

Abstract
Background
Mutations in SPINK5, encoding the serine protease inhibitor LEKTI, cause Comèl-Netherton syndrome, an autosomal-recessive disease characterized by congenital ichthyosis, bamboo hair, and atopic diathesis. Despite increased frequency of infections, the immunocompetence of Comèl-Netherton syndrome patients has not been extensively investigated.
Objective
To define Comèl-Netherton syndrome as a primary immunodeficiency and to explore the benefit of IVIG replacement therapy.
Methods
We enrolled nine patients with Comèl-Netherton syndrome, sequenced SPINK5, and analyzed LEKTI expression by immunohistochemistry. Immune function was assessed by measuring cognate immunity, serum cytokine-levels and natural killer cell cytotoxicity.
Results
All patients presented with recurrent skin infections caused predominantly by Staphylococcus aureus. All but one reported recurrent respiratory tract infections; 78% had sepsis and/or pneumonia; 67% suffered from recurrent gastroenteritis and failure to thrive. Mutations in SPINK5 – including six novel mutations- were identified in eight patients. LEKTI expression was decreased or absent in all patients.
Immunologic evaluation revealed reduced memory B cells and defective responses to vaccination with Pneumovax® and bacteriophage phiX174, characterized by impaired antibody amplification and class-switching. Immune dysregulation was suggested by a skewed TH1-phenotype and elevated proinflammatory cytokine levels, while serum concentrations of the chemokine RANTES and NK cell cytotoxicity were decreased.
Treatment with intravenous immunoglobulin substitution resulted in remarkable clinical improvement and temporarily increased NK cell cytotoxicity.
Conclusion
These data provide new insights into the immunopathology of Comèl-Netherton syndrome and demonstrate that this multisystem disorder, characterized by lack of LEKTI expression in epithelial cells, is complicated by cognate and innate immunodeficiency that responds favorably to IVIG therapy.
Keywords: Comèl-Netherton Syndrome, SPINK5, LEKTI, immune deficiency, NK cell cytotoxicity, selective antibody deficiency, IVIG, ichthyosis, bamboo hair, atopic diathesis
Introduction
Comèl-Netherton syndrome is a rare autosomal recessive disease characterized by congenital ichthyosis, trichorrhexis invaginata (bamboo hair), and atopic diathesis(1–4), with a 20% fatality rate in the first year of life.(5)
Patients present shortly after birth with generalized rashes that develop into severe ichthyosis.(1)Bamboo hair is pathognomonic indicating a structural defect of the hair shaft.(6) A broad spectrum of atopic diseases includes eczema, reactive airway disease, food allergy, and angioedema.(3, 7, 8) Enteropathy, failure to thrive, hypernatremia, hypoalbuminemia, aminoaciduria, developmental delay, and recurrent infections have been reported.(5, 7–10)
Most patients have mutations in the SPINK5 gene, located on chromosome 5q32, resulting in a loss or reduced expression of the multi-domain serine protease inhibitor LEKTI (lymphoepithelial Kazal-type-5 serine protease inhibitor).(4, 11) LEKTI has been proposed to negatively regulate desquamation and matrix maturation.(12) LEKTI is expressed by epithelial cells of skin, mucosae, and Hassall’s corpuscles(13) raising the possibility that LEKTI affects T-cell maturation.
Several previous studies recognized the increased infection rate and postulated an underlying immune defect, but reported findings were not consistent with a well-defined immune deficiency.(7–10, 14) Thus, Comèl-Netherton syndrome is generally not listed as a primary immunodeficiency disorder.(15, 16)
We studied nine patients with Comèl-Netherton syndrome for SPINK5 mutations, LEKTI expression, and immune abnormalities. Our results strongly suggest that Comèl-Netherton syndrome is a multisystem disorder associated with dysfunctional innate and cognate immunity.
Methods
Subjects
Nine unrelated children (three girls and six boys; age 6 months to 9 years) with diverse ethnic backgrounds were enrolled. Institutional Review Board approval and informed consent were obtained. Diagnostic criteria for Comèl-Netherton syndrome included the presence of congenital ichthyosis, bamboo hair, elevated serum IgE levels, allergies, mutations in SPINK5, and/or decreased or absent LEKTI expression by skin and/or buccal mucosal epithelial cells. None of the patients had systemic infections, or received systemic steroids or immunosuppressive treatment, for at least four weeks prior to immunological evaluation.
SPINK5 mutations
DNA was prepared from heparinized blood using QIAamp DNA Mini Kit (QIAGEN, Valencia, CA). The 33 exons of the SPINK5 gene including the intron-exon boundaries, the proximal promoter region (1000 bp upstream of the first exon) and the first polyadenylation-site were sequenced using the Big Dye Terminator kit (Applied Biosystems, Foster City, CA) and analyzed with a 3730xl DNA Analyzer (Applied Biosystems) as previously described.(17) Mutations are reported as recommended.(18) Primer sequences are available upon request.
