

Abstract
Despite decades of research, primary brain tumors, gliomas, lack effective treatment options and present a huge clinical challenge. Particularly, the most malignant subtype, Glioblastoma multiforme, proliferates extensively and cells often undergo incomplete cell divisions, resulting in multinucleated cells. We now present evidence that multinucleated glioma cells result from the functional loss of transient receptor potential canonical 1 (TRPC1) channels, plasma membrane proteins involved in agonist-induced calcium entry and reloading of intracellular Ca2+ stores. Pharmacological inhibition or shRNA mediated suppression of TRPC1 causes loss of functional channels and store-operated calcium entry in D54MG glioma cells. This is associated with reduced cell proliferation and, frequently, with incomplete cell division. The resulting multinucleated cells are reminiscent of those found in patient biopsies. In a flank tumor model, tumor size was significantly decreased when TRPC1 expression was disrupted using a doxycycline inducible shRNA knockdown approach. These results suggest that TRPC1 channels play an important role in glioma cell division most likely by regulating calcium signaling during cytokinesis.
Keywords: TRPC channels, glioma, proliferation, multinucleation, calcium
INTRODUCTION
Gliomas account for 40% of all central nervous system tumors (Schwartzbaum et al., 2006) and 78% of malignant brain tumors. The most malignant Glioblastoma multiforme (GBM, grade IV), carry an extraordinarily poor prognosis with a median survival under 1 year and a 5-year survival of only 4% (Butowski et al., 2006). GBM tumors proliferate extensively (Cheng et al., 2009), and histological hallmarks include anaplastic cells, extensive proliferation, necrosis, angiogenesis, and nuclear atypia (Kleihues et al., 2002). The latter signifies mutlinucleated cells that have undergone incomplete cytokinesis and are a pathological diagnostic criterion for GBMs. Indeed, a subset of GBMs, ‘‘giant’’ cell glioblastomas, present almost exclusively with atypical, large, multinucleated cells (Palma et al., 1989).
Because proliferation is the principle process driving tumor expansion, much research has focused on studying proteins involved in cell-cycle progression (Martin and Hussaini, 2005). Over the past decade, ion channels have been added to the list of molecular candidates involved in normal and aberrant cell proliferation (Kunzelmann, 2005), particularly channels that flux Ca2+ (Bodding, 2007; Landsberg and Yuan, 2004; Taylor et al., 2008). Ca2+ permeable ion channels include the family of transient receptor potential (TRP) ion channels, nonselective cation channels involved in signal transduction (Pedersen et al., 2005). The canonical family (TRPC), has seven members that assemble as homo- or heterotetramers (Putney, 2005; Schaefer, 2005). TRPC channels may be activated directly by diacylglycerol (Dietrich et al., 2005; Kress et al., 2008) or indirectly through calcium release from the endoplasmic reticulum following stimulation of the inositol triphosphate receptor (Salido et al., 2009a; Sours-Brothers et al., 2009). Recent studies suggest that TRPC channels play a role in cellular growth control. For example, Ca2+ entry via TRPC channels is essential for the proliferation of pulmonary artery myocytes (Golovina et al., 2001) and pharmacological TRPC channel inhibition arrest proliferation of human ovarian cancer cells (Yang et al., 2009). Downregulation of TRPC channels using siRNA arrested the growth of human corneal epithelial cells (Golovina et al., 2001; Yang et al., 2005) and cultured rat astrocytes (Golovina, 2005) via reduced store-operated calcium entry (SOCE; Malarkey et al., 2008).
In a recent study, we demonstrated TRPC subunit expression profiles within various human malignant gliomas by Western blot and showed the presence of Ca2+ permeable transient receptor potential canonical 1 (TRPC1) channels biophysically (Bomben and Sontheimer, 2008). We have now generated human glioma lines in which TRPC1 channel expression can be manipulated by shRNA knockdown. With these, we provide in vitro and in vivo evidence, suggesting that TRPC1 function is essential for normal proliferation and its loss causes incomplete cell divisions, leading to multi-nucleated ‘‘giant’’ cells reminiscent of those seen in patient biopsies. We show that loss of TRPC1 function impairs tumor growth in nude mice.
MATERIALS AND METHODS
Cell Culture
Experiments were done using a human grade IV glioma cell line D54MG, a gift by Dr. D. Bigner (Duke University, Durham, N.C. obtained 2001). The cell line has not recently been authenticated. Cells were maintained as described in Bomben and Sontheimer (2008).
Drugs and Solutions
The inhibitors SKF96365, MRS-1845, and 2-aminoe-thoxydiphenylborane (2-APB) were obtained from Sigma Aldrich as was puromycin, doxycycline, and cyclopiazonic acid (CPA). Recordings were done in the following bath solution (in mM): 130 NaCl, 5 KCl, 1 CaCl2, 10.5 D-glucose, 32.5 HEPES, and pH adjusted to 7.4 with NaOH. For calcium imaging, bath solutions consisted of (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 1 CaCl2, 1.6 Na2HPO4, 0.4 NaH2PO4, 10.5 D-glucose, 32.5 HEPES, and pH adjusted to 7.4 with NaOH. Pipette solutions contained (in mM): 145 KCl, 1 MgCl2, 0.2 CaCl2, 10 EGTA, 10 HEPES sodium salt, and pH adjusted to 7.2 with Tris-base.
