GH has anti-apoptotic effects on crypt epithelial cells (Figure 2). This is despite the fact that in GH-treated or transgenic animals, circulating IGF1 was increased to similar or greater levels than observed in IGF1 transgenic or IGF1-treated animals. Even though IGF1 has more potent growth effects in preclinical models, GH and not IGF1 is in clinical trial as enterotrophic therapy. In part this reflects the fact that recombinant human growth hormone (rhGH) has been used to treat growth delay in more than 50,000 children over relatively long periods (average 3 years), with a favorable safety profile [44**].
Figure 2. Growth effects of GH and IGF1 on small intestine in transgenic overexpression models or when given systemically in TPN-fed animals.
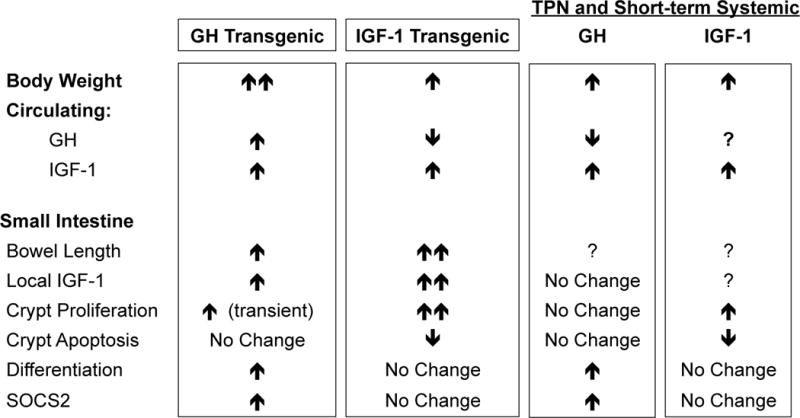
In GH or IGF1 transgenic mice and GH or IGF1 infused animals, GH and IGF1 lead to similar increases in circulating IGF1. As indicated, IGF1 more potently increases bowel length and growth than GH, while GH induces cellular differentiation and suppressors of cytokine signaling (SOCS), which limit growth and promote differentiation.
In animal models of TPN, systemic GH, but not IGF1 induces suppressor of cytokine signaling 2 (SOCS2) in the small intestine [45]. Findings in SOCS2 null mice suggest that SOCS2 normally limits the proliferative and anti-apoptotic actions of IGF1 on small intestinal epithelium [46], but also protects against tumor development or growth in the small intestine and colon of ApcMin/+ mice [47*]. GH was also shown to induce SOCS3 in the intestine of a rat model of Crohn’s disease, but not non-inflamed controls [48]. This suggests that GH interacts with pro-inflammatory cytokines to induce intestinal SOCS3. Evidence in humans and preclinical models indicates that SOCS3 protects against inflammation-associated intestinal cancer [49**, 50], has potent anti-inflammatory activities [51], and limits inflammation-induced intestinal fibrosis [48], a major problem in Crohn’s disease [52]. Together these studies indicate that GH-induction of SOCS2 or SOCS3 may limit the magnitude of trophic responses to GH or GH-induced IGF1 in intestinal epithelium. However, the downside of this diminished growth response is counterbalanced by the ability of SOCS to protect against undesirable tumor promoting or profibrogenic effects of GH or GH-induced IGF1, and possibly, the anti-inflammatory effects of SOCS. These preclinical findings suggest that evaluation of SOCS in patients with SBS or Crohn’s disease treated with GH would be of considerable interest to assess if there is an association between SOCS expression and growth or mucosal healing responses.
Growth failure in children with Crohn’s disease, has been linked to hepatic GH resistance and reduced circulating IGF1 [28**]. TPN has been linked to intestinal GH resistance [53]. In these settings, GH therapy may at least in part abrogate GH resistance and increase circulating IGF1 towards normal levels. It seems possible that GH combined with low dose IGF1 in children with Crohn’s disease might represent a strategy to maximize impact on linear growth, growth of intestinal epithelium, or mucosal healing, while still preserving the anti-tumorigenic, anti-fibrotic and anti-inflammatory actions of GH-induced SOCS. This will clearly require formal testing in preclinical models, and more information about safety of IGF1 therapy in humans.
