

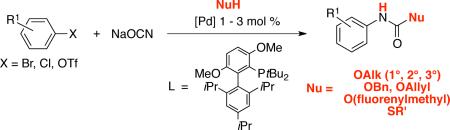
Abstract
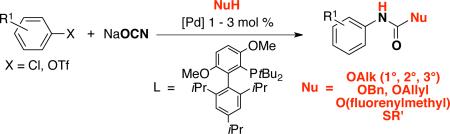
An efficient synthesis of aryl carbamates was achieved by introducing alcohols into the reaction of palladium-catalyzed cross-coupling of ArX (X = Cl, OTf) with sodium cyanate. The use of aryl triflates as electrophilic components in this transformation allowed for an expanded substrate scope for direct synthesis of aryl isocyanates. This methodology provides direct access to major carbamate protecting groups, S-thiocarbamates, and diisocyanate precursors to polyurethane materials.
Carbamates are found both in biologically active compounds1 and in polymers,2 and are valuable protecting groups in organic synthesis.3 As a result, many methods are available for their synthesis,4 including the aminolysis of acid anhydrides5 and chloroformates,6 the coupling of amines with CO2 and alkyl halides,7 and the reaction of alcohols with isocyanates. These are often generated in situ in the presence of alcohols via the Hofmann,8 Curtius,9 Lossen,10 and Schmidt11 rearrangements or the reductive carbonylation of nitroaromatic compounds.12 These methods, however, suffer from a limited substrate scope, harsh reaction conditions, multiple-step preparations or the lack of readily available starting materials.
The shortcomings of the existing methods have prompted the development of new approaches to synthesize these important building blocks. Transition-metal catalyzed reactions of the isocyanate anion with aryl electrophiles or nucleophiles have shown potential for accessing the corresponding aryl isocyanate intermediates in the carbamate synthesis. Previously, Tkatchenko showed that Ni(0) catalysts promote the cross coupling of simple aryl halides with potassium cyanate.13 While this was the first example of using an isocyanate anion in a cross-coupling reaction, the yields of the carbamate products were low to moderate. More recently, a copper-catalyzed reaction of boronic acids with the same cyanate source has been reported by Baghersad.14 While the method allowed for improved yields of the carbamates, the substrate scope was limited in both the alcohols as well as the aromatic nucleophiles. During the completion of this work a related copper-catalyzed method for the in situ generation of aryl isocyanates has been introduced by Ma, where aryl bromides and iodides were used as electrophiles.15 While the substrate scope with respect to the aryl halide was broad, the authors reported that they were unable to prepare N-Boc substituted amines. Furthermore, the range of other alcohols utilized was narrow.
Recently, we reported that the palladium-catalyzed cross-coupling of aryl chlorides and triflates with sodium cyanate affords aryl isocyanates that can be trapped in situ with amines to produce the corresponding ureas in a one-pot process.16 We felt that this could be extended to the synthesis of aryl carbamates by using alcohols as the nucleophilic trapping agents [Eq. (1)]. We now describe a general one-pot method for the palladium-catalyzed synthesis of aryl carbamates - including six of the most important carbamate protecting groups - from aryl chlorides and triflates.
 |
(1) |
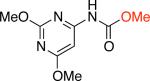
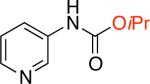
For the synthesis of N-aryl ureas we demonstrated that the addition of phenol as a nucleophile to form a phenyl carbamate intermediate broadened the substrate scope of the process. We thought that the use of other alcohols would lead to general conditions for the synthesis of N-aryl carbamates.16 On this basis we began our current work by examining the Pd-catalyzed cross-coupling of 2,4-dimethoxy-5-chloropyrimidine with sodium cyanate using methanol as the nucleophilic trapping agent. No product of direct C–O cross-coupling reactions (alkyl-aryl ether) was observed and the desired N–aryl–O-alkyl carbamate was obtained in 86% yield (Table 1, entry 1a).
Table 1.