Transfections of shRNA and Control Plasmids
To knockdown TRPC1, we obtained pGIPZ-lentiviral shRNAmir vectors containing either nonsilencing (NS) scrambled sequence or one of two hairpin sequences targeting TRPC1 (Open Biosystems, Huntsville, AL). Plasmids were catalog numbers RHS4346 (NS), RHS4430-98486752 (shRNA1), and RHS4430-99292249 (shRNA2). The pGIPZ vectors also expressed GFP to identify transfected cells. For inducible knockdown, pTRIPZ-lentiviral vectors were obtained (catalog numbers RHS4743 and RHS4696-99683013) for NS and shRNA1 plasmids, respectively, and TurboRed® expression indicated induction of shRNA. Cells were transfected as described in Weaver et al. (2006). To generate stable lines, 1 μg/mL puromycin treatment began 96 h after transfection. After selection, cells were passed (density: 0.5 cells/100 μL) into 96 well plates and scored for single colonies.
Calcium Imaging
Cells were loaded with Fura-2-acetoxymethylester (5 μmol/L, TEFLABS) reconstituted in 20% w/v pluronic acid in DMSO (Invitrogen, Carlsbad, CA). For SOCE, cells were in normal bath (containing calcium) and placed on microscope to equilibrate. Recordings were obtained with an Olympus Disk Spinning Unit fluorescent imaging microscope where cells were alternately excited at 340 and 380 nm. Emitted light was collected at >520 nm. Slidebook software (Intelligent Imaging Innovations, Denver, CO) digitized images and obtained 340:380 nm ratios. After obtaining baselines, cells were rinsed twice and calcium-free bath was applied. Resuming imaging, we collected baseline data then added 10 μM CPA (first peak). After 10 min, extracellular calcium was restored to 1 mM (second peak) and imaging continued for 10 min.
Western Blots
Blots were run as described in Bomben and Sontheimer (2008). We obtained antibodies from Alomone Labs, Jerusalem, Israel (TRPC1, 3, 4, 5, STIM1, Orai1) and Sigma, St. Louis, MO (actin).
Immunocytochemistry
D54MG cells were seeded and stained as described in Bomben and Sontheimer (2008). FITC-tubulin antibody was obtained from Abcam (Cambridge, MA).
Electrophysiology
Recordings of whole-cell currents were made using an Axopatch 200A amplifier (Axon instruments, Foster City, CA) following standard recording techniques (Hamill et al., 1981) and described in Bomben and Sontheimer (2008).
Proliferation Assays
For proliferation assays, pTRIPZ or pGIPZ-transfected cells were assayed as described in Bomben and Sontheimer (2008).
Live/Dead Cell Viability Assay
Nonsilencing and TRPC1 shRNA cells were treated for 0–5 days with doxycycline, and the Live/Dead Viabiltiy/Cytotoxicity kit was used as described in Bomben and Sontheimer (2008). Positive controls were achieved by 50% ethanol treatment (5 min) and >30% cells were killed (data not shown).
Flank Tumor Protocol
Nude mice were randomized and half received pre-treatment of 40 mg/kg doxycycline food (Bioserv, French-town, NJ) before surgery for 4 days. Mice were anesthetized and their flanks prepared for tumor injections with alcohol pads. Transfected cells were prepared (density: 1 × 107) in serum-free media. An equal volume of Matrigel (Fisher Scientific, Pittsburgh, PA) was added (density: 5 × 106 cells). One-milliliter syringes were loaded and 200 μL injected into individual mice using 27G needles. All experiments were done in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Alabama-Birmingham Institutional Animal Care and Use Committee.
Time-Lapse Imaging
pTRIPZ-transfected D54MG cells were plated as described in Bomben and Sontheimer (2008). Before time-lapse, control cells and TRPC1 shRNA cells were treated with or without 1 μg/mL doxycycline and equilibrated on the microscope. Time-lapse images were acquired as described in Bomben and Sontheimer (2008). Phase and fluorescent images were obtained at intervals of 20 min for 48–96 h, and three fields per treatment condition were imaged at each time point, experiments were repeated in triplicate.
Data Analysis
Results were analyzed using Origin (v.6.0, MicroCal Software, Northhampton, MA). Significance was determined by one-way ANOVA or Student’s t-tests, as appropriate, because all data showed normal distribution. All data reported are mean ± SEM and * denotes P < 0.05 unless otherwise stated.
RESULTS
TRPC Channel Inhibitors Decrease Glioma Proliferation and Result in Multinucleated Cells
In light of previous reports suggesting a role for TRPC channels in proliferation (Golovina, 2005; Yang et al., 2005), we first ascertained whether pharmacological TRPC inhibitors, namely SKF96365, 2-APB, and MRS1845 inhibited glioma proliferation. Using D54MG glioma cells as a model system for human GBM tumors, in our hands, all three drugs inhibited small voltage-independent currents indicative of TRPC channels. Figure 1A shows mean data derived from electrophysiological recordings where drug sensitive currents for at least six cells were determined, normalized to membrane capacitance, and plotted as a function of voltage. Patch-clamp recordings were obtained under conditions where large conductance K+ channels were inhibited with paxilline (Basrai et al., 2002), applying 100-ms voltage step ranging from −80 to 140 mV from a holding potential of −40 mV. Subtraction of currents before and after drug application isolated the drug-sensitive current. These experiments established concentrations of 25 μM for SKF96365 and 100 μM for 2-APB and MRS-1845 as most effective drug dose. Growth curves were established for sister cultures treated continuously with each of these inhibitors (Fig. 1B), which all reduced glioma cell growth significantly (P < 0.05) with SKF96365 and 2-APB causing complete growth arrest. TRPC inhibition resulted in large multinucleated cells (representative example, Fig. 1C) not seen in untreated controls. Sister cultures were fixed and stained for α-tubulin (green), TRPC1 channels (red), and the nuclei with DAPI (blue) and show cells undergoing incomplete cytokinesis.