Immunologic assessment
White blood cell and differential counts, lymphocyte subsets, serum immunoglobulin levels and lymphocyte proliferation to mitogens and specific antigens were analyzed using standard protocols. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque™ plus (Bioscience AB, Uppsala, Sweden). Lymphocyte subsets were identified by multicolor flow cytometry(19, 20) using the following conjugated monoclonal antibodies: anti-CD27-APC, anti-CD31-Biotin, anti-CD8-Alexa700 (eBioscience, San Diego, CA), anti-CD45RA-FITC, anti-IgD-Biotin, anti-CD19-ChyChr (BD Bioscience, San Jose, CA), anti-CD38-FITC, anti-CD4-CyC5 (Immunotech, Fullerton, CA), anti-IgM-PE (Southern Biotechnology, Birmingham, Alabama), and steptavidine APC-Cy7 and PE-Cy7 (eBioscience). Regulatory T cells (CD4+CD25+FOXP3+) were assessed with anti-CD4-PE-Cy5/CD25-PE cocktail and Alexa Fluor 488 conjugated anti-FOXP3 monoclonal antibody after exposure to fix/perm solution (all of BioLegend, San Diego, CA). Samples were measured on an LSRII (BD Bioscience) and analyzed with FlowJo (TreeStar, Ashland, OR). The TCR β variable (TCRBV) gene repertoire on CD4+ cells was determined with a panel of 22 antibodies.(21) Immunization with bacteriophage phiX174 was performed following a previously described protocol.(22) Serum cytokines were measured with the Luminex 100 system using the Human Cytokine Twenty-Five-Plex Antibody Bead Kit (BioSource International, Inc., Camarillo, CA). NK cell cytotoxicity of ficoll-hypaque isolated PBMCs was evaluated by 51Cr-release assay using K562 erythroleukemia target cells.(23)
LEKTI expression
Buccal mucosa epithelial cells were collected with a Cytobrush Plus GT (Medscand Medical AB, Malmoe, Sweden), spread on glass slides, fixed with acetone, permeabilized with 0.1% Triton-X 100 (Boehringer Mannheim, Mannheim, GER) and 0.5% H2O2, and stained with anti-LEKTI monoclonal antibody (Zymed Laboratories, Inc., San Francisco, CA). Peroxidase-based immunohistochemical staining was performed with the Elite ABC Kit (Vector Laboratories, Burlingame, CA) using aminoethylcarbazol substrate-chromogen (DakoCytomation, Carpinteria, CA). After counterstaining with hematoxylin (Sigma-Aldrich, St. Loiuse, MO), two hundred cells from each subject were evaluated microscopically for LEKTI expression. Paraffin sections of tissues were similarly stained after pretreatment with CitriSolv (Decon Labs, Inc., King of Prussia, PA) and ethanol.
Statistics
Statistical analyses of responses to phiX174 were performed on log-transformed control K values (KV) that are expressed as geometric means and 95% confidence limit. Cytokine levels of patients and 16 age matched controls are shown as medians with interquartile ranges and analyzed with the non-parametric Mann-Whitney U Test. A two-sided p-value < 0.01 was considered significant. For NK cell cytotoxicity, serum immunoglobulin levels and LEKTI expression patient results were compared to the arithmetic mean +/−2SD of controls; lymphocyte subset values were compared to the geometric mean and the 95% confidence limit of normal control data. The student t-test was used to compare control versus patient group.
Results
Clinical presentation
The clinical triad of Comèl-Netherton syndrome - congenital ichthyosis, bamboo hair, and allergic diathesis - was found in all patients except one who lacked bamboo hair (Table 1). Recurrent or persistent Staphylococcus aureus skin infections, frequently Methicillin-resistant, occurred in all patients once skin lesions had developed.
Table 1.
Clinical findings and gene analysis in nine patients with Comèl-Netherton Syndrome
| Patient # | Age12 (years) | Sex | Congenital Ichthyosis |
Bamboo hair | Atopic manifestations |
Recurrent or persistent infections of: |
Severe invasive infections11 |
Additional findings |
Mutation in the gene SPINK5 |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Skin | Gastro-intestinal tract | Respiratory tract | |||||||||
| 1 | 0.5 | F | Yes | Yes | Eczema, food allergy | Yes1–3 | YesND | Yes3 | Sepsis1,5 | Hypernatremic dehydration, failure to thrive | Compound heterozygous 1) c.354_357delTTGT C119AfsX25 (exon 5)* 2) IVS 15 + 13 G > A10 |
| 2 | 3 | M | Yes | No | Eczema | Yes1–3,8 | YesND | Yes1,3 | Pneumonia, sepsis1,8 | Hypernatremic dehydration, failure to thrive | Homozygous c.1346_1352insT C451LfsX5 (exon 15)* |
| 3 | 6 | M | Yes | Yes | Rhinitis, food allergy, eczema, asthma | Yes1–4 | Yes6 | Yes1,3,7 | Pneumonia | Failure to thrive | Homozygous c.2459-2468delA K823RfsX100 (exon 26) |
| 4 | 9 | M | Yes | Yes | Rhinitis, food allergy, angioedma, egg-induced anaphylaxis, eczema, asthma | Yes1, | Yes6 | Yes1,3 | Sepsis1 | Hypernatremic dehydration | Compound heterozygous: 1) IVS 15 + 13 G > A10 2) not determined |
| 5 | 0.5 | M | Yes | Yes | Eczema, food allergy | Yes1–5 | YesND | Yes1 | Sepsis1,9 | Failure to thrive | Compound heterozygous 1) c.377-8delAT Y126X (exon 5) 2) c.2473-4delGA E825GfsX1 (exon 26)* |
| 6 | 7 | M | Yes | Yes | Rhinitis, eczema | Yes1,3 | No | YesND | No | No | Homozygous c.2459-2468delA K823RfsX100 (exon 26) |
| 7 | 6 | M | Yes | Yes | Rhinitis, food allergy, eczema | Yes1 | YesND | YesND | Pneumonia, sepsis1,13 | Cardiomyopathy with acute heart failure, failure to thrive | No mutation identified |
| 8 | 1 | F | Yes | Yes | Eczema, food allergy | Yes3 | No | No | No | Hypernatremic dehydration, failure to thrive | Compound heterozygous 1) IVS5 + 1 G > A 2) c.2098G > T G700X (Exon 22)* |
| 9 | 7 | F | Yes | Yes | Rhinitis, food allergy | YesND | No | YesND | Pneumonia | No | Compound heterozygous 1) IVS4 + 1 G > A* 2) IVS8 + 1 G > A* |
| Percent of patients positive for symptom of column | 100% | 89% | 100% | 100% | 67% | 89% | 78% | 78% | – | ||
Infectious agents
Stapylococcus aureus
Stapylococcus aureus Methicillin resistant (MRSA)
Pseudomonas aeruginosa
Streptococcus viridans
Klebsiella oxytoca
Rotavirus
Proteus mirabilis
Enterococcus supp.