Synthesis of aryl carbamates.a
| ||
|---|---|---|
|
|
|
|
1ab,c 86% X = Cl |
1bd 92% X = Cl |
1cd 82% X = Cl |
|
|
|
|
|
1gb,e 88% X = Cl |
1ed 90% X = Cl |
1fd 89% X = Cl |
|
|
|
|
1gb,e 86% X = Cl |
1hd 86% X = Cl |
1id 79% X = OTf |
|
|
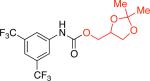
|
|
1jb 74% X = OTf |
1kb,f 90% X = Cl |
1lg 92% X = Br |
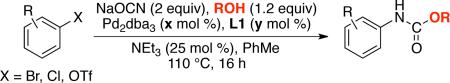
Reaction conditions (isolated yields, average of 2 runs): ArX (1 mmol), NaOCN (2 mmol), ROH (1.2 mmol), Pd2dba3 (x mol %), L1 (y mol %), NEt3 (0.25 mmol), toluene (2 mL). The Pd2(dba)3 and L1 were preheated in toluene at 120 °C for 3 minutes.
Pd2dba3 (0.75 mol %), L1 (1.8 mol %)
ROH (2 mmol), 130 °C
Pd2dba3 (0.5 mol %), L1 (1.2 mol %)
NEt3 (1.2 mmol), toluene (6 mL)
toluene (8 mL)
Pd2dba3 (1.5 mol %), L1 (3.6 mol %), 90 °C.
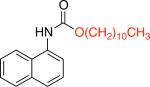
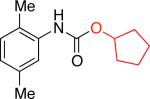
It was necessary to preheat Pd2dba3 with L1 prior to their introduction to the reaction mixture16,17 and to use triethylamine as an additive16 in order to obtain a high-yielding process. Using longer chain primary alcohols did not affect the course of the reaction and the corresponding carbamates were obtained in 82 - 92% yields (Table 1, entries 1b, 1c). Alcohols bearing trialkylsilyl functional groups could be employed, providing access to Teoccarbamates (Table 1, entries 1d, 1e).
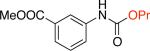
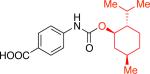
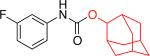
Moreover, the carbamate products were obtained in good yields (79 - 89%) when secondary alcohols were used (Table 1, entries 1f - 1i), including more sterically demanding alcohols with branching at the b-positions, such as 2-adamantyl alcohol and L-menthol (Table 1, entries 1g, 1h). Phenols could also be employed in the reaction,16 however, the stability of the resulting products is significantly lower. Isolation of N–aryl–O-phenyl carbamates was generally problematic as these products underwent significant decomposition during chromatographic purification on silica gel. However, with 4-fluorophenol the more stable carbamate could be isolated in 90% yield (Table 1, entry 1k).18
The above-described cross-coupling conditions could be applied to an array of aryl electrophiles. Aryl chlorides bearing an ester, an unprotected acid or a secondary amide (Table 1, entries 1b, 1g and 1k) were successfully converted to the corresponding N–aryl–O-alkyl carbamates. In our work on urea formation we showed that transmetallation of the isocyanate anion to the Pd-X intermediate was the most difficult step of the catalytic cycle.16 Thus, aryl bromides were found to be less reactive than the corresponding aryl chlorides or triflates. Here, too, the same trend was observed, although we were able to couple an electron-deficient aryl bromide, 3,5-bis(trifluoromethyl)bromobenzene; the corresponding carbamate product was obtained in 93% yield (Table 1, entry 1l).
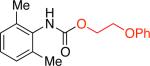
Aryl chlorides with one or two bulky ortho-substituents were generally less reactive under our standard conditions, producing the corresponding aryl carbamates in lower yields. Believing transmetallation to be the difficult step of the catalytic cycle, we felt that switching to aryl triflates might ameliorate this situation.16,19 In fact, the coupling reaction of 2,6-dimethylphenyl trifluoromethanesulfonate in the presence of 2-phenoxyethanol provided the carbamate product in 74% yield, while no product was observed with the corresponding aryl chloride (Table 1, entry 1j).