Fig. 1.

TRPC inhibitors decrease glioma proliferation and store-operated calcium entry. A: Whole-cell patch-clamp recordings were used to isolate currents sensitive to TRPC inhibitors. The drug-sensitive currents were plotted as a function of applied voltage. Currents were linear and reversed at a potential of ~−25 mV. About 100 μM 2-APB (n = 6), 100 μM MRS1845 (n = 6), and 25 μM SKF96365 (n = 12) blocked identical currents. B: Proliferation assays (n = 5) revealed that channel inhibition with each of three inhibitors decreased glioma proliferation. By day 5, control cells (0.1% DMSO) grew to 42.24 ± 4.19-fold their starting cell number, whereas MRS1845 treatment reduced growth to 24.5 ± 6.5-fold. 2-APB and SKF treatments resulted in complete growth inhibition (0.54 ± 0.16-fold and 1.19 ± 0.24-fold, respectively). C: Immunocytochemical staining of D54MG cells using α-tubulin (green), TRPC1 (red), and DAPI (blue) staining and treated with either SKF or without drug for 2 days. SKF treatment resulted in larger cells with frequent multinucleation. Scale bars are 20 μm. D: Plots of Fura-2 imaging using the SOCE protocol (see Methods section). Two minutes before calcium addition, we applied either 0.1% DMSO (control) (n = 121) or the TRPC inhibitors 2-APB (n = 88), MRS1845 (n = 109), and SKF (n = 108). SOCE was significantly decreased (P < 0.05) with all TRPC inhibitors. The SOCE protocol was repeated with a TRPC1-blocking antibody applied for the entire imaging protocol (n = 108), and SOCE was reduced (P < 0.05) when compared with no drug controls (n = 100) or a nonblocking TRPC5 antibody (n = 75).
TRPC Channel Inhibitors Decrease SOCE in Human Malignant Gliomas
Because TRPC channel inhibition decreased proliferation, we hypothesize that TRPC channels participate in Ca2+ dynamics during cell division and that disruption of function impairs this process. TRPC channels are suggested to participate in store-operated calcium entry (SOCE) (Salido et al., 2009a), whereby depletion of intra-cellular Ca2+ stores triggers influx of extracellular Ca2+. To determine whether TRPC channels participate in SOCE Ca2+ flux, we used FURA-2 for quantitative intra-cellular Ca2+ imaging. To visualize calcium entry through store-operated channels, the endoplasmic reticulum stores were first depleted using 10 μM CPA in extracellular bath without calcium. After 10 min, 1 mM extracellular calcium was applied and calcium entry monitored. Cumulative imaging data using this protocol on at least 88 cells are illustrated in Fig. 1D in the presence and absence of TRPC inhibitors. The initial increase in [Ca2+]i, following store depletion with CPA, was followed by rapid entry of Ca2+ upon application of 1 mM extracellular Ca2+ (second peak). This second peak was attenuated by 92.5%, 48.1%, and 86.1%, respectively, in the presence of 25 μM SKF96365, 100 μM MRS-1845, or 100 μM 2-APB when compared with vehicle (0.1% DMSO). The almost complete inhibition of Ca2+ entry with two inhibitors suggests that, in gliomas, TRPC channels constitute the major SOCE pathway. A similar finding was reported for astrocytes (Malarkey et al., 2008) where TRPC1 channels were identified as participating in SOCE. Indeed, our immunocytochemistry (Fig. 1C), biotinylations, and western analysis (Supp. Info. Fig. 1) suggest that TRPC1 subunits compose TRPC channels on glioma plasma membranes. In light of this, we applied a rabbit polyclonal TRPC1-blocking antibody, shown to block TRPC1 channels in rat astrocytes (Malarkey et al., 2008). Incubating cells with the TRPC1 antibody for the entire experiment reduced SOCE by 25.2% (n = 108) (P < 0.05) compared with controls (n = 100) (Fig. 1D). To account for nonspecific effects, we utilized a nonblocking rabbit polyclonal TRPC5 antibody and saw no difference in SOCE (n = 75). This data suggest that TRPC channels, particularly, TRPC1 channels, contribute to glioma SOCE.
Constitutive TRPC1 shRNA Plasmids Decrease TRPC1 Expression and Channel Function
To more unequivocally show that TRPC1 channels play a major role in SOCE, we turned to shRNA knockdown approaches whereby cell lines were generated with TRPC1 suppressed. Specifically, we transfected glioma cells with pGIPZ vectors containing either one of two shRNA constructs directed against different TRPC1 gene sequences or scrambled shRNA control. Transfected cells were selected by puromycin selection. Whole-cell lysates were collected and compared by Western blot. Representative blots show that both shRNA constructs decreased TRPC1 expression (Fig. 2A) and densitometry (normalization to actin) showed a 50–60% reduction (n = 4) (Fig. 2B). We did not see changes in TRPC5, STIM1, or Orai1 expression. Therefore, although it cannot be ruled out that decreased TRPC1 expression affects heteromeric channel assembly, shRNA constructs did not affect expression of other known SOCE components. To assess whether suppression of TRPC1 expression resulted in loss of functional channels, we performed whole-cell recordings on pGIPZ transfected cells, specifically isolating TRPC inhibitor SKF96365-sensitive currents. In control nonsilencing transfected D54MG cells, SKF-sensitive currents were prominent (n = 12). In contrast, both TRPC1 shRNA constructs essentially eliminated SKF-sensitive currents (Fig. 2C, n = 9 each). This indicates that TRPC1 channels mediate SKF sensitive currents in D54MG cells or at least are an essential subunit for SKF-sensitive channels.