IGF1 as a mediator of GLP2 action
The evidence for the enterotrophic effects of GLP2 and longer-acting derivatives in preclinical models or clinical trials is summarized in a recent, excellent review [54*]. Compelling evidence that IGF1 is a required mediator of these effects of GLP2 stems from recent observations that GLP2 is ineffective at increasing small bowel mass, crypt depth, or villus height in mice with targeted disruption of both IGF1 alleles [55] and mice with inducible deletion of IGF1R specifically in IEC [56**]. The receptor for GLP2 is not expressed at detectable levels in IEC, but is expressed in intestinal mesenchymal cells, including subepithelial myofibroblasts, where it directly induces the synthesis of IGF1 [57]. Expression of the GLP2 receptor is restricted to the gastrointestinal tract except for low-level expression in lung and hypothalamus [54*], minimizing undesirable off-target growth effects of GLP2 or GLP2-induced IGF1. However, while the available evidence suggests that GLP2 therapies reduce the need for parenteral nutrition, relatively few GLP2-treated patients are completely weaned from parenteral nutrition, and this is true also for GH [41**–43**]. Studies in preclinical models of SBS and TPN indicate that GLP2-induced increases in small intestine mass are reversed after cessation of GLP2 treatment [17**]. Interestingly, this contrasts with IGF1 where acute treatment with IGF1 in a rat SBS/TPN model facilitated transition to enteral feeding and caused sustained increases in mass of small bowel epithelium [33]. Thus in preclinical models of SBS, exogenous IGF1 appears to better sustain adaptive growth of small intestine than GLP2, or GLP2-induced endogenous IGF1. In an ICR model, GLP2 was able to increase endogenous IGF1 expression, IESC expansion, and crypt fission only when given in the immediate period after resection [16*]. While limited to preclinical evidence, these findings suggest that after bowel resections that pose risk of SBS early treatment with GLP2 or other factors that induce IGF1 may be critically important to sustained therapeutic adaptive growth response. This is not current clinical practice, since SBS patients are typically treated with trophic therapies months or years after bowel loss [11*, 58]. Also, the possibility that IGF1 might more effectively sustain adaptive growth than GLP2, if given at later times after bowel resection or in SBS, due to its ability to directly expand IESC is worthy of consideration, and is currently under test in preclinical models in our laboratories [Lund PK, Helmrath M, unpublished data].
Despite recent reports indicating benefits of GLP2 in Crohn’s disease, the mechanisms are not defined. Interestingly, in a preclinical model of IBD, GLP2 actually reduced crypt proliferation, reduced apoptosis and had anti-inflammatory effects; these effects were associated with increases in both local IGF1 and SOCS3 [59]. As with GH therefore, anti-inflammatory and possibly anti-proliferative effects of GLP2 in a setting of IBD may be linked to SOCS3 induction. However, more direct evidence about the roles of GLP2-induced SOCS and IGF1 in a setting of intestinal inflammation is clearly needed.
As highlighted in a recent review, other mediators including the ligands for the ErbB2 receptor and ErbB signaling have been linked to trophic effects of GLP2 in small intestine [54*]. Space does not permit a full consideration of how these findings may be reconciled with lack of enterotrophic response of small intestine to GLP2 in mice with IGF1 gene deletion or IEC-specific IGF1R gene deletion except to note that IGF1 is known to interact with other growth factors including members of the epidermal growth factor family to exert additive or potent synergistic effects on IEC proliferation [60], which may explain the dual roles of IGF1R and ErbB receptors in mediating GLP2 action [54*].
IGF binding proteins (IGFBPs)
A family of IGF binding proteins (IGFBPs) have major effects on levels of bioavailable circulating and locally expressed IGF1. These IGFBPs may be altered with nutritional status (reviewed in [61]), inflammation [62, 63], or resection [64, 65], as well as following TPN or rhGH administration [64]. A discussion of IGFBPs is beyond the scope of this review, but evaluation of the role of IGFBPs in IGF1, GLP2, or GH action in the intestine is an area for additional research.
What are the barriers to clinical testing of IGF1 as enterotrophic therapy?
The extensive evidence that elevated circulating or locally expressed IGF1 or insulin-like growth factor 2 (IGF2) may promote risk of gastrointestinal or other cancers (reviewed in [66]) is a major barrier to use of IGF1 as enterotrophic therapy. IGF1 can induce growth of many organs, in addition to the small intestine, and these off target effects are a significant clinical concern. IGF1 therapy could potentially exacerbate fibrosis during intestinal inflammation or injury, which would be a potential concern in Crohn’s disease [52], and in radiation-induced intestinal injury. IGF1 potently induces collagen synthesis in intestinal mesenchyme in vitro or in vivo [67]. Recent findings that mice heterozygous for IGF1 gene deletion exhibit reduced fibrosis in the trinitrobenzensulfonic acid (TNBS) colitis model provide direct in vivo evidence that endogenous IGF1 contributes to inflammation-induced fibrosis [68]. Despite these concerns, and as noted in a recent review [11*], the potential benefits of IGF1 therapy in certain situations deserve some consideration. There is growing clinical literature that rhIGF1 therapy is effective and safe in children with primary IGF1 deficiency due to genetic or acquired GH resistance [69]. Malnutrition due to small intestinal malabsorptive or inflammatory disorders, including Crohn’s disease, is commonly associated with deficits in linear growth in children, which are linked to hepatic GH resistance and reduced circulating IGF1 levels [28**, 70*]. In such circumstances it is possible that IGF1 given at doses which normalize rather than induce supraphysiological levels of circulating IGF1, could improve linear growth and also promote growth or repair of small bowel epithelium without undesired or excessive growth of other organs, or other adverse effects. Overall, more information is needed to establish the potential benefits and risks of therapeutically administered IGF1, as well as enterotrophic factors that induce IGF1 in SBS, Crohn’s disease or other small bowel disorders. In particular, the impact of short-term, or low dose IGF1 therapy on intestinal epithelial growth or mucosal healing, responses versus detrimental pro-tumorigenic or pro-fibrotic responses should be studied in preclinical models. Information about whether effects of IGF1 differ in young versus adult animals or humans is also needed.