Salmonella cholerae-suis
putative splice site upstream (see Raghunath et al. 24)
defined by requiring i.v. antibiotic treatment
age when studied
Acinetobacter
novel mutation
no identification of pathogens
All but one patient had histories of recurrent upper and lower respiratory tract infections, most frequently recurrent otitis media and/or externa. Four patients suffered recurrent lower respiratory tract disease, including multiple episodes of pneumonia and allergy or infection related respiratory distress. Four infants developed Staphylococcus aureus sepsis with one also having Salmonella cholerae-suis sepsis. At 6 years of age, patient #7 developed acute heart failure with cardiomyopathy possibly associated with recurrent Staphylococcus aureus sepsis. He recovered after removal of a central line, presumably the nidus for infection. Polymerase chain reaction failed to detect common viruses during heart failure. Overall, Staphylococcus aureus was the most frequent infectious argent, followed by Pseudomonas aeruginosa and Klebsiella oxytoca.
The majority of our patients suffered recurrent acute gastroenteritis resulting in failure to thrive and requiring repeated hospitalization. Three patients developed hypernatremic dehydration, and three needed short-term parenteral nutrition. No patient had clinical signs of autoimmunity such as hemolytic anemia or thrombocytopenia, arthritis or vasculitic skin lesions.
Mutations of SPINK5 and LEKTI expression
SPINK5 mutations, including six novel mutations, were identified in all but patient #7 (Table 1). Most mutations were located in the coding region and included short deletions or insertions of up to four base pairs resulting in frame shift and early termination of translation. Of the four intronic mutations, three (including two novel) were found at highly conserved intron-exon boundaries. The intronic mutation IVS15+13a > g, observed in two patients, has been previously described and creates an alternative splice site leading to premature termination within exon 16.(24) Two unrelated patients (#3 and #6), both of Polynesian origin but from different Pacific islands, had an identical homozygous mutation in exon 26 suggesting a founder effect in the Polynesian population. Patient #2 who is homozygous for a single nucleotide insertion is the only patient with a family history of consanguinity.
LEKTI protein expression was absent or present as small immunoreactive foci in fewer than 2% of epithelial cells from skin biopsies and/or buccal mucosa from all nine patients, including one (#4) with a detectable mutation in only one allele and one patient (#7) without detectable SPINK5 mutations. In contrast, controls [n=20] had a mean of 43% LEKTI-positive cells (95%-confidence limit: 21%-64%) which showed diffuse cytoplasmic staining. Heterozygous parents of three patients showed a mean of 42% (range 40%-49%) LEKTI-positive buccal mucosa cells.
Immunological studies
Except for eosinophilia, leukocyte and differential counts were normal. There were no significant alterations of T, B, and NK cell numbers. The TCRBV repertoire was normally distributed in both patients investigated, and the percentage of recent thymic emigrant cells (CD4+CD31+CD27+CD45RA+) was within the 95% confidence limit of healthy controls in six patients investigated (data not shown).
As a group the geometric mean of NKT cells (CD3+CD56+; patient geometric mean 11.0% of lymphocytes; control geometric mean 4.0% with 95% confidence limit 1.4%-10.7%) were significantly increased (p < 0.001). The geometric mean of unswitched memory B cells (CD19+CD27+IgM+IgD+) were significantly decreased (p < 0.0001) in the patient group compared to controls (patient geometric mean 3.0% of CD19+ cells, control geometric mean 8.7%; 95% confidence interval 2.6%-29.3%) with three of seven patients below the 95% confident limit. Similarly, switched memory B cells (CD19+CD27+IgM−IgD−) were decreased (p < 0.01) with two of seven patients being below the 95% confident limit (patient geometric mean 4.5% of CD19+ cells, control geometric mean 7.3 %; 95% confidence interval 2.2%-24.0%). Mean percentages of γδ-T cells and Tregs (CD4+CD25+FOXP3+) were within the 95% confidence limit of healthy controls (data not shown).
Lymphocyte proliferation to mitogens and antigens was normal and random serum antibody titers to tetanus and diphtheria were protective in the six patients investigated (data not shown). NADPH oxidase activity of neutrophils by dihydrorhodamine (DHR) studies was normal in the four patients evaluated (data not shown).
Serum IgE were significantly elevated in all and IgM and IgA in three and four patients, respectively (Figure 1A). Serum IgG was elevated in two and reduced in one patient; IgG subclasses were normal in the five patients studied (data not shown).
Figure 1. Immunological assessment of patients with Comèl-Netherton Syndrome.