Fig. 2.

Constitutive TRPC1 shRNA plasmids decrease TRPC1 protein expression, SKF-sensitive currents, and store-operated calcium entry. A: Representative Western blots of puromycin selected D54MG cells transfected with NS or TRPC1 shRNA plasmids showed that TRPC1 protein expression is decreased. Antibodies to TRPC5, Orai1, and Stim1 revealed no change in the protein level of other purported SOCE channel members. B: Quantification of TRPC1 protein knockdown (n = 4) revealed a 50–60% decrease in TRPC1 expression. C: Using whole-cell patch-clamp recordings, we isolated SKF-sensitive currents from TRPC1 shRNA cells transfected cells. The NS control plasmid (n = 12) displayed a prominent SKF-sensitive current while either two shRNA plasmids (n = 9 each), by contrast, did not display currents. D: Fura-2 imaging on transfected D54MG cells using SOCE protocol (see Methods section). Transient transfections were used and indicated by GFP expression. There was no difference in SOCE between control plasmid transfected (n = 71), control plasmid nontransfected (n = 109), shRNA1 nontransfected (n = 117), or shRNA2 nontransfected (n = 114). Cells transfected with TRPC1 shRNA (n = 45) or TRPC1 shRNA2 (n = 42) had a decrease (P < 0.05) in SOCE. Peak fluorescence data indicated SOCE decreased by 25–30% when TRPC1 shRNA was transfected.
If TRPC1 channels were responsible for SOCE, we would expect decreased Ca2+ influx through these channels upon store depletion. Indeed, each of the two silencing constructs significantly (P < 0.05) reduced SOCE (Fig. 2D) with mean data from 45 cells (shRNA1) and 42 cells (shRNA2) showing a 25–30% SOCE reduction. SOCE was unaffected in nontransfected sister cells from transient transfections.
We then examined the proliferative ability of these knockdowns cells. The challenge was that stably transfected D54MG cells containing silencing constructs for TRPC1 grew very slowly, which was expected given TRPC inhibitors’ profound effect on cell growth (Fig. 1B). To nevertheless ascertain cell growth, we transiently transfected cells with the shRNA plasmids to determine the relative numbers of transfected cells that divided compared with cells transfected with the control plasmid and found a selective reduction in the growth rate for the TRPC1 knockdown cells (Supp. Info. Fig. 2). Notably, cells transfected with either shRNA plasmids showed frequent appearance of enlarged, multinucleated cells, as we had observed in our pharmacological assays (Supp. Info. Fig. 2B). However, further studies would have been cumbersome given the inability to propagate these cells. To overcome these issues, we turned to an inducible shRNA vector system.
Inducible TRPC1 shRNA Decreases Expression, SKF-Sensitive Currents, and SOCE
Glioma cells were transfected with an inducible TRPC1 shRNA vector or nonsilencing vector. In the presence of doxycycline, shRNA and TurboRed®, a fluorescent indicator, were transcribed. We generated stable cell lines and tested them, with or without doxycycline, for decreased TRPC1 expression. Representative Western blots (Fig. 3A) show a nonsilencing control clone and a TRPC1 shRNA clone. Using doxycycline to turn on shRNA transcription, we confirmed that TRPC1 expression decreased following 5 days of doxycycline treatment. Moreover, whole-cell recordings obtained 4–6 days following doxycycline treatment showed a reduction of SKF-sensitive current in TRPC1 shRNA cells treated with doxycycline compared with cells without doxycycline (Fig. 3B). To assure that neither transfection nor treatment with doxycycline had unspecific effects, we validated that paxilline-sensitive Ca2+-activated K+ (BK) currents mediated by BK channels were unaffected by these treatments (Fig. 3C). We also found that TRPC1 knockdown cells were much larger than their TRPC1 expressing sister cells as judged by whole-cell capacitance (Fig. 3D). There was no change in resting membrane potentials (Supp. Info. Fig. 3A).
Fig. 3.

Inducible TRPC1 shRNA plasmids decrease TRPC1 protein expression and channel function. A: Representative Western blot displaying reduced TRPC1 protein expression in shRNA transfected cells when doxycycline treated for 5 days. B: Using whole-cell patch-clamp electro-physiology, SKF-sensitive currents from TRPC1 shRNA cells induced with doxycycline for 4–6 days (n = 19) were decreased (P < 0.05) when compared with control plasmid noninduced (n = 17), control plasmid induced (n = 16), or TRPC1 shRNA plasmid noninduced (n = 14). C: Mean electro-physiological data from the same cells revealed no change in paxilline-sensitivity (BK channels). D: Doxycycline treatment on TRPC1 shRNA transcription resulted in larger cells, measured by whole-cell capacitance (P < 0.05). Control plasmid noninduced were 44.33 ± 1.56 pF, control plasmid induced were 46.93 ± 1.61 pF, and shRNA plasmid noninduced were 39.2 ± 1.47 pF. The induced TRPC1 shRNA cells were 86.89 ± 3.7 pF.
We used the SOCE protocol on our inducible shRNA vector system to ascertain that both shRNA methods decreased SOCE. Cells were plated, treated for 4–5 days with or without doxycycline, and loaded with FURA-2. Following store depletion, we observed that TRPC1 shRNA cells treated with doxycycline had decreased SOCE by 24.1% (n = 255) following application of extracellular calcium (Fig. 4A,B). Untreated TRPC1 shRNA cells (n = 228) were not different from doxycycline-treated control cells (n = 234) or untreated control cells (n = 215). We concluded that doxycycline itself did not affect SOCE.