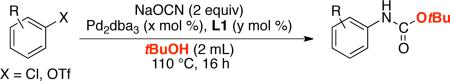
We next sought to apply our method to the synthesis of the widely used tert-butyl carbamate (Boc) functional group. While the first set of conditions allowed for the efficient carbamate synthesis using primary and secondary alcohols, the use of 1.2 – 5-fold excess of tert-butanol afforded the corresponding Boc-protected anilines only in low yields (<50%). This result is consistent with the significantly lower reactivity of isocyanates towards tertiary alcohols.20 However, employing tert-butanol as the solvent afforded the desired tert-butylcarbamates in 60 - 94% yield (Table 2).
Table 2.
Synthesis of Boc-carbamates.a
| ||
|---|---|---|
|
|
|
|
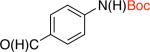
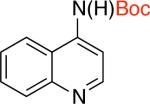
2ab,c 76% X = Cl |
2bb 87% X = Cl |
2cb 83% X = Cl |
|
|
|
|
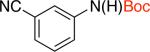
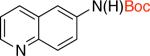
2db 94% X = Cl |
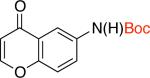
2eb 72% X = Cl |
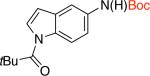
2fd,e 73% X = Cl |
|
|
|
|
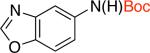
2gd 60% X = Cl |
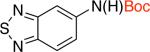
2hb 70% X = Cl |
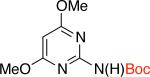
2ib,c 93% X = Cl |
| ||
|
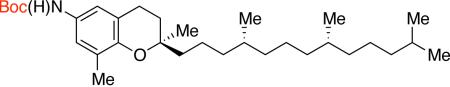
2jb,e 71% X = OTf | ||
Reaction conditions (isolated yields, average of 2 runs): ArX (1 mmol), NaOCN (2 mmol), Pd2dba3 (x mol %), L1 (y mol %), tBuOH (2 mL).
Pd2dba3 (1.5 mol %), L1 (3.6 mol %)
90 °C
Pd2dba3 (1.5 mol %), L1 (3.6 mol %)
130 °C
fPd2dba3 (0.75 mol %), L1 (1.8 mol %), 24 h.
Additionally there was no need either to preheat Pd2dba3 and L1 or to use triethylamine as an additive. These reaction conditions tolerated aldehyde and cyano groups as well as a variety of heteroaryl electrophiles (Table 2, entries 2c-2i).
Although the conditions described above (Tables 1 and 2) provided a process that manifested a broad substrate scope with respect to both aryl electrophile and the alcohol nucleophile, they were not suitable for use with certain alcohols, such as benzyl alcohols or trichloroethanol, and in these cases reduced yields of the carbamate products were observed. Additionally, byproducts resulting from direct C–S cross-coupling (thioethers) rather than aryl carbamates were observed when we attempted to use thiols as substrates. Thus, we attempted to utilize a phenyl carbamate, as before, followed by displacement with the alcohol or thiol (Scheme 1a).16 Unfortunately, this approach did not lead to a general protocol.
Scheme 1.
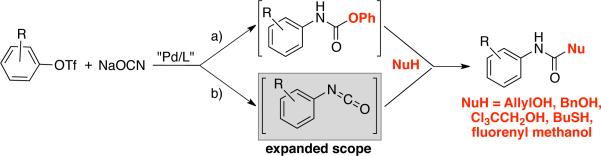
Synthesis of N-aryl carbamates, involving a) O-phenyl carbamate or b) isocyanate intermediates.