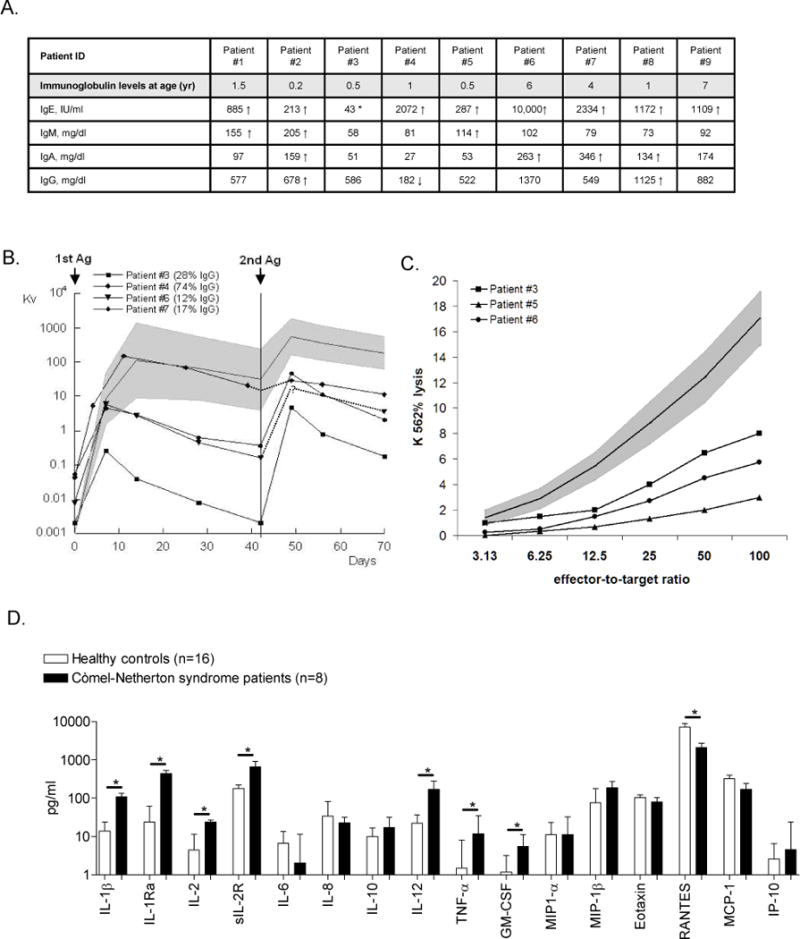
Panel A: Serum immunoglobulin levels measured before IVIG was started; arrows indicate 2SD below (↓) or above (↑) age matched control value.(30) (*) Patient #3 had a serum IgE level of 1119 IU/ml (2SD above age matched controls) at 6 years of age.
Panel B: Decreased antibody responses following primary and secondary immunization with the neoantigen, bacteriophage phiX174, which was injected twice six weeks apart. Neutralizing antibody titres were determined in serially obtained serum samples and expressed as rate of phage inactivation or K value (Kv)22 plotted on a log scale (solid line in gray area, geometric mean and 95% confidence limit measured in 50 normal controls). The mean percentage of phage-specific IgG antibody in serum collected two weeks after the second immunization was identified as being resistant to treatment with 2-Mercaptoethanol (2-ME) (values of 50 normal controls: 48% +/− 23% 1SD). For patient #6, only one sample was collected post-secondary immunization.
Panel C: NK cell cytotoxicity of ficoll-hypaque isolated PBMCs against K562 cells measured before IVIG treatment. The mean percent of lysis observed in at least three independent experiments preformed in each of the three patients studied (solid line in gray area, mean and +/− 2SD measured in 18 normal controls).
Panel D: Median ± inter quartile ranges (IQR) of serum cytokine levels observed in eight patients with Comèl-Netherton syndrome (filled columns) are compared to those of sixteen age-matched normal controls (open columns); * p-values < 0.01. Median values of IL-4, IL-5, IL-7, IL-13, IL-15, IL-17, INF-α, INF-γ, and MIG [monokine induced by interferon gamma] were below detection limit in the majority of patients and control subjects (data not shown).
Primary and/or secondary antibody responses to bacteriophage phiX174 were quantitatively depressed in all four patients studied, with impaired isotype switching in three (Figure 1B). Protective anti-pneumoccocal polysaccharide antibody titers (protection defined as ≥ 1 ug/ml) in random samples were reduced in five patients tested (range 0-36% of up to 12 serotypes tested, mean 21%). Both patients immunized with Pneumovax® showed poor responses: patient #6 responded to 3 of 14 and patient #7 to 3 of 12 serotypes tested.
Although the absolute numbers of NK cells (CD3−CD56+) were normal or elevated in all patients, NK-cell cytotoxicity, measured in at least thee independent experiments in three patients prior to monthly IVIG infusion, was consistently below the 95% confidence limit of healthy controls.
Serum analysis revealed significantly increased pro-inflammatory [IL-1β, IL-12, TNF-α, GM-CSF, IL-1 receptor antagonist (IL-1Ra)] and anti-inflammatory cytokines [IL-2 and soluble IL-2 receptor (sIL-2R)] compared to age-matched controls (Figure 1D). In both patients and controls, TH2-associated cytokines IL-4 and IL-5 were undetectable or detected at low amounts (data not shown).Only the chemokine RANTES (Regulated on Activation, Normally T cell Expressed and Secreted) was significantly diminished in patients compared to controls (Figure 1D).
Response to IVIG therapy
Symptomatic treatment with allergy control, moisturizers, steroid creams, and antibiotics had uniformly limited effect. Intravenous immunoglobulin (IVIG) replacement therapy (0.4 g/kg/month) was initiated because of abnormal antibody responses to bacteriophage in the four patients studied (Pts. #3, 4, 6, 7; Figure 1B) and in one additional patient (#5) because of severe failure to thrive. All five families reported remarkable clinical benefit including decreased inflammation and itching of the skin, thicker hair with less breaking of hair shafts and healthier scalps than observed with conventional treatment. Most dramatic improvement occurred in patient #5, the youngest and most severely affected child (Figure 2). In response to a survey on the utility of IVIG, parents of the five children treated estimated that the number of missed school days and doctors’ visits was reduced, infections were lessened, and their overall assessment of quality of life improved. Over two years’ observation on IVIG, patient #3 advanced from the 3rd to the 5th percentile for height and from the 50th to the 75th percentile in weight. One year after starting IVIG, patient #5, whose height and weight were far below the 3rd percentile is approaching the 3rd percentile for both parameters. Patient #6 is reaching the 3rd percentile for height and his weight increased from the 50th to the 90th percentile 6 months after IVIG was started.