Fig. 4.

Doxycycline-induced TRPC1 shRNA impairs store-operated calcium entry and glioma proliferation. A: Fura-2 imaging on pTRIPZ transfected cells using SOCE protocol (see Methods section) showed no difference between control plasmid noninduced (n = 215), control plasmid induced (n = 234), or TRPC1 shRNA noninduced (n = 228). When TRPC1 shRNA cells were treated with doxycycline for 4–5 days (n = 255), SOCE was reduced (P < 0.05). B: Peak fluorescence data indicated a 24.1% decrease in SOCE when TRPC1 shRNA was induced with doxycycline. C: Proliferation assays (n = 6) with inducible control and shRNA plasmids were performed (see Methods section). After 6 days of treatment with or without doxycycline, cells proliferated 46.75 ± 4.28-fold (noninduced control), 40.77 ± 4.03-fold (induced control), and 39.24 ± 3.12-fold (noninduced TRPC1 shRNA) their starting cell number. Induced TRPC1 shRNA cells only proliferated to 7.26 ± 1.03-fold their starting cell number.
Decreased Functional TRPC1 Channels Reduce Glioma Proliferation
Because we were now able to disrupt TRPC1 channel expression using an inducible knockdown, we ascertained the specific role that TRPC1 function had on glioma proliferation. Nonsilencing shRNA and TRPC1 shRNA cells were plated on six well plates and individual wells harvested daily then normalized to starting cell number. On day 0, half of the transfected cells of both cell lines received doxycycline. Beginning at day 3, TRPC1 shRNA cells treated with doxycycline had significantly (P < 0.05) decreased cell numbers from their sister cells without doxycycline as well as control cells with or without doxycycline (Fig. 4C). Indeed, following 6 days of doxycycline treatment, the induced TRPC1 shRNA cells only reached 7.26 ± 2.53-fold above their starting cell number. Considering that their sister cells reached 39.24 ± 7.64-fold, this was less than 20% the total relative cell number. This decrease was not due to cell death as judged by a cell-viability assay (Supp. Info. Fig. 3B).
Because enlarged, multinucleated cells appeared following pharmacological inhibition of TRPC channels, we examined mitosis and multinucleation in cells with disrupted TRPC1 channel expression. Specifically, sister cultures treated with or without doxycycline for 5 days were stained with FITC-tubulin to visualize cells with mitotic spindles. Cells in which the shRNA was induced were indicated by TurboRed® reporter expression. We readily visualized cells that had mitotic spindles in control cells and noninduced TRPC1 shRNA cells (Fig. 5A). In contrast, induced TRPC1 shRNA cells undergoing mitosis were rarely identified, but instead larger, multinucleated cells were frequently found (Fig. 5A). A quantitative analysis showed that induced TRPC1 shRNA cells had significantly fewer cells (P < 0.05) with mitotic spindles versus noninduced TRPC shRNA cells (Fig. 5B). There was no difference in control cells with or without doxycycline; hence, this effect was specific to gene silencing. Intriguingly, as with pharmacological inhibition, the percentage of multinucleated cells was significantly increased (P < 0.05) with TRPC1 channel inhibition (Fig. 5C). This suggests that TRPC1 is crucial for proper cell division.
Fig. 5.

Interference with TRPC1 channel function results in higher percentages of multinucleated cells. Representative stainings of D54MG cells with FITC-tubulin identified the mitotic spindle within a (A) noninduced control cell, induced control cell, or noninduced TRPC1 shRNA cell. FITC-tubulin staining of induced TRPC1 shRNA cells revealed a different morphology including multinucleated cells. Scale bars are 20 μm. B: There were fewer cells in mitosis (P < 0.05), with mitotic spindles, when TRPC1 shRNA was induced when compared with noninduced shRNA cells. There was no difference between control plasmid cells treated with or without doxycycline. C: Induced TRPC1 shRNA cells had significantly more (P < 0.05) multinucleated cells (13% ± 2%) when compared with control noninduced (1% ± 0.1%), control induced (1% ± 0.1%), or TRPC1 shRNA noninduced (1% ± 0.1%). D: We observed normal mitosis from TRPC1 shRNA cells without doxycycline where, in positions 1–5, they started flat, the cell rounded up into mitosis, the cell began to divide, and two daughter cells emerged (4 and 5). In induced TRPC1 shRNA cells, we observed instances of incomplete cell division where, in positions 1–5, they started flat, the cell rounded up into mitosis, the cell began to divide, and one cell emerged (4) and bizarre nuclei were observed (5). Scale bars are 20 μm.
To visualize cell division in real time, we obtained time-lapse video recordings of TRPC1 shRNA transfected cells treated with or without doxycycline. We identified incidents where a cell treated with doxycycline rounded up, attempted to divide, but did not become two cells (Supp. Info. Movie 2). In contrast, untreated TRPC1 shRNA cells were able to undergo normal cell divisions (Supp. Info. Movie 1). Still pictures from these movies (Fig. 5D) show distinct differences in division with TRPC1 expression impairment. A representative-noninduced TRPC1-expressing cells are shown beginning as flat cell, rounding up into mitosis, dividing, and becoming two daughter cells (positions 1–5). By contrast, a representative-induced TRPC1-knockdown cell began flat, rounded into mitosis, attempted to divide, and remained one cell with bizarre nuclei (4 and 5). Scale bars are 20 μm.