IGF1 receptor (IGF1R) as the primary mediator of IGF1 action
Current views indicate that the IGF1R is a primary mediator of body and organ growth since IGF1R null mice die in early embryogenesis due to severe growth defects. However, as yet, there is no definitive evidence that in the small intestinal epithelium IGF1R is a required mediator of IGF1 action. In a recent report, inducible, conditional deletion of IGF1R using tamoxifen-inducible Villin-Cre recombinase led to no overt phenotype in the basal state even though the trophic effects of GLP2 were lost [56**]. This suggests either that IGF1R is not required for normal intestinal homeostasis or that another receptor can compensate for loss of IGF1R in IEC. As depicted in the schematic in Figure 3, IGF1R is highly homologous to the IR with 45–65% homology in the ligand binding domain and 60–85% homology in the tyrosine kinase and substrate recruitment domains [71]. IGF1R binds IGF1 with the highest affinity but also has high affinity for IGF2 and can bind insulin at elevated concentrations [71–73**]. IR exists as two isoforms, IR-A and IR-B derived from alternative pre-mRNA splicing. These isoforms differ by the inclusion or exclusion of exon 11, which alters the ligand binding affinity and downstream signaling properties of the IR [71–74]. Inclusion of exon 11 yields IR-B, which is highly expressed in known insulin-target organs, such as liver, muscle, and adipose tissue and has a high affinity for insulin [71–73**]. IR-A lacks exon 11, has higher affinity for IGF2 and insulin than IGF1, but binds all ligands. IR-A is expressed at high levels during embryonic development when IGF2 is also highly expressed and is up-regulated in cancer cells [72, 73**, 75]. The expression patterns and respective roles of IGF1R, versus IR-A or IR-B, in small intestinal epithelium are unknown. This is relevant to obesity, a major public health problem, which leads to hyperinsulinemia, and has recently been associated with increased growth of small intestine [76, 77], as well as increased risk of gastrointestinal and other cancers [71–73**]. Obesity/hyperinsulinemia may promote intestinal growth via insulin acting on IR, IR-A or IR-B, or because elevated insulin liberates IGF1 from IGFBPs for interaction with IGF1R [71–73**]. A complete understanding of IGF1 action in small intestine will require more information about ability of IGF1 to signal through IR-A or IR-B as well as IGF1R.
Figure 3. Schematic of structure, ligand binding affinity and putative roles of IGF1R and insulin receptor (IR) isoforms (IR-A and IR-B).
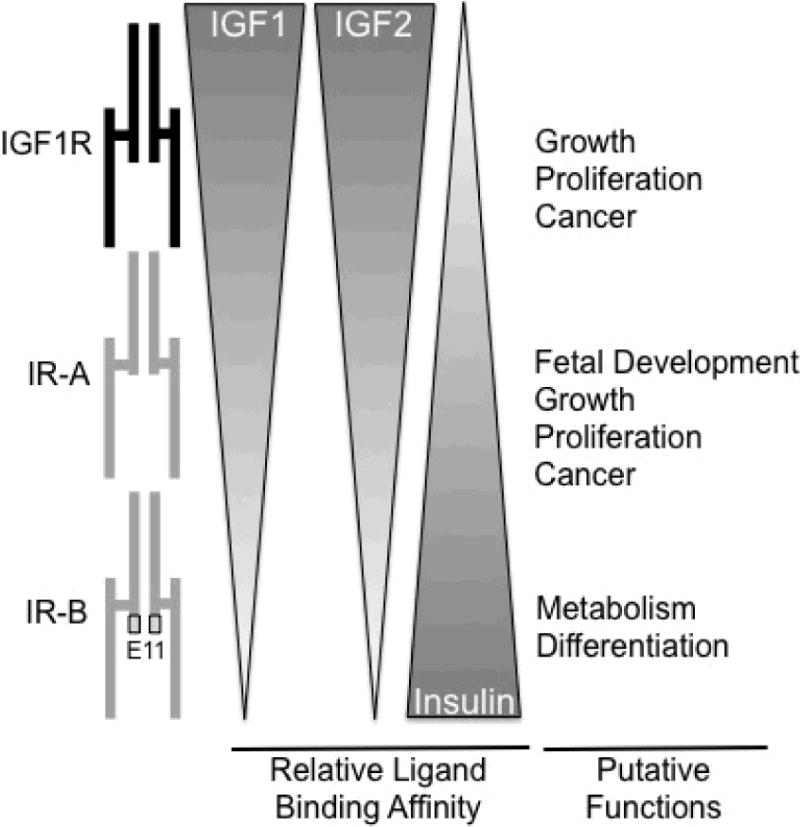
High sequence homology allows all three receptors to bind IGF1, IGF2, and insulin, but with differing affinities, indicated by gradients. IR-A and IR-B differ by inclusion of exon 11 (E11) only in IR-B. Different ligand binding affinities and downstream signaling intermediates, confer the different functional outcomes indicated. For small intestine these are still putative outcomes since neither expression patterns nor specific functions of IGF1R, IR-A and IR-B are defined.
Concluding Remarks
IGF1 potently induces small intestinal growth, mediates the actions of multiple enterotrophic factors and drives IESC expansion and crypt fission that sustain adaptive growth responses after resection. Off-target growth effects and concerns about enhanced risk of cancer or fibrosis pose barriers to clinical use of IGF1 as enterotrophic therapy. Full understanding of the roles of IGF1 in mediating the benefits or potential risks of therapy with, GH, GLP2 and analogues, the roles of SOCS in these responses, and the receptor(s) that mediate IGF1 action in small intestine is critical to identification of safe and effective enterotrophic and growth-promoting therapies in SBS, Crohn’s disease, and potentially other situations of compromised intestinal growth.