Figure 2.
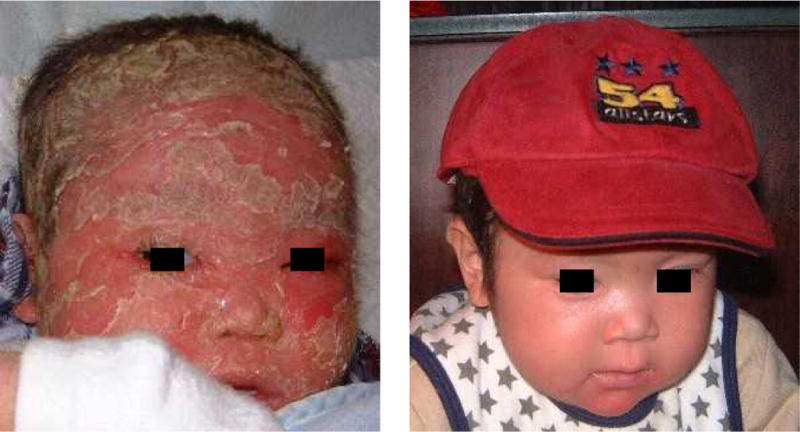
Toddler (patient #5) with Comèl-Netherton Syndrome before and three months after IVIG treatment.
To evaluate objectively the impact of IVIG therapy on the immune system, we explored the effect on NK cell function prior to, immediately after, and six to seven days after a monthly infusion. NK cell cytotoxicity has been reported to increase after IVIG therapy.(25) We found that NK cell cytotoxicity did not improve immediately after IVIG infusion but was temporarily restored six to seven days later to the normal range in patient #3 and increased by 80% in patient #6. In patient #5, there was no baseline NK cell cytotoxicity detectable prior to receiving monthly IVIG infusion but 6 days post infusion, NK cell cytotoxicity was present, although at reduced intensity.
Discussion
The initial presentation of Comèl-Netherton syndrome is early onset severe chronic skin disease requiring expert dermatologic care. However, Comèl-Netherton syndrome has been recognized not only as a disorder of skin and hair but also as a complex systemic disease including atopic diathesis, recurrent infections, failure to thrive, and high fatality rate in early childhood.(3, 5)
The observation of recurrent infections has suggested an immune defect which led to limited investigations.(7–10, 14, 16) Findings, however, were too inconsistent to define Comèl-Netherton syndrome as a primary immunodeficiency disease, and is generally not listed as such.(15) Unlike earlier reports that estimated a 30% rate of recurrent infections(7, 8) we observed this complication in all of our patients. Bacterial infections involving the skin, respiratory and gastrointestinal tract often resulted in severe failure to thrive and were life threatening in 78% (Table 1). Infections included neonatal sepsis as reported by others.(9, 26, 27) Cardiomyopathy, which developed in one of our patients following Staphylococcus aureus sepsis, is a complication with unknown etiology, reported previously in two patients.(28)
All patients with sepsis were culture positive for Staphylococcus aureus, a major pathogen present in normal skin flora. Disruption of the skin and gut barrier in Comèl-Netherton syndrome likely contributes to susceptibility to Staphylococcus aureus skin infections and sepsis, and salmonella sepsis, respectively.(29) However multiple immune defects reported here, from antibody deficiency to impaired NK cell function, and the favorable response to IVIG treatment imply that a defective skin barrier cannot entirely explain the susceptibility to infections.
The consistently abnormal antibody responses to bacteriophage are similar to those observed in other primary immunodeficiencies affecting both cognate(30–33) and innate(34, 35) immunity suggesting abnormal T or B cell development or defective costimulatory signaling resulting in reduced isotype switching and defective immunologic memory.
As a group, memory B cells, especially IgM+ memory B cells, were significantly decreased compared to a normal control group. IgM+ memory B cells, also referred to as unswitched memory B cells or splenic marginal zone B cells, play a role in defense against encapsulated bacteria,(36) supported by the finding that splenectomized/asplenic patients have reduced numbers of IgM+ memory B cells and decreased responses to Pneumovax®.(37) As reported by others,(7, 14) protective type-specific anti-polysaccharide antibodies were reduced in the five patients tested and the two patients challenged with Pneumovax® failed to respond to most serotypes.
Reports of skin cancer in young adults with Comèl-Netherton syndrome(38–40) raised consideration of defective cellular immunity. None of our pediatric patients, all younger than 10 years of age, developed a malignancy, nor was a clearly defined T-cell defect demonstrated. Although LEKTI is expressed in normal Hassall’s corpuscles,(13) those patients tested (n=2) had normal TCRBV repertoires and all but one exhibited normal thymic output judged by the presence of a normal circulating pool of recent thymic emigrant cells (CD4+CD31+CD27+CD45RA+). Increased T-cell activation is suggested by elevated serum levels of pro-inflammatory cytokines with compensatory elevated anti-inflammatory cytokines. There was no skewing towards TH2-cytokines unlike other diseases associated with allergies and elevated serum IgE.(41)
RANTES, the only chemokine significantly decreased in patient sera, is secreted by T cells, monocytes and especially NK cells activated in the context of FcR engagement upon antibody exposure.(41, 42) Since NK cell numbers in the peripheral blood were normal in all but one patient, reduced levels of RANTES cannot be accounted for by quantitatively low NK cells. More likely, RANTES deficiency results from the decreased NK cell function observed in all patients studied. Reduced RANTES levels may directly effect NK cell migration preventing NK cells from reaching their destination in peripheral lymphoid tissue where final maturation occurs.(43)
NK cells are critical in host defense and impaired NK-cell function has been observed in several primary immunodeficiencies.(44) The combination of a NK cell and specific antibody deficiency is exemplified in the Wiskott-Aldrich syndrome (WAS). Increased susceptibility to infections and malignancies observed in WAS patients(23, 45) are also characteristic of Comèl-Netherton syndrome. Unlike in WAS, the diminished NK cytotoxicity we observed in our patients seems not to be caused by an intrinsic cellular defect, but may reflect impaired NK cell maturation that typically requires contact with epithelial cells.(46) Abnormal epithelium caused by LEKTI deficiency may result in aberrant NK cell-epithelial cell interaction. Although the mechanisms by which LEKTI mutations result in the Comèl-Netherton phenotype are unknown, the strong LEKTI expression in normal epidermis and epithelial cells within tonsils and thymus (Figure 3) suggests that LEKTI secondarily affects development of the cognate immune system, underscoring the critical link between epithelial cells and the development and function of the immune system.