Decreased TRPC1 Protein Expression Is Observed in Patient Biopsy Lysates with Increasing Levels of Giant, Multinucleated Phenotype
Although most GBMs have some cells with multiple nuclei, multinucleated ‘‘giant’’ cells specifically define a GBM subtype called giant cell glioblastoma. These cells are enlarged and have multiple, up to 10, bizarre nuclei (Palma et al., 1989). Because we observed a similar, large, multinucleated phenotype when TRPC1 channels were impaired in vitro, we investigated whether patient biopsy lysates from patients diagnosed with discretely different levels of giant, multinucleated cells in the pathological report had a change in TRPC1 protein expression. The relative percentage of large, multinucleated cells in glioblastoma tissue was certified by pathology reports accompanying tumor samples. In Fig. 6, we show that patients with an increased number of large, multinucleated had decreased amounts of TRPC1 channel protein expression. This correlated well with the above data suggesting that interference with TRPC1 channel expression and function increased the occurrence of multinucleation.
Fig. 6.
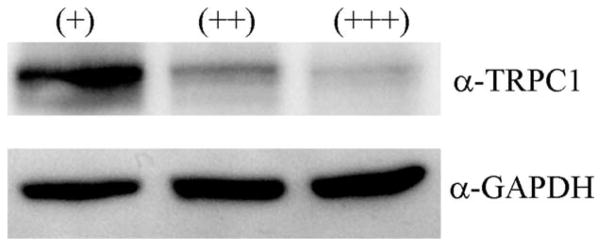
Decreased TRPC1 protein expression correlates with an increased percentage of multinucleated cells in GBM patient biopsies. Glioblastoma patient biopsy lysates were analyzed using western blot for TRPC1 expression in tissue from patients with a small (+), medium (++), or large (+++) percentage of large, multinucleated cells. As the number of large, multinucleated cells increased, TRPC1 channel expression decreased when compared with GAPDH-loading control.
Decreased TRPC1 Channel Function Inhibits In Vivo Tumor Growth
Although in vitro studies are necessary to determine the cellular effects of TRPC1 channel inhibition, we explored whether TRPC1 knockdown affected in vivo tumor growth. Therefore, nude mice were injected with 1 × 106 inducible TRPC1 shRNA cells in the flank and either fed regular chow or doxycycline-supplemented chow. Calipers were used to measure tumor length and width to determine tumor volume twice weekly over 5 weeks. Mice on doxycycline (n = 12) showed consistently smaller tumor volumes than their litter mates (n = 11), and this difference was significant (P < 0.05) after 31 days (Fig. 7A,B). After 5 weeks, mice fed doxy-cycline had a smaller range of tumor sizes, and the smallest tumors were shallow (Fig. 7C, tumor examples). Mice were sacrificed at 5 weeks, and the tumor was excised to accurately measure the depth of the tumor (Fig. 7D). The excised tumor volume for doxycy-cline fed mice was significantly (P < 0.05) smaller than the controls. This data suggest that TRPC1 channel function is important for tumor growth in vivo.
Fig. 7.

Decreased TRPC1 channel function impairs in vivo tumor growth. (A) Scatter and (B) line plot of flank tumor size of nude mice fed regular or doxycycline supplemented chow. After 31 days, the mice fed doxycycline to silence TRPC1 (n = 12) had significantly smaller tumors (P < 0.05) than their litter-mates fed regular food (n = 11). These tumors were still smaller (P < 0.05) at 35 days when the animals were sacrificed. C: Photographs show representative mice from noninduced and induced conditions of the largest and smallest tumors of each group. Some mice fed doxycycline had shallow tumors. D: The excised tumor from mice was measured again and a depth measurement obtained. Tumors from mice fed doxycycline were 37% smaller (P < 0.05).
DISCUSSION
Over the past two decades, ion channels have been implicated as important contributors to cell proliferation of normal and cancerous cells (Kunzelmann, K., 2005). TRPC channels have recently been added to the list of ion channels that may be affected in cancer (Bodding, M., 2007). TRPC6 channel dysfunction has been linked to prostate cancer (Thebault et al., 2006) and TRPC-1, -4, and -6 have been linked to ovarian cancer (Yang et al., 2009). The findings in this study support the notion that TRPC1 channels are intrinsically involved in proliferation of human malignant gliomas. Parallel use of pharmacological inhibitors, constitutive TRPC1 shRNA plasmids, and inducible TRPC1 shRNA plasmids make a strong case for a TRPC1 channel role in normal mitosis. All three approaches inhibited TRPC1 channels as judged from patch-clamp recordings and FURA-2 imaging. Western analysis confirms the selective loss of TRPC1 channels in cells transfected with shRNA. Furthermore, our studies reveal an essential role for TRPC1 channels in normal cytokinesis, because blockade of TRPC1 channels causes cells to undergo incomplete cell division. This resulted in large, bizarre multinucleated cells replicating the pathological phenotype observed in many glioblastoma biopsies. Although found frequently in most GBMs, multinucleated ‘‘giant’’ cells specifically define a GBM subtype typically found in pediatric cases called giant cell glioblastoma, which are enlarged containing up to 10 nuclei (Palma et al., 1989). It is conceivable that a deletion or loss of function mutation in the TRPC1 gene may underlie the multinucleated phenotype. The human TRPC1 gene has been mapped to 3q22–24 (Abramowitz. and Birnbaumer, 2007), an area found to present with genetic alterations in some glio-blastoma patients (Dahlback et al., 2009). The limited genomic data available for giant cell glioblastomas suggest that these pediatric cases present with chromosomal imbalances distinct from adult cases and genetic loci affected include 1q, 3q, and 16q (Rickert et al., 2001). In the limited number of samples, we examined, decreased TRPC1 channel expression correlated with an increase in the percentage of large, multinucleated cells. Hence, it appears plausible that a loss of function in the TRPC1 gene may accompany some of these pediatric GBMs, a theory warranting further study. If, in fact, TRPC1 expression is necessary for glioma proliferation as our study implies, then functional inhibition of TRPC1 channels once gliomas are established may result in this phenotype of large, multinucleated cells that have difficulty further proliferating.