Key points.
Growth of small intestinal epithelium changes in response to nutrient restriction, resection, inflammation or injury.
Growth factors and hormones may enhance intestinal growth and limit need for parenteral nutrition after resection and promote mucosal healing or normalize linear body growth in Crohn’s disease.
Improved measures of early response of intestinal epithelial stem cells (IESC) to growth factors and sustained intestinal epithelial growth in response to trophic therapies are needed.
IGF1 is a key mediator of the actions of clinically used trophic factors, GH and GLP2, and may preferentially target IESC.
GH or GLP2-induction of SOCS proteins may limit risk of undesirable or excessive IGF1-mediated growth.
Acknowledgments
The authors would like to thank Michael Helmrath for input on this manuscript. Sarah Bortvedt is also funded by F31 AG040943.
Funding NIH DK047769-12 and NIH DK040247-19
References
- 1.Lund PK. Insulin-like growth factors: gene structure and regulation. In: Kostyo Jack L, Goodman H Maurice., editors. Handbook of Physiology, Section 7, The Endocrine System. Vol. 5. New York, NY: American Physiological Society/Oxford University Press; 1999. pp. 537–71. (Hormonal Control of Growth). [Google Scholar]
- 2.Ramocki NM, Wilkins HR, Magness ST, et al. Insulin receptor substrate-1 deficiency promotes apoptosis in the putative intestinal crypt stem cell region, limits Apcmin/+ tumors, and regulates Sox9. Endocrinology. 2008;149(1):261–7. doi: 10.1210/en.2007-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dekaney CM, Fong JJ, Rigby RJ, et al. Expansion of intestinal stem cells associated with long-term adaptation following ileocecal resection in mice. American journal of physiology Gastrointestinal and liver physiology. 2007;293(5):G1013–22. doi: 10.1152/ajpgi.00218.2007. [DOI] [PubMed] [Google Scholar]
- 4.Buchman AL, Moukarzel AA, Bhuta S, et al. Parenteral nutrition is associated with intestinal morphologic and functional changes in humans. JPEN Journal of parenteral and enteral nutrition. 1995;19(6):453–60. doi: 10.1177/0148607195019006453. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez G, Velasco N, Wainstein C, et al. Gut mucosal atrophy after a short enteral fasting period in critically ill patients. Journal of critical care. 1999;14(2):73–7. doi: 10.1016/s0883-9441(99)90017-5. [DOI] [PubMed] [Google Scholar]
- 6.Pironi L, Paganelli GM, Miglioli M, et al. Morphologic and cytoproliferative patterns of duodenal mucosa in two patients after long-term total parenteral nutrition: changes with oral refeeding and relation to intestinal resection. JPEN Journal of parenteral and enteral nutrition. 1994;18(4):351–4. doi: 10.1177/014860719401800413. [DOI] [PubMed] [Google Scholar]
- 7.Duran B. The effects of long-term total parenteral nutrition on gut mucosal immunity in children with short bowel syndrome: a systematic review. BMC nursing. 2005;4(1):2. doi: 10.1186/1472-6955-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joly F, Mayeur C, Messing B, et al. Morphological adaptation with preserved proliferation/transporter content in the colon of patients with short bowel syndrome. American journal of physiology Gastrointestinal and liver physiology. 2009;297(1):G116–23. doi: 10.1152/ajpgi.90657.2008. [DOI] [PubMed] [Google Scholar]
- 9.Schaart MW, de Bruijn AC, Bouwman DM, et al. Epithelial functions of the residual bowel after surgery for necrotising enterocolitis in human infants. Journal of pediatric gastroenterology and nutrition. 2009;49(1):31–41. doi: 10.1097/MPG.0b013e318186d341. [DOI] [PubMed] [Google Scholar]
- 10.Vieten D, Corfield A, Ramani P, Spicer R. Proliferative response in necrotising enterocolitis is insufficient to prevent disease progression. Pediatric surgery international. 2006;22(1):50–6. doi: 10.1007/s00383-005-1588-1. [DOI] [PubMed] [Google Scholar]
- 11*.McMellen ME, Wakeman D, Longshore SW, et al. Growth factors: possible roles for clinical management of the short bowel syndrome. Seminars in pediatric surgery. 2010;19(1):35–43. doi: 10.1053/j.sempedsurg.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an excellent, comprehensive review of the effects of growth factors on intestinal adpation following massive bowel resection in animal models and human patients.
- 12.Drozdowski L, Thomson AB. Intestinal hormones and growth factors: effects on the small intestine. World journal of gastroenterology : WJG. 2009;15(4):385–406. doi: 10.3748/wjg.15.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein N, Dulai M. Contemporary morphologic definition of backwash ileitis in ulcerative colitis and features that distinguish it from Crohn disease. American journal of clinical pathology. 2006;126(3):365–76. doi: 10.1309/UAXMW3428PGN9HJ3. [DOI] [PubMed] [Google Scholar]
- 14*.Baert F, Moortgat L, Van Assche G, et al. Mucosal healing predicts sustained clinical remission in patients with early-stage Crohn’s disease. Gastroenterology. 2010;138(2):463–8. doi: 10.1053/j.gastro.2009.09.056. quiz e10–1. [DOI] [PubMed] [Google Scholar]; This clinical study describes the importance of mucosal healing in Crohn’s disease patients as predictive of sustained remission.