Figure 3. Expression of LEKTI in different human tissues.
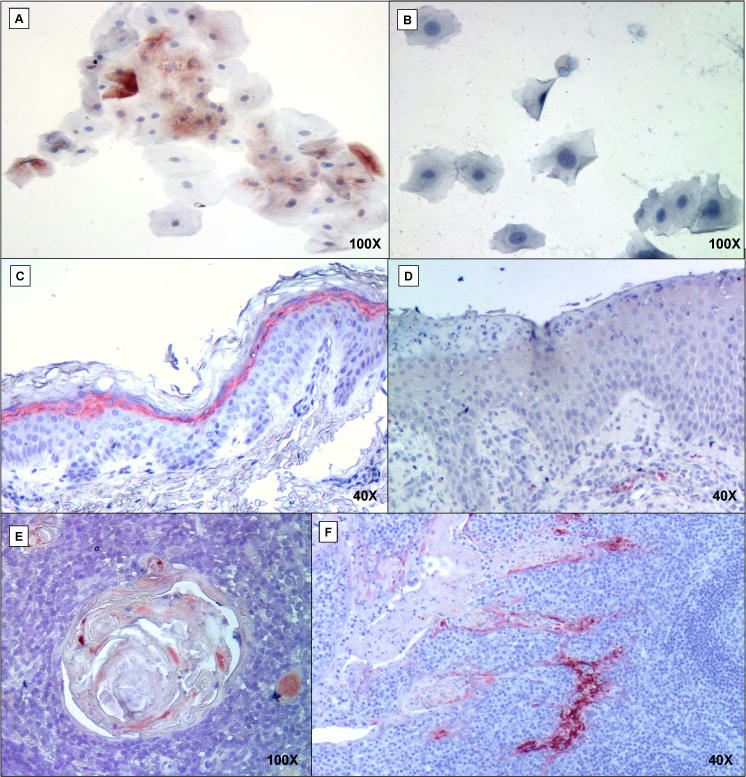
Immunohistochemistry counterstained with hematoxylin shows LEKTI protein in red, if present. Panel detail: buccal mucosal cells of healthy individual (A) and patient #3 (B); skin biopsies of healthy child with LEKTI expression in superficial epidermal squamous layer (C) and patient #4 (D) lacking LEKTI; LEKTI expression in Hassall’s corpuscles (E) and tonsillar crypts (F) of normal controls.
The decision to initiate IVIG treatment was based on the observed abnormal antibody responses to bacteriophage and Pneumovax®. Monthly IVIG infusions may improve the clinical course of Comèl-Netherton syndrome by providing high affinity neutralizing and opsonizing antibody required for phagocytosis and killing of bacteria. In addition IVIG is known to reduce inflammation in patients with chronic inflammatory disorders(47) and to increase NK cytotoxicity temporarily.(25) Although regular IVIG infusions at a dose recommended for replacement therapy in primary immunodeficient patients (0.4g/kg/months) benefited all five patients treated to date, only controlled clinical trials will define the role of IVIG treatment in Comèl-Netherton syndrome.
Acknowledgments
We thank the patients, their families and their physicians especially Uwe Ermer, MD, Annette Jansson, MD, and Felicitas Nagel, MD for their contributions, Kathey Mohan and Theresa Gettmann for their help with patient care, Stephanie Anover-Sombke, Qili Zhu, MD, Arumugam Jayakumar, PhD, and Vitaliy Starosta, PhD for methodical assistance, data acquisition and critical discussions. We thank Philip Fleckman, MD, Principal Investigator National Registry for Ichthyosis and Related Disorders who is supported by University of Washington General Clinical Research Center, NIH M01-RR-00037 and the Foundation for Ichthyosis and Related Skin Types and the Pachyonychia Congenita Fund.
Funding:
This work was supported by the Fritz Thyssen Foundation (Az. 10.07.1.159), the American Academy of Allergy, Asthma and Immunology’s Strategic Training in Allergy Research Award 2007 (EDR), NIH grants AI-063267 (TRT), AI-47040 and AI 54843 (MLM), AI-067946 and AI-079731 (JSO), HD017427 (HDO) and support from the Jeffrey Modell Foundation (HDO and JSO).