One main function of TRPC channels is a replenishment of intracellular Ca2+-stores following agonist-induced release of Ca2+ from the endoplasmic reticulum, which occurs, for example, following growth factor stimulation. This phenomenon, called SOCE (Ambudkar et al., 2007), involves either TRPC1 channels forming heteromers with Orai1 (Liao et al., 2007) or assembly with other TRPC channels into SOCE channels (Wu et al., 2004). The requirement for an association with Orai1 may be cell-type specific [reviewed in Salido et al. (2009b)]. Although pharmacology and shRNA plasmids may also affect heteromeric channels, our data strongly implicate that TRPC1 is a major component of SOCE in glioma cells.
During the cell cycle, calcium oscillations occur during both the G1/S and G2/M phase and play a role in centro-some duplication and separation (Matsumoto and Maller, 2002). Additionally, through effector molecules such as calmodulin and calmodulin kinase, calcium has been shown to affect proliferation components from transcription to cytokinesis (Corcoran and Means, 2001; Liu et al., 1992; Matsumoto and Maller, 2002). Cytokinesis requires actomyosin contractile ring formation, which requires calcium (Miller et al., 1993). Additionally, myosin-II is a component of the contractile ring (Fujiwara and Pollard, 1976; Yumura, 2001), and one activation mechanism is dependent on myosin light chain kinase (Matsumura, 2005), which is dependent on calcium/calmodulin (Iwasaki et al., 2001). However, there are many other mechanisms that regulate cytokinesis including RhoA, rho kinase, and other signaling pathways (Piekny et al., 2005). We hypothesize based on our observation that TRPC1 participates preferentially in Ca2+ entry during late M-phase, possibly Ca2+ required for cytokinesis. This remains a hypothesis, because the interdependence of Ca2+ influx via TRPC1 was not directly examined.
Given the dire prognosis of patients with glioblastoma, the search for new pharmacological targets is ongoing. Our in vivo studies disrupting expression TRPC1 channel expression suggest that pharmacological inhibition of these channels may slow tumor growth. Although the resulting tumors may change their phenotype to ‘‘Giant cell glioblastomas,’’ these cancers carry a more favorable prognosis, which agrees with the slowed tumor growth observed in our animal model. In spite of having grade IV cancers, many patients with giant cell glioblastoma experienced life expectancies as long as 7–9 years compared with <1 year typical for GBMs (Becker et al., 1967; Klein et al., 1998). We therefore feel that future studies exploring TRPC1 as a therapeutic target in glioma are warranted.
Supplementary Material
Acknowledgments
The authors thank Naomi Logsdon for her help with animal experiments.
Grant sponsor: National Institutes of Health; Grant numbers: F31-NS063688, RO1-NS031234.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Abramowitz J, Birnbaumer L. Know thy neighbor: A survey of diseases and complex syndromes that map to chromosomal regions encoding TRP channels. Handb Exp Pharmacol. 2007:379–408. doi: 10.1007/978-3-540-34891-7_23. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Ong HL, Liu X, Bandyopadhyay BC, Cheng KT. TRPC1: The link between functionally distinct store-operated calcium channels. Cell Calcium. 2007;42:213–223. doi: 10.1016/j.ceca.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Basrai D, Kraft R, Bollensdorff C, Liebmann L, Benndorf K, Patt S. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. Neuroreport. 2002;13:403–407. doi: 10.1097/00001756-200203250-00008. [DOI] [PubMed] [Google Scholar]
- Becker DP, Benyo R, Roessmann U. Glial origin of monstrocellular tumor. Case report of prolonged survival. J Neurosurg. 1967;26:72–77. doi: 10.3171/jns.1967.26.1part1.0072. [DOI] [PubMed] [Google Scholar]
- Bodding M. TRP proteins and cancer. Cell Signal. 2007;19:617–624. doi: 10.1016/j.cellsig.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Bomben VC, Sontheimer HW. Inhibition of transient receptor potential canonical channels impairs cytokinesis in human malignant gliomas. Cell Prolif. 2008;41:98–121. doi: 10.1111/j.1365-2184.2007.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowski NA, Sneed PK, Chang SM. Diagnosis and treatment of recurrent high-grade astrocytoma. J Clin Oncol. 2006;24:1273–1280. doi: 10.1200/JCO.2005.04.7522. [DOI] [PubMed] [Google Scholar]
- Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma—Animal models and therapeutic challenges. Brain Pathol. 2009;19:112–120. doi: 10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran EE, Means AR. Defining Ca2+/calmodulin-dependent protein kinase cascades in transcriptional regulation. J Biol Chem. 2001;276:2975–2978. doi: 10.1074/jbc.R000027200. [DOI] [PubMed] [Google Scholar]