We felt that a more reactive intermediate, that could still be generated efficiently in situ, was needed in order to extend the scope of these reactions. Aryl isocyanates would be the ideal reactive intermediates needed for these reactions.21 Unfortunately in our previous work their synthesis was of limited success for sterically hindered aryl chlorides and with many heteroaryl chlorides.16
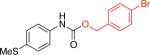
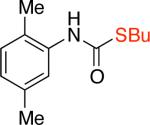
In fact, switching from aryl chlorides to aryl triflates allowed access to a broader range of aryl isocyanates (Scheme 1b). These include substrates bearing a variety of ortho-substituents (methyl, phenyl and isopropyl groups; Table 3, entries 3b, 3e-3g), as well as other less reactive aryl-based electrophiles12 (Table 3, entries 3c, 3d). Trichloroethanol and benzyl alcohol were used to produce Troc- and Cbz-carbamates in 75% – 84% yields (Table 3, entries 3a-3d). Butanethiol could also be used to provide the corresponding S-thiocarbamate in 86% yield (Table 3, entry 3e). Additionally, the two-step approach enables the use of nucleophiles bearing otherwise reactive functional groups, such as aryl bromides, which could interfere with the cross-coupling process in the one-step procedure (Table 3, entry 3d), thus allowing for further functionalization of the initial carbamate products.
Table 3.
| |||
|---|---|---|---|
|
|
|
|
|
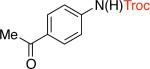
3ac,d 75% X = Cl |
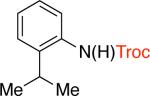
3be,f,g 91% X = OTf |
3cf,h 79% X = OTf |
|
|
|
|
|
|
3de,f 75% X = OTf |
3ec,f,g,j 86% X = OTf |
3fe,f,i 85% X1 = Cl, X3 = OTf |
3gc,f,j 83% X = OTf |

|
|
|
|
|
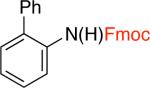
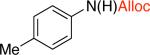
3hd,f,j,k 67% X = OTf |
3id,f,l 63% X = Cl |
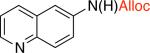
3jd,f,l 67% X = OTf |
|
Reaction conditions (isolated yields, average of 2 runs). Step 1: ArX (1 mmol), NaOCN (2 mmol), Pd2dba3 (x mol %), L1 (y mol %), toluene (2 mL). The Pd2(dba)3 and L1 were preheated in toluene at 120 °C for 3 minutes. Step 2: NuH (2 mmol), NEt3 (0.1 mmol).
Troc - 2,2,2-trichloroethoxycarbonyl, Cbz - carboxybenzyl, Fmoc - fluorenylmethyloxycarbonyl, Alloc - allyloxycarbonyl
NR3 = NEt3 (25 mol %)
Pd2dba3 (0.5 mol %), L1 (1.2 mol %)
Td2dba3 (1 mol %), L1 (2.4 mol %)
NR3 = TDA (10 mol %)
130 °C
Pd2dba3 (0.75 mol %), L1 (1.8 mol %)
NaOCN (3 mmol)), toluene (3 mL). Step 2: BnOH (3 mmol), NEt3 (0.2 mmol).
No NEt3 was used in Step 2.
0.5 mmol scale.
Step 2: PhI (0.1 mmol), 30 min at rt, then AllylOH (2 mmol), NEt3 (0.1 mmol).
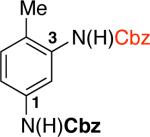
Aryl diisocyanates are key intermediates in the industrial production of polyurethanes and are usually produced by phosgenation of the corresponding dianilines.22 To examine the potential of accessing these building blocks using our methodology, 5-chloro-2-methylphenyl trifluoromethanesulfonate was subjected to the one-pot two-step procedure. The in situ generated aryl diisocyanate was reacted with benzyl alcohol to afford the bis-Cbz-protected dianiline in 85% yield (92%/each carbamate formed; Table 3, entry 3f).
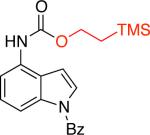
9-fluorenylmethyl carbamate (Fmoc) is a protecting group for amino acids widely used in peptide synthesis. It can be easily cleaved by weak bases, such as piperidine and triethylamine.23 Therefore, it is unsurprising that our standard conditions that employed triethylamine led to deprotection of the resulting carbamate and the formation of anilines as the major products. To overcome this problem, tris(2-(2-methoxyethoxy)ethyl)amine (TDA) was used in lieu of triethylamine.24 The use of this milder base did not affect the stability of the resulting carbamate products and the Fmoc-protected amines 3g and 3h were obtained in 83% and 67% yields (Table 3). TDA was also employed for the cross-coupling of certain aryl triflates, as it was found to improve the yields of the corresponding carbamate products (Table 3, entries 3b, 3c, 3e).
