

ABSTRACT
St. Louis encephalitis virus (SLEV) is the prototypic mosquito-borne flavivirus in the Americas. Birds are its primary vertebrate hosts, but amplification in certain mammals has also been suggested. The place and time of SLEV emergence remain unknown. In an ecological investigation in a tropical rainforest in Palenque National Park, Mexico, we discovered an ancestral variant of SLEV in Culex nigripalpus mosquitoes. Those SLEV-Palenque strains form a highly distinct phylogenetic clade within the SLEV species. Cell culture studies of SLEV-Palenque versus epidemic SLEV (MSI-7) revealed no growth differences in insect cells but a clear inability of SLEV-Palenque to replicate in cells from birds, cotton rats, and free-tailed bats permissive for MSI-7 replication. Only cells from nonhuman primates and neotropical fruit bats were moderately permissive. Phylogeographic reconstruction identified the common ancestor of all epidemic SLEV strains to have existed in an area between southern Mexico and Panama ca. 330 years ago. Expansion of the epidemic lineage occurred in two waves, the first representing emergence near the area of origin and the second involving almost parallel appearances of the virus in the lower Mississippi and Amazon delta regions. Early diversification events overlapped human habitat invasion during the post-Columbian era. Several documented SLEV outbreaks, such as the 1964 Houston epidemic or the 1990 Tampa epidemic, were predated by the arrival of novel strains between 1 and 4 years before the outbreaks. Collectively, our data provide insight into the putative origins of SLEV, suggesting that virus emergence was driven by human invasion of primary rainforests.
IMPORTANCE
St. Louis encephalitis virus (SLEV) is the prototypic mosquito-transmitted flavivirus of the Americas. Unlike the West Nile virus, which we know was recently introduced into North America from the Old World, the provenience of SLEV is obscure. In an ecological investigation in a primary rainforest area of Palenque National Park, Mexico, we have discovered an ancestral variant of SLEV. The ancestral virus was much less active than the epidemic virus in cell cultures, reflecting its incomplete adaptation to hosts encountered outside primary rainforests. Knowledge of this virus enabled a spatiotemporal reconstruction of the common ancestor of all SLEVs and how the virus spread from there. We can infer that the cosmopolitan SLEV lineage emerged from Central America in the 17th century, a period of post-Columbian colonial history marked by intense human invasion of primary rainforests. Further spread followed major bird migration pathways over North and South America.
Introduction
St. Louis encephalitis virus (SLEV) is the major representative of the Japanese encephalitis serocomplex (genus Flavivirus, family Flaviviridae) in the Americas. Since the first identification of SLEV in St. Louis, MO, in 1933, more than 50 outbreaks and numerous epidemics have occurred in the United States and southern Canada (1–4). The mortality rates of symptomatic patients in epidemics range from 5 to 20%, with a disproportionate impact on the elderly (5, 6). Although SLEV has been known to occur in Argentina and Brazil since the 1960s, human cases of SLEV infection have been reported only sporadically (7–10). The first confirmed outbreak of SLEV outside North America was reported in 2005 in Argentina and was followed by an outbreak in 2006 in Brazil (11–13). Most recently, infections associated with febrile illness and encephalitis in South America have increasingly been attributed to SLEV on the basis of viral isolation or serology (8, 11–13).
The geographic and ecological origins of SLEV remain obscure. Neither is there any knowledge of transhemisphere introduction, such as for West Nile virus, nor have ancestral variants of SLEV ever been found, such as for dengue or yellow fever virus, whose emergence can be traced back to sylvatic reservoirs (14, 15). In North America, where most of the investigations have been done, the epizootic and epidemic transmission cycles of SLEV involve Culex mosquitoes as vectors and passeriform and columbiform birds as amplificatory hosts. In the neotropics, small birds (family Formicariidae) and a variety of mammals (cingulates, bats, folivores, rodents, marsupials) have been found infected (16–20). Alternative transmission cycles involving mammals instead of birds, as well as atypical mosquito vectors belonging to the genera Aedes, Coquillettidia, Deinocerites, Mansonia, Psorophora, Sabethes, and Wyeomyia, have been reported (5, 16, 21–25). It is unclear whether these alternative hosts represent ecological dead ends for the virus or whether they can enable efficient virus maintenance.
SLEV strains generally cluster according to their geographic origin and can be divided into eight genotypes (I to VIII) (25, 26). Genotypes I and II circulate mainly in North America, and genotypes III to VIII circulate mainly in Central and South America. Genotype V is also prevalent in Florida (27). The strict geographic segregation of major phylogenetic clades has long been noted, and a phylogeographic analysis has suggested the Gulf of Mexico in an area between 15 and 30°N latitude to have acted as a common source of gene flow into North America (28). Other authors have suggested that genotype II, typical of the southeast Atlantic forest system in Brazil, may have been the first phylogenetic clade to emerge in the Americas (25). In this study, we characterized important biological properties of an unusual clade of SLEV identified during studies in a primary tropical ecosystem in Central America and conducted advanced phylogeographic analyses. In summary, our result suggest the discovery of ancestral SLEV in Central America and virus emergence in North and South America during colonial times.
RESULTS
Identification of novel SLEV strains.