- Dahlback HS, Brandal P, Meling TR, Gorunova L, Scheie D, Heim S. Genomic aberrations in 80 cases of primary glioblastoma multiforme: Pathogenetic heterogeneity and putative cytogenetic pathways. Genes Chromosomes Cancer. 2009;48:908–924. doi: 10.1002/gcc.20690. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Schnitzler M, Kalwa H, Storch U, Gudermann T. Functional characterization and physiological relevance of the TRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:257–265. doi: 10.1007/s00210-005-1052-8. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Pollard TD. Fluorescent antibody localization of myosin in the cytoplasm, cleavage furrow, and mitotic spindle of human cells. J Cell Biol. 1976;71:848–875. doi: 10.1083/jcb.71.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol. 2005;564(Pt 3):737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP, enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280:H746–H755. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Iwasaki T, Murata-Hori M, Ishitobi S, Hosoya H. Diphosphory-lated MRLC is required for organization of stress fibers in interphase cells and the contractile ring in dividing cells. Cell Struct Funct. 2001;26:677–683. doi: 10.1247/csf.26.677. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- Klein R, Molenkamp G, Sorensen N, Roggendorf W. Favorable outcome of giant cell glioblastoma in a child. Report of an 11-year survival period. Childs Nerv Syst. 1998;14:288–291. doi: 10.1007/s003810050228. [DOI] [PubMed] [Google Scholar]
- Kress M, Karasek J, Ferrer-Montiel AV, Scherbakov N, Haberberger RV. TRPC channels and diacylglycerol dependent calcium signaling in rat sensory neurons. Histochem Cell Biol. 2008;130:655–667. doi: 10.1007/s00418-008-0477-9. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. 2004;19:44–50. doi: 10.1152/nips.01457.2003. [DOI] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Williams JG, Clarke M. Inducible expression of calmodulin antisense RNA in Dictyostelium cells inhibits the completion of cytokinesis. Mol Biol Cell. 1992;3:1403–1413. doi: 10.1091/mbc.3.12.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Martin PM, Hussaini IM. PKC eta as a therapeutic target in glioblastoma multiforme. Expert Opin Ther Targets. 2005;9:299–313. doi: 10.1517/14728222.9.2.299. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Maller JL. Calcium, calmodulin, and CaMKII requirement for initiation of centrosome duplication in Xenopus egg extracts. Science. 2002;295:499–502. doi: 10.1126/science.1065693. [DOI] [PubMed] [Google Scholar]
- Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005;15:371–377. doi: 10.1016/j.tcb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Miller AL, Fluck RA, McLaughlin JA, Jaffe LF. Calcium buffer injections inhibit cytokinesis in Xenopus eggs. J Cell Sci. 1993;106 (Pt 2):523–534. doi: 10.1242/jcs.106.2.523. [DOI] [PubMed] [Google Scholar]
- Palma L, Celli P, Maleci A, Di Lorenzo N, Cantore G. Malignant monstrocellular brain tumours. A study of 42 surgically treated cases. Acta Neurochir (Wien) 1989;97:17–25. doi: 10.1007/BF01577735. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G, Nilius B. TRP channels: An overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Piekny A, Werner M, Glotzer M. Cytokinesis: Welcome to the Rho zone. Trends Cell Biol. 2005;15:651–658. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Putney JW. Physiological mechanisms of TRPC activation. Pflugers Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- Rickert CH, Strater R, Kaatsch P, Wassmann H, Jurgens H, Dockhorn-Dworniczak B, Paulus W. Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol. 2001;158:1525–1532. doi: 10.1016/S0002-9440(10)64103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salido GM, Sage SO, Rosado JA. Biochemical and functional properties of the store-operated Ca2+ channels. Cell Signal. 2009a;21:457–461. doi: 10.1016/j.cellsig.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Salido GM, Sage SO, Rosado JA. TRPC channels and store-operated Ca2+ entry. Biochim Biophys Acta. 2009b;1793:223–230. doi: 10.1016/j.bbamcr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Schaefer M. Homo- and heteromeric assembly of TRP channel subunits. Pflugers Arch. 2005;451:35–42. doi: 10.1007/s00424-005-1467-6. [DOI] [PubMed] [Google Scholar]
- Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- Sours-Brothers S, Ding M, Graham S, Ma R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med (Maywood) 2009;234:673–682. doi: 10.3181/0809-RM-279. [DOI] [PubMed] [Google Scholar]
- Taylor JT, Zeng XB, Pottle JE, Lee K, Wang AR, Yi SG, Scruggs JA, Sikka SS, Li M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J Gastroenterol. 2008;14:4984–4991. doi: 10.3748/wjg.14.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thebault S, Flourakis M, Vanoverberghe K, Vandermoere F, Roudbar-aki M, Lehen’kyi V, Slomianny C, Beck B, Mariot P, Bonnal JL, Mau-roy B, Shuba Y, Capiod T, Skryma R, Prevarskaya N. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006;66:2038–2047. doi: 10.1158/0008-5472.CAN-05-0376. [DOI] [PubMed] [Google Scholar]
- Weaver AK, Bomben VC, Sontheimer H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia. 2006;54:223–233. doi: 10.1002/glia.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J Biol Chem. 2004;279:43392–43402. doi: 10.1074/jbc.M408959200. [DOI] [PubMed] [Google Scholar]
- Yang H, Mergler S, Sun X, Wang Z, Lu L, Bonanno JA, Pleyer U, Reinach PS. TRPC4 knockdown suppresses epidermal growth factor-induced store-operated channel activation and growth in human corneal epithelial cells. J Biol Chem. 2005;280:32230–32237. doi: 10.1074/jbc.M504553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SL, Cao Q, Zhou KC, Feng YJ, Wang YZ. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene. 2009;28:1320–1328. doi: 10.1038/onc.2008.475. [DOI] [PubMed] [Google Scholar]
- Yumura S. Myosin II dynamics and cortical flow during contractile ring formation in Dictyostelium cells. J Cell Biol. 2001;154:137–146. doi: 10.1083/jcb.200011013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.