Abbreviations
- SPINK5
serine protease inhibitor Kazal-type-5
- LEKTI
lymphoepithelial Kazal-type-5 serine protease inhibitor
- IVIG
Intravenous immunoglobulin
- NK cell
Natural killer cell
- NKT cell
Natural killer T cell
- RANTES
Regulated on Activation, Normally T cell Expressed and Secreted
- TCRBV repertoire
T cell receptor ß-chain variable repertoire
- WAS
Wiskott-Aldrich Syndrome
Footnotes
Clinical implications
Comèl-Netherton Syndrome, a multi-system disorder, is associated with abnormal antibody responses and decreased NK cell function. IVIG substitution reduces infections, increases NK cell function and improves quality of life
References
- 1.Comel M. Ichthyosis linearis circumflexa. Dermatologica. 1949;98(3):133–6. [PubMed] [Google Scholar]
- 2.Netherton EW. A case for diagnosis. AMA Arch Derm Syphilol. 1951;64(1):84–5. [PubMed] [Google Scholar]
- 3.Wilkinson RD, Curtis GH, Hawk WA. Netherton’s Disease; Trichorrhexis Invaginata (Bamboo Hair), Congenital Ichthyosiform Erythroderma and the Atopic Diathesis. A Histopathologic Study Arch Dermatol. 1964;89:46–54. doi: 10.1001/archderm.1964.01590250052010. [DOI] [PubMed] [Google Scholar]
- 4.Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–2. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 5.Jones SK, Thomason LM, Surbrugg SK, Weston WL. Neonatal hypernatraemia in two siblings with Netherton’s syndrome. Br J Dermatol. 1986;114(6):741–3. doi: 10.1111/j.1365-2133.1986.tb04885.x. [DOI] [PubMed] [Google Scholar]
- 6.Netherton EW. A unique case of trichorrhexis nodosa; bamboo hairs. AMA Arch Derm. 1958;78(4):483–7. doi: 10.1001/archderm.1958.01560100059009. [DOI] [PubMed] [Google Scholar]
- 7.Smith DL, Smith JG, Wong SW, deShazo RD. Netherton’s syndrome: a syndrome of elevated IgE and characteristic skin and hair findings. J Allergy Clin Immunol. 1995;95(1 Pt 1):116–23. doi: 10.1016/s0091-6749(95)70159-1. [DOI] [PubMed] [Google Scholar]
- 8.Greene SL, Muller SA. Netherton’s syndrome. Report of a case and review of the literature. J Am Acad Dermatol. 1985;13(2 Pt 2):329–37. [PubMed] [Google Scholar]
- 9.Van Gysel D, Koning H, Baert MR, Savelkoul HF, Neijens HJ, Oranje AP. Clinico-immunological heterogeneity in Comel-Netherton syndrome. Dermatology. 2001;202(2):99–107. doi: 10.1159/000051607. [DOI] [PubMed] [Google Scholar]
- 10.Judge MR, Morgan G, Harper JI. A clinical and immunological study of Netherton’s syndrome. Br J Dermatol. 1994;131(5):615–21. doi: 10.1111/j.1365-2133.1994.tb04971.x. [DOI] [PubMed] [Google Scholar]
- 11.Hachem JP, Wagberg F, Schmuth M, Crumrine D, Lissens W, Jayakumar A, et al. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol. 2006;126(7):1609–21. doi: 10.1038/sj.jid.5700288. [DOI] [PubMed] [Google Scholar]
- 12.Jayakumar A, Kang Y, Henderson Y, Mitsudo K, Liu X, Briggs K, et al. Consequences of C-terminal domains and N-terminal signal peptide deletions on LEKTI secretion, stability, and subcellular distribution. Arch Biochem Biophys. 2005;435(1):89–102. doi: 10.1016/j.abb.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Magert HJ, Kreutzmann P, Standker L, Walden M, Drogemuller K, Forssmann WG. LEKTI: a multidomain serine proteinase inhibitor with pathophysiological relevance. Int J Biochem Cell Biol. 2002;34(6):573–6. doi: 10.1016/s1357-2725(01)00179-0. [DOI] [PubMed] [Google Scholar]
- 14.Stryk S, Siegfried EC, Knutsen AP. Selective antibody deficiency to bacterial polysaccharide antigens in patients with Netherton syndrome. Pediatr Dermatol. 1999;16(1):19–22. doi: 10.1046/j.1525-1470.1999.99005.x. [DOI] [PubMed] [Google Scholar]
- 15.Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120(4):776–94. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ming JE, Stiehm ER, Graham JM., Jr Syndromes associated with immunodeficiency. Adv Pediatr. 1999;46:271–351. [PubMed] [Google Scholar]
- 17.Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105(5):1881–90. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 18.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15(1):7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 19.Kimmig S, Przybylski GK, Schmidt CA, Laurisch K, Mowes B, Radbruch A, et al. Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med. 2002;195(6):789–94. doi: 10.1084/jem.20011756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agematsu K, Futatani T, Hokibara S, Kobayashi N, Takamoto M, Tsukada S, et al. Absence of memory B cells in patients with common variable immunodeficiency. Clin Immunol. 2002;103(1):34–42. doi: 10.1006/clim.2001.5197. [DOI] [PubMed] [Google Scholar]
- 21.Cossarizza A, Poccia F, Agrati C, D’Offizi G, Bugarini R, Pinti M, et al. Highly active antiretroviral therapy restores CD4+ Vbeta T-cell repertoire in patients with primary acute HIV infection but not in treatment-naive HIV+ patients with severe chronic infection. J Acquir Immune Defic Syndr. 2004;35(3):213–22. doi: 10.1097/00126334-200403010-00001. [DOI] [PubMed] [Google Scholar]
- 22.Ochs HD, Davis SD, Wedgwood RJ. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J Clin Invest. 1971;50(12):2559–68. doi: 10.1172/JCI106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orange JS, Ramesh N, Remold-O’Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci U S A. 2002;99(17):11351–6. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghunath M, Tontsidou L, Oji V, Aufenvenne K, Schurmeyer-Horst F, Jayakumar A, et al. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J Invest Dermatol. 2004;123(3):474–83. doi: 10.1111/j.0022-202X.2004.23220.x. [DOI] [PubMed] [Google Scholar]