The synthesis of allyl carbamates typically requires the use of allyl chloroformate or allyl alcohol as the solvent, both of which are particularly hazardous substances. Therefore, applying the developed methodology to the synthesis of allyl carbamates would be advantageous, since it requires only a slight excess of the nucleophile. Allyl-esters and carbamates are known to easily undergo palladium(0)-catalyzed deprotection25 in the presence of nucleophiles. Formation of the palladium p-allyl complex from the allyl carbamate generates the deprotonated carbamic acid, which readily decarboxylates to give the free amine.26 In order to circumvent the undesired p-allyl pathway, the reactive palladium(0) species needed to be removed from the reaction mixture prior to the addition of allyl alcohol. Filtration of the reaction mixture through a plug of celite or silica gel did not remove the palladium species completely, which lead to diminished yields of the allyl carbamates. We envisioned that addition of an iodobenzene to the reaction mixture would provide an operationally simple procedure to rapidly convert the Pd(0) present to Pd(II), thus minimizing cleavage of the allyl carbamates without significantly altering the reaction conditions.27 Indeed, stirring the reaction mixture with 10 mol % of iodobenzene prior to the introduction of allyl alcohol provided carbamates 3i and 3j in 63% and 67% yields (Table 3) using only 2-fold excess of the allyl alcohol.
In summary, we have shown that using alcohols as nucleophiles in the palladium-catalyzed cross-coupling of aryl chlorides and triflates with sodium isocyanate provides a broad range of aryl carbamate products in good yields. The substrate scope of the direct isocyanate coupling producing aryl isocyanates in situ was extended by using aryl triflates instead of aryl chlorides, allowing for the use of alcohols and thiols not compatible with cross-coupling conditions.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM58160) for financial support of this project. N.H.P. acknowledges a National Science Foundation Graduate Research Fellowship. This activity was supported in part by an educational donation provided by Amgen. We thank James Colombe (M.I.T.) for helpful discussions and the Pentelute group (M.I.T.) for help in obtaining HRMS spectra. MIT has patents on the ligand used in this paper from which S.L.B. receives royalty payments.
Footnotes
Supporting Information Available. Experimental procedures, characterizations and spectral data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Baldinger SL, Shore ET. Formulary. 1996;31:1029. [Google Scholar]; b Nicolai E, Curtet S, Sicsic J, Lezoualc'h, Fischmeister R, Langlois M, Maillet M, Launay M. 2007 U.S. Patent 7238693.; c Li W, Li J, Wu Y, Wu J, Hotchandani R, Cunningham K, McFadyen I, Bard J, Morgan P, Schlerman F, Xu X, Tam S, Goldman SJ, Williams C, Sypek J, Mansour TS. J. Med. Chem. 2009;52:1799. doi: 10.1021/jm900093d. [DOI] [PubMed] [Google Scholar]
- 2.a Lee BS, Chun BC, Chung Y-C, Sul KI, Cho JW. Macromolecules. 2001;34:6431. [Google Scholar]; b Król P. Prog. Mater Sci. 2007;52:915. [Google Scholar]
- 3.Kocienski PJ. In: Protecting Groups: Thieme foundations of organic chemistry series. Enders D, Noyori R, Trost BM, editors. Thieme; Stuttgart; New York: 1994. pp. 192–209. [Google Scholar]
- 4.Chaturvedi D. Tetrahedron. 2012;68:15–45. and references therein. [Google Scholar]
- 5.Perron V, Abbott S, Moreau N, Lee D, Penney C, Zacharie B. Synthesis. 2008:283. [Google Scholar]
- 6.Carpino LA. Acc. Chem. Res. 1987;20:401. [Google Scholar]
- 7.Salvatore RN, Shin SI, Nagle AS, Jung KW. J. Org. Chem. 2001;66:1035. doi: 10.1021/jo001140u. [DOI] [PubMed] [Google Scholar]
- 8.Keillor JW, Huang X. Org. Syntheses. 2002;78:234. [Google Scholar]
- 9.Smith PAS. Org. React. 1946;3:337. [Google Scholar]
- 10.Dubé P, Fine Nathel NF, Vetelino M, Couturier M, Larrivée Aboussafy C, Pichette S, Jorgensen ML, Hardink M. Org. Lett. 2009;11:5622. doi: 10.1021/ol9023387. [DOI] [PubMed] [Google Scholar]
- 11.Wolff H. Org. React. 1946;3:307. [Google Scholar]
- 12.Paul F. Coord. Chem. Rev. 2000;203:269. [Google Scholar]
- 13.Tkatchenko I, Jaouhari R, Bonnet M, Dawkins G, Lecolier S. 19861988 FR2575467 U.S. Patent 4749806.