- 15*.Krishnan K, Arnone B, Buchman A. Intestinal growth factors: potential use in the treatment of inflammatory bowel disease and their role in mucosal healing. Inflamm Bowel Dis. 2011;17(1):410–22. doi: 10.1002/ibd.21316. [DOI] [PubMed] [Google Scholar]; This is a comprehensive review summarizing recent work examining the therapeutic efficacy of growth factors in promoting mucosal healing in inflammatory bowel disease.
- 16*.Garrison AP, Dekaney CM, von Allmen DC, et al. Early but not late administration of glucagon-like peptide-2 following ileo-cecal resection augments putative intestinal stem cell expansion. American journal of physiology Gastrointestinal and liver physiology. 2009;296(3):G643–50. doi: 10.1152/ajpgi.90588.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a novel study showing that early adminsitation of GLP2 is necessary for induction of intestinal stem cell expansion following ileo-cecal resection.
- 17**.Koopmann MC, Chen X, Holst JJ, Ney DM. Sustained glucagon-like peptide-2 infusion is required for intestinal adaptation, and cessation reverses increased cellularity in rats with intestinal failure. American journal of physiology Gastrointestinal and liver physiology. 2010;299(6):G1222–30. doi: 10.1152/ajpgi.00367.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an exceptional study showing the sustained GLP2 administation is required for sustained intestinal adaptation following massive ileo-cecal resection. In the presence of GLP2, bowel mass and cellularity increased, whereas withdrawal of GLP2 resulted in loss of additional bowel mass in a rat model of short bowel syndrome.
- 18.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 19.Gracz AD, Ramalingam S, Magness ST. Sox9 expression marks a subset of CD24-expressing small intestine epithelial stem cells that form organoids in vitro. American journal of physiology Gastrointestinal and liver physiology. 2010;298(5):G590–600. doi: 10.1152/ajpgi.00470.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sangiorgi E, Capecchi MR. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(17):7101–6. doi: 10.1073/pnas.0902508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Flier LG, van Gijn ME, Hatzis P, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136(5):903–12. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 22.Van Landeghem L, Santoro MA, Krebs AE, et al. Insulin-Like Growth Factor-I Promotes Intestinal Stem Cell Expansion During Crypt Regeneration and Mucosal Healing Following Radiation. Gastroenterology. 2011;140(5):S-39. [Google Scholar]
- 23*.Speck KE, Garrison AP, Rigby RJ, et al. Inflammation enhances resection-induced intestinal adaptive growth in IL-10 null mice. The Journal of surgical research. 2011;168(1):62–9. doi: 10.1016/j.jss.2009.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study showing that inflammation drives crypt fission following ileo-cecal resection. This article emphasizes the importance of inflammation in triggering intestinal adaptation.
- 24.Breynaert C, Vermeire S, Rutgeerts P, Van Assche G. Dysplasia and colorectal cancer in inflammatory bowel disease: a result of inflammation or an intrinsic risk? Acta gastro-enterologica Belgica. 2008;71(4):367–72. [PubMed] [Google Scholar]
- 25.Liu X, Murali SG, Holst JJ, Ney DM. Enteral nutrients potentiate the intestinotrophic action of glucagon-like peptide-2 in association with increased insulin-like growth factor-I responses in rats. American journal of physiology Regulatory, integrative and comparative physiology. 2008;295(6):R1794–802. doi: 10.1152/ajpregu.90616.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taqi E, Wallace LE, de Heuvel E, et al. The influence of nutrients, biliary-pancreatic secretions, and systemic trophic hormones on intestinal adaptation in a Roux-en-Y bypass model. Journal of pediatric surgery. 2010;45(5):987–95. doi: 10.1016/j.jpedsurg.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 27.Theiss AL, Fruchtman S, Lund PK. Growth factors in inflammatory bowel disease: the actions and interactions of growth hormone and insulin-like growth factor-I. Inflamm Bowel Dis. 2004;10(6):871–80. doi: 10.1097/00054725-200411000-00021. [DOI] [PubMed] [Google Scholar]
- 28**.D’Mello S, Trauernicht A, Ryan A, et al. Innate dysfunction promotes linear growth failure in pediatric Crohn’s disease and growth hormone resistance in murine ileitis. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21689. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an excellent study that utilized a pediatric population of Crohn’s disease patients and card15-deficient mice to link growth hormone resistance to growth failure, demonstrating that innate immune dysfunction leads to growth failure in pediatric Crohn’s disease patients and hepatic growth hormone restistance in mice with ileitis.