- 25.Finberg RW, Newburger JW, Mikati MA, Heller AH, Burns JC. Effect of high doses of intravenously administered immune globulin on natural killer cell activity in peripheral blood. J Pediatr. 1992;120(3):376–80. doi: 10.1016/s0022-3476(05)80900-x. [DOI] [PubMed] [Google Scholar]
- 26.Hausser I, Anton-Lamprecht I. Severe congenital generalized exfoliative erythroderma in newborns and infants: a possible sign of Netherton syndrome. Pediatr Dermatol. 1996;13(3):183–99. doi: 10.1111/j.1525-1470.1996.tb01202.x. [DOI] [PubMed] [Google Scholar]
- 27.De Wolf K, Ferster A, Sass U, Andre J, Stene JJ, Song M. Netherton’s syndrome: a severe neonatal disease. A case report Dermatology. 1996;192(4):400–2. doi: 10.1159/000246431. [DOI] [PubMed] [Google Scholar]
- 28.Hoeger PH, Adwani SS, Whitehead BF, Finlay AY, Harper JI. Ichthyosiform erythroderma and cardiomyopathy: report of two cases and review of the literature. Br J Dermatol. 1998;139(6):1055–9. doi: 10.1046/j.1365-2133.1998.02565.x. [DOI] [PubMed] [Google Scholar]
- 29.Cork MJ, Robinson DA, Vasilopoulos Y, Ferguson A, Moustafa M, MacGowan A, et al. New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene-environment interactions. J Allergy Clin Immunol. 2006;118(1):3–21. doi: 10.1016/j.jaci.2006.04.042. quiz 22–3. [DOI] [PubMed] [Google Scholar]
- 30.Stiehm ER, Ochs HD, Winkelstein JA. Immunodeficiencey disorders: General considerations. In: Stiehm ER, Ochs HD, Winkelstein JA, editors. Immunologic Disorders in Infants & Childrens. 5. Vol. 2004. Philadelphia: Elsevier Saunders; pp. 289–355. [Google Scholar]
- 31.Seyama K, Nonoyama S, Gangsaas I, Hollenbaugh D, Pabst HF, Aruffo A, et al. Mutations of the CD40 ligand gene and its effect on CD40 ligand expression in patients with X-linked hyper IgM syndrome. Blood. 1998;92(7):2421–34. [PubMed] [Google Scholar]
- 32.Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, Wedgwood RJ. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55(2):243–52. [PubMed] [Google Scholar]
- 33.Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, et al. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol. 1998;161(8):3925–9. [PubMed] [Google Scholar]
- 34.Ochs HD, Nonoyama S, Zhu Q, Farrington M, Wedgwood RJ. Regulation of antibody responses: the role of complement and adhesion molecules. Clin Immunol Immunopathol. 1993;67(3 Pt 2):S33–40. [PubMed] [Google Scholar]
- 35.Day N, Tangsinmankong N, Ochs H, Rucker R, Picard C, Casanova JL, et al. Interleukin receptor-associated kinase (IRAK-4) deficiency associated with bacterial infections and failure to sustain antibody responses. J Pediatr. 2004;144(4):524–6. doi: 10.1016/j.jpeds.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, et al. Human blood IgM "memory" B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood. 2004;104(12):3647–54. doi: 10.1182/blood-2004-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruetzmann S, Rosado MM, Weber H, Germing U, Tournilhac O, Peter HH, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003;197(7):939–45. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folster-Holst R, Swensson O, Stockfleth E, Monig H, Mrowietz U, Christophers E. Comel-Netherton syndrome complicated by papillomatous skin lesions containing human papillomaviruses 51 and 52 and plane warts containing human papillomavirus 16. Br J Dermatol. 1999;140(6):1139–43. doi: 10.1046/j.1365-2133.1999.02892.x. [DOI] [PubMed] [Google Scholar]
- 39.Krasagakis K, Ioannidou DJ, Stephanidou M, Manios A, Panayiotides JG, Tosca AD. Early development of multiple epithelial neoplasms in Netherton syndrome. Dermatology. 2003;207(2):182–4. doi: 10.1159/000071791. [DOI] [PubMed] [Google Scholar]
- 40.Katugampola RP, Finlay AY, Harper JI, Dojcinov S, Maughan TS. Primary cutaneous CD30+ T-cell lymphoproliferative disorder following cardiac transplantation in a 15-year-old boy with Netherton’s syndrome. Br J Dermatol. 2005;153(5):1041–6. doi: 10.1111/j.1365-2133.2005.06839.x. [DOI] [PubMed] [Google Scholar]
- 41.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338(7):436–45. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 42.Roda JM, Parihar R, Lehman A, Mani A, Tridandapani S, Carson WE., 3rd Interleukin-21 enhances NK cell activation in response to antibody-coated targets. J Immunol. 2006;177(1):120–9. doi: 10.4049/jimmunol.177.1.120. [DOI] [PubMed] [Google Scholar]
- 43.Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol. 2007;179(1):89–94. doi: 10.4049/jimmunol.179.1.89. [DOI] [PubMed] [Google Scholar]
- 44.Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol. 2006;6(6):399–409. doi: 10.1097/ACI.0b013e3280106b65. [DOI] [PubMed] [Google Scholar]
- 45.Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–38. doi: 10.1016/j.jaci.2006.02.005. quiz 739. [DOI] [PubMed] [Google Scholar]
- 46.Cooley S, Xiao F, Pitt M, Gleason M, McCullar V, Bergemann TL, et al. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood. 2007;110(2):578–86. doi: 10.1182/blood-2006-07-036228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in autoimmune and chronic inflammatory disorders. Ann Pharmacother. 2007;41(5):812–23. doi: 10.1345/aph.1K037. [DOI] [PubMed] [Google Scholar]