- 14.Kianmehr E, Baghersad MH. Adv. Synth. Catal. 2011;353:2599. [Google Scholar]
- 15.Yang X, Zhang Y, Ma D. Adv. Synth. Catal. 2012;354:2443. doi: 10.1002/adsc.201100831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vinogradova EV, Fors BP, Buchwald SL. J. Am. Chem. Soc. 2012;134:11132. doi: 10.1021/ja305212v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For other uses of pre-mixing or pre-heating the palladium-source and ligand to access the active catalyst species prior to introduction into the reaction mixture, see: Ueda S, Su M, Buchwald SL. J. Am. Chem. Soc. 2012;134:700. doi: 10.1021/ja2102373.Kotecki BJ, Fernando DP, Haight AR, Lukin KA. Org. Lett. 2009;11:947. doi: 10.1021/ol802931m.
- 18.The electronic nature of the aryl alcohol does affect the efficiency of the transformation. The reactivity of substituted phenols was evaluated in experiments with 4-chlorotoluene. Using more electron-rich 4-methoxyphenol did not significantly alter the carbamate yield. Both phenyl p-tolylcarbamate and 4-methoxyphenyl p-tolylcarbamate were obtained in >95% yield, determined by 1H NMR spectroscopy. However, no carbamate products were observed in the reactions with extremely electron-poor phenols, such as 4-cyanophenol, pentafluorophenol and 4-nitrophenol.
- 19.Hicks JD, Hyde AM, Cuezva AM, Buchwald SL. J. Am. Chem. Soc. 2009;131:16720. doi: 10.1021/ja9044357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis TL, Farnum JM. J. Am. Chem. Soc. 1934;56:883. [Google Scholar]
- 21.Isocyanates are versatile intermediates, which can be used in a number of processes. For other examples, showing the extent of the isocyanate reactions, see aza-Wittig cyclyzations: Marsden SP, McGonagle AE, McKeever-Abbas B. Org. Lett. 2008;10:2589. doi: 10.1021/ol800921n. Rh(III)-catalyzed amidations: Hesp KD, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2011;133:11430. doi: 10.1021/ja203495c. 1,3-dipolar cycloadditions: Holt J, Fiksdahl A. J. Heterocyclic Chem. 2007;44:375.
- 22.Seymour RB, Kauffman GB. J. Chem. Educ. 1992;69:909. [Google Scholar]
- 23.Fields GB. In: Methods in Molecular Biology. Pennington MW, Dunn BM, editors. Vol. 35. Humana Press Inc.; Totowa, NJ: 1994. pp. 17–27. [DOI] [PubMed] [Google Scholar]
- 24.TDA has been previously shown to facilitate the cross coupling of aryl chlorides with sodium cyanate: [12], supporting information.
- 25.Jellerichs BG, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2003;125:7758. doi: 10.1021/ja0301469. [DOI] [PubMed] [Google Scholar]
- 26.As expected, the presence of palladium in the reaction medium resulted in the deprotection and the formation of the corresponding anilines, in spite of the relatively mild conditions employed for the second step of the sequence (15 h at 45 °C).
- 27.Palladium(0)-complexes bearing biarylphosphine supporting ligands are known to undergo oxidative addition with sp2 carbon-halogen bonds at room temperature. Halogen = Cl, Br: Wolfe JP, Singer RA, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1999;121:9550.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.