- 29.Elis S, Courtland HW, Wu Y, et al. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25(6):1257–66. doi: 10.1002/jbmr.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stratikopoulos E, Szabolcs M, Dragatsis I, et al. The hormonal action of IGF1 in postnatal mouse growth. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(49):19378–83. doi: 10.1073/pnas.0809223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters TD, Griffiths AM. Mechanisms of growth impairment in pediatric Crohn’s disease. Nature reviews Gastroenterology & hepatology. 2009;6(9):513–23. doi: 10.1038/nrgastro.2009.124. [DOI] [PubMed] [Google Scholar]
- 32.Pucilowska JB, McNaughton KK, Mohapatra NK, et al. IGF-I and procollagen alpha1(I) are coexpressed in a subset of mesenchymal cells in active Crohn’s disease. American journal of physiology Gastrointestinal and liver physiology. 2000;279(6):G1307–22. doi: 10.1152/ajpgi.2000.279.6.G1307. [DOI] [PubMed] [Google Scholar]
- 33.Gillingham MB, Dahly EM, Murali SG, Ney DM. IGF-I treatment facilitates transition from parenteral to enteral nutrition in rats with short bowel syndrome. American journal of physiology Regulatory, integrative and comparative physiology. 2003;284(2):R363–71. doi: 10.1152/ajpregu.00247.2002. [DOI] [PubMed] [Google Scholar]
- 34.Knott AW, Juno RJ, Jarboe MD, et al. Smooth muscle overexpression of IGF-I induces a novel adaptive response to small bowel resection. American journal of physiology Gastrointestinal and liver physiology. 2004;287(3):G562–70. doi: 10.1152/ajpgi.00438.2003. [DOI] [PubMed] [Google Scholar]
- 35.Hunninghake GW, Doerschug KC, Nymon AB, et al. Insulin-like growth factor-1 levels contribute to the development of bacterial translocation in sepsis. American journal of respiratory and critical care medicine. 2010;182(4):517–25. doi: 10.1164/rccm.200911-1757OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene. 2010;29(11):1622–32. doi: 10.1038/onc.2009.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkins HR, Ohneda K, Keku TO, et al. Reduction of spontaneous and irradiation-induced apoptosis in small intestine of IGF-I transgenic mice. American journal of physiology Gastrointestinal and liver physiology. 2002;283(2):G457–64. doi: 10.1152/ajpgi.00019.2002. [DOI] [PubMed] [Google Scholar]
- 38.Tee CT, Wallis K, Gabe SM. Emerging treatment options for short bowel syndrome: potential role of teduglutide. Clinical and experimental gastroenterology. 2011;4:189–96. doi: 10.2147/CEG.S13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Vortia E, Kay M, Wyllie R. The role of growth hormone and insulin-like growth factor-1 in Crohn’s disease: implications for therapeutic use of human growth hormone in pediatric patients. Current opinion in pediatrics. 2011;23(5):545–51. doi: 10.1097/MOP.0b013e32834a7810. [DOI] [PubMed] [Google Scholar]; This review summarizes the therapeutic efficacy of recombinant human growth hormone (rhGH) in reducing growth deficits in pediatric Crohn’s disease patients, but limited and inconsistent effects on clinical or mucosal measures of disease activity. The work highlights a need for a better understanding of the mechanisms mediating rhGH-induced growth and any potential benefits to reduced disease activity.
- 40.Wales PW, Nasr A, de Silva N, Yamada J. Human growth hormone and glutamine for patients with short bowel syndrome. Cochrane Database Syst Rev. 2010;(6):CD006321. doi: 10.1002/14651858.CD006321.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Goulet O, Dabbas-Tyan M, Talbotec C, et al. Effect of recombinant human growth hormone on intestinal absorption and body composition in children with short bowel syndrome. JPEN Journal of parenteral and enteral nutrition. 2010;34(5):513–20. doi: 10.1177/0148607110362585. [DOI] [PubMed] [Google Scholar]; This is a clinical study showing efficacy of recombinant human growth hormone (rhGH) in reducing need for parenteral nutrition in pediatric short bowel syndrome patients. Patients experienced a decreased need for parenteral nutrition, improved nutrient uptake, and increased energy balance; however some patients returned to parenteral nutrition after discontinuation of rhGH administration. Results highlight the need for further study of rhGH efficacy as a treatment for short bowel syndrome patients, and better understanding of mechanisms.
- 42.Buchman AL, Katz S, Fang JC, et al. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm Bowel Dis. 2010;16(6):962–73. doi: 10.1002/ibd.21117. [DOI] [PubMed] [Google Scholar]
- 43**.Jeppesen PB, Gilroy R, Pertkiewicz M, et al. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut. 2011;60(7):902–14. doi: 10.1136/gut.2010.218271. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a clinical study examining the effects of teduglutide (a glucagon-like peptide 2 analogue) on short bowel syndrome patients. The results show that teduglutide was well-tolerated, reduced the need for parenteral nutrition, and increased intestinal adaptive responses. This work emphasizes the need for further studies examining the effects of GLP2 and analogues in promoting intestinal adaptation following massive resection and limiting need for parenteral nutrition.
- 44**.Bell J, Parker KL, Swinford RD, et al. Long-term safety of recombinant human growth hormone in children. The Journal of clinical endocrinology and metabolism. 2010;95(1):167–77. doi: 10.1210/jc.2009-0178. [DOI] [PubMed] [Google Scholar]; This is a comprehensive, long-term study examining the safety of recombinant human growth hormone (rhGH) in over 50,000 pediatric patients. Findings revealed that rhGH did not increase risk of leukemia, but did increase risk of second malignancy in some populations. This work suggests that rhGH is a safe therapy for many children, but caution should be exercised in patients with prior malignancies.
- 45.Miller ME, Michaylira CZ, Simmons JG, et al. Suppressor of cytokine signaling-2: a growth hormone-inducible inhibitor of intestinal epithelial cell proliferation. Gastroenterology. 2004;127(2):570–81. doi: 10.1053/j.gastro.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 46.Michaylira CZ, Simmons JG, Ramocki NM, et al. Suppressor of cytokine signaling-2 limits intestinal growth and enterotrophic actions of IGF-I in vivo. American journal of physiology Gastrointestinal and liver physiology. 2006;291(3):G472–81. doi: 10.1152/ajpgi.00218.2005. [DOI] [PubMed] [Google Scholar]
- 47*.Newton VA, Ramocki NM, Scull BP, et al. Suppressor of cytokine signaling-2 gene disruption promotes Apc(Min/+) tumorigenesis and activator protein-1 activation. The American journal of pathology. 2010;176(5):2320–32. doi: 10.2353/ajpath.2010.090684. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows a protective role for SOCS2 (which is induced by GH and limits IGF1 action) in preventing spontaneous small intestine and colon tumors. These results indicate that induction of SOCS by trophic therapies may protect again dysplasia.
- 48.Theiss AL, Fuller CR, Simmons JG, et al. Growth hormone reduces the severity of fibrosis associated with chronic intestinal inflammation. Gastroenterology. 2005;129(1):204–19. doi: 10.1053/j.gastro.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 49**.Li Y, de Haar C, Chen M, et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59(2):227–35. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]; This study describes the loss of SOCS3 expression, as well as increases in IL-6 and pSTAT3 expressing cells in ulcerative colitis patients with dysplasia and cancer. These findings suggest a role for SOCS3 in protecting against abnormal cell proliferation and IL-6/STAT3 as mediators of dysplasia in the presence of inflammation.
- 50.Rigby RJ, Simmons JG, Greenhalgh CJ, et al. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26(33):4833–41. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- 51.Tamiya T, Kashiwagi I, Takahashi R, et al. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arteriosclerosis, thrombosis, and vascular biology. 2011;31(5):980–5. doi: 10.1161/ATVBAHA.110.207464. [DOI] [PubMed] [Google Scholar]
- 52.Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. American journal of physiology Gastrointestinal and liver physiology. 2011;300(5):G677–83. doi: 10.1152/ajpgi.00104.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dahly EM, Miller ME, Lund PK, Ney DM. Postreceptor resistance to exogenous growth hormone exists in the jejunal mucosa of parenterally fed rats. The Journal of nutrition. 2004;134(3):530–7. doi: 10.1093/jn/134.3.530. [DOI] [PubMed] [Google Scholar]
- 54*.Rowland KJ, Brubaker PL. The “cryptic” mechanism of action of glucagon-like peptide-2. American journal of physiology Gastrointestinal and liver physiology. 2011;301(1):G1–8. doi: 10.1152/ajpgi.00039.2011. [DOI] [PubMed] [Google Scholar]; This is a recent and comprehensive review of GLP2 action in the small intestine. Studies show that GLP2 aids in intestinal adaptation following injury by increasing proliferation and absorptive surface area. However, some studies have also shown that GLP2 increases colorectal cancer risk in sporadic models of colon cancer, highlighting the need for a thorough understanding of the downstream mediators of GLP2 action.
- 55.Dube PE, Forse CL, Bahrami J, Brubaker PL. The essential role of insulin-like growth factor-1 in the intestinal tropic effects of glucagon-like peptide-2 in mice. Gastroenterology. 2006;131(2):589–605. doi: 10.1053/j.gastro.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 56**.Rowland KJ, Trivedi S, Lee D, et al. Loss of Glucagon-Like Peptide-2-Induced Proliferation Following Intestinal Epithelial Insulin-Like Growth Factor-1-Receptor Deletion. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.09.014. [DOI] [PubMed] [Google Scholar]; This novel study shows that the GLP2-mediated intestinal growth requires the IGF1R and inducible IGF1R deletion has no impact on normal intestinal homesostasis. These findings underscore the importance of IGF1 signaling in mediating GLP2-induced intestinal growth, as well as a need for further understanding the roles of IGF1R in normal intestinal epithelial homeostasis.
- 57.Leen JL, Izzo A, Upadhyay C, et al. Mechanism of action of glucagon-like peptide-2 to increase IGF-I mRNA in intestinal subepithelial fibroblasts. Endocrinology. 2011;152(2):436–46. doi: 10.1210/en.2010-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dehmer JJ, Fuller MK, Helmrath MA. Management of pediatric intestinal failure. Advances in pediatrics. 2011;58(1):181–94. doi: 10.1016/j.yapd.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Ivory CP, Wallace LE, McCafferty DM, Sigalet DL. Interleukin-10-independent anti-inflammatory actions of glucagon-like peptide 2. American journal of physiology Gastrointestinal and liver physiology. 2008;295(6):G1202–10. doi: 10.1152/ajpgi.90494.2008. [DOI] [PubMed] [Google Scholar]
- 60.Simmons JG, Hoyt EC, Westwick JK, et al. Insulin-like growth factor-I and epidermal growth factor interact to regulate growth and gene expression in IEC-6 intestinal epithelial cells. Mol Endocrinol. 1995;9(9):1157–65. doi: 10.1210/mend.9.9.7491108. [DOI] [PubMed] [Google Scholar]
- 61.Estivariz CF, Ziegler TR. Nutrition and the insulin-like growth factor system. Endocrine. 1997;7(1):65–71. doi: 10.1007/BF02778066. [DOI] [PubMed] [Google Scholar]
- 62.Baricevic I, Jones DR, Nikolic JA, Nedic O. Gastrointestinal inflammation and the circulating IGF system in humans. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2006;38(1):22–7. doi: 10.1055/s-2006-924972. [DOI] [PubMed] [Google Scholar]
- 63.Corkins MR, Gohil AD, Fitzgerald JF. The insulin-like growth factor axis in children with inflammatory bowel disease. Journal of pediatric gastroenterology and nutrition. 2003;36(2):228–34. doi: 10.1097/00005176-200302000-00014. [DOI] [PubMed] [Google Scholar]
- 64.Justova V, Lacinova Z, Melenovsky V, et al. The changes of IGF binding proteins after rhGH administration to patients totally dependent on parenteral nutrition. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society. 2001;11(6):407–15. doi: 10.1054/ghir.2001.0257. [DOI] [PubMed] [Google Scholar]
- 65.Ziegler TR, Mantell MP, Chow JC, et al. Intestinal adaptation after extensive small bowel resection: differential changes in growth and insulin-like growth factor system messenger ribonucleic acids in jejunum and ileum. Endocrinology. 1998;139(7):3119–26. doi: 10.1210/endo.139.7.6097. [DOI] [PubMed] [Google Scholar]
- 66.Gallagher EJ, LeRoith D. Minireview: IGF, Insulin, and Cancer. Endocrinology. 2011;152(7):2546–51. doi: 10.1210/en.2011-0231. [DOI] [PubMed] [Google Scholar]
- 67.Fruchtman S, Simmons JG, Michaylira CZ, et al. Suppressor of cytokine signaling-2 modulates the fibrogenic actions of GH and IGF-I in intestinal mesenchymal cells. American journal of physiology Gastrointestinal and liver physiology. 2005;289(2):G342–50. doi: 10.1152/ajpgi.00413.2004. [DOI] [PubMed] [Google Scholar]
- 68.Mahavadi S, Flynn RS, Grider JR, et al. Amelioration of excess collagen IalphaI, fibrosis, and smooth muscle growth in TNBS-induced colitis in IGF-I(+/−) mice. Inflamm Bowel Dis. 2011;17(3):711–9. doi: 10.1002/ibd.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Backeljauw P, Bang P, Clayton PE, et al. Diagnosis and management of primary insulin-like growth factor-I deficiency: current perspectives and clinical update. Pediatric endocrinology reviews : PER. 2010;7(Suppl 1):154–71. [PubMed] [Google Scholar]
- 70*.Thayu M, Denson LA, Shults J, et al. Determinants of changes in linear growth and body composition in incident pediatric Crohn’s disease. Gastroenterology. 2010;139(2):430–8. doi: 10.1053/j.gastro.2010.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a clinical study examining the associations between linear body growth and composition and plasma TNFα, IL-6, and lipopolysaccride binding protein (LBP), as well as growth factors in Crohn’s disease patients. Decreases in TNFα, IL-6, and LBP, as well as increases in IGF1 were associated with increases in growth and body composition. Results underscore a need for trophic therapies that also have anti-inflammatory effects.
- 71.Frasca F, Pandini G, Sciacca L, et al. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Archives of physiology and biochemistry. 2008;114(1):23–37. doi: 10.1080/13813450801969715. [DOI] [PubMed] [Google Scholar]
- 72.Belfiore A, Genua M, Malaguarnera R. PPAR-gamma Agonists and Their Effects on IGF-I Receptor Signaling: Implications for Cancer. PPAR research. 2009;2009:830501. doi: 10.1155/2009/830501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73**.Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocrine-related cancer. 2011;18(4):R125–47. doi: 10.1530/ERC-11-0074. [DOI] [PubMed] [Google Scholar]; This is an excellent, comprehensive review comparing the role of the insulin receptor isoforms (IR-A and IR-B) with IGF1R in growth and cancer of many organs. This work provides a detailed discussion of the putative role for IR-A in promoting growth and underscores the importance of understanding this novel isoform in the intestine.
- 74.Giudice FS, Dal Vechio AM, Abrahao AC, et al. Different expression patterns of pAkt, NF-kappaB and cyclin D1 proteins during the invasion process of head and neck squamous cell carcinoma: an in vitro approach. Journal of oral pathology & medicine : official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2011;40(5):405–11. doi: 10.1111/j.1600-0714.2010.00960.x. [DOI] [PubMed] [Google Scholar]
- 75.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Molecular and cellular biology. 1999;19(5):3278–88. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gniuli D, Castagneto-Gissey G, Iaconelli A, et al. Fat mass largely contributes to insulin mediated glucose uptake in morbidly obese subjects. Int J Obes (Lond) 2010;34(12):1726–32. doi: 10.1038/ijo.2010.99. [DOI] [PubMed] [Google Scholar]
- 77.Verdam FJ, Greve JW, Roosta S, et al. Small intestinal alterations in severely obese hyperglycemic subjects. The Journal of clinical endocrinology and metabolism. 2011;96(2):E379–83. doi: 10.1210/jc.2010-1333. [DOI] [PubMed] [Google Scholar]