In Palenque National Park, Mexico, 3,491 female mosquitoes were trapped in primary rainforest and adjacent disturbed habitat types (Fig. 1). Mosquitoes were tested by reverse transcription (RT)-PCR assay (371 pools consisting of 3,491 mosquitoes) for the genus Flavivirus. Insect-specific flaviviruses were detected in 152 pools (40.9%). Two pools yielded SLEV-related sequences. Retesting of all of the RNA pools with three different SLEV-specific RT-PCR assays revealed no further SLEV-positive pools. Screening of individual mosquitoes contained in those two SLEV-positive pools identified SLEV-related sequences in three individual Culex nigripalpus mosquitoes, two originating from primary rainforest (Palenque-A770 and Palenque-A772) and one originating from the forest edge (Palenque-C475). Mosquito species were confirmed by sequencing of cytochrome c oxidase I genes and phylogenetic analysis (see Data set S1 in the supplemental material). The concentrations of viral genome copies in mosquitoes ranged from 8 × 105 to 2 × 109 copies per ml, as measured by real-time RT-PCR (Table 1).
FIG 1.
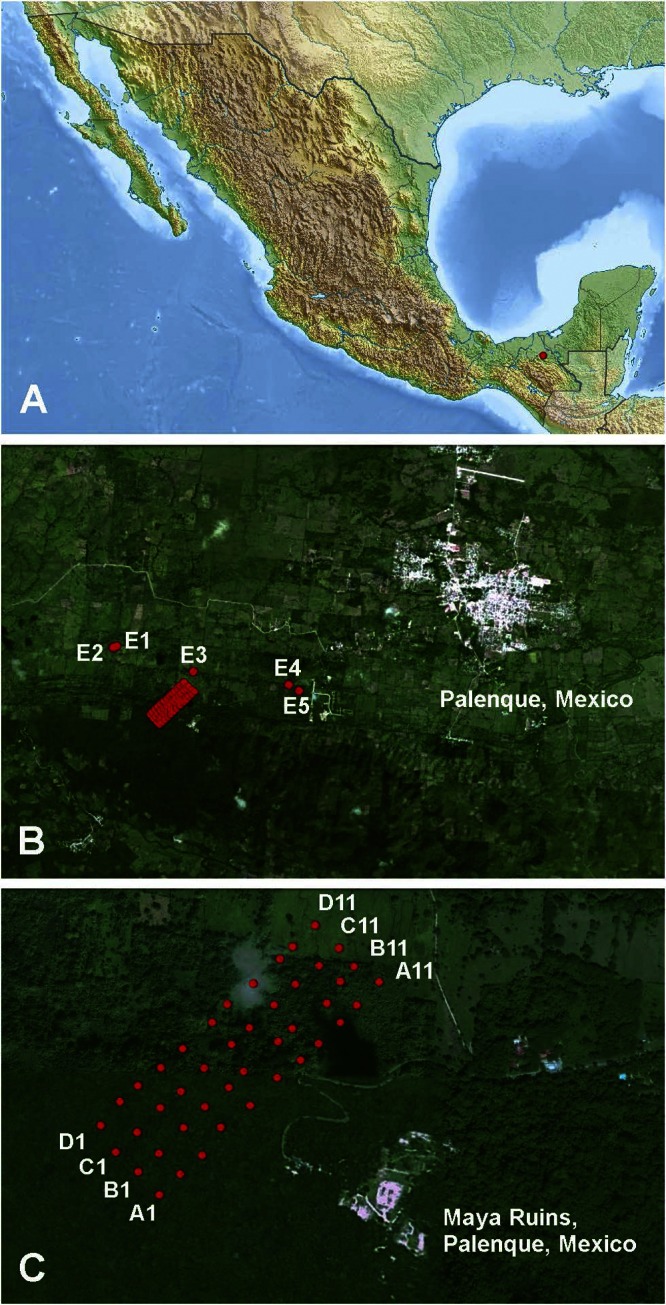
Study area. Locations in Palenque National Park, Mexico (A), mosquito sampling points in highly modified areas (B), and sampling points along parallel transects from natural to modified areas (C) are indicated by red dots. Map data: Google, Sanborn.
TABLE 1.
SLEV-Palenque strains detected in and around Palenque National Park, Mexico
| Strain | Trapping locationa | Habitat type | Host | No. of viral genome copies/ml |
|---|---|---|---|---|
| Palenque-A770 | 600679/1933486 | Primary forest | Culex nigripalpus | 2.5 × 109 |
| Palenque-A772 | 600679/1933486 | Primary forest | Culex nigripalpus | 8.1 × 105 |
| Palenque-C475 | 600624/1933711 | Forest edge | Culex nigripalpus | 1.9 × 108 |
Universal transverse Mercator coordinates, longitude/latitude.
The full genomes of Palenque-C475 and Palenque-A770, as well as the envelope and NS5 genes of Palenque-A772, were sequenced. The entire genome of the representative Palenque-A770 strain, here named SLEV-Palenque, comprised 10,938 nucleotides (nt) coding for a polyprotein of 3,430 amino acids (aa) flanked by a 5′ noncoding region (NCR) of 98 nt and a 3′ NCR of 550 nt (see Table S1A in Data set S2 in the supplemental material). SLEV-Palenque was distinct from all presently known SLEV strains, showing 94.2 to 95.7% amino acid identity within the predicted polyprotein. This level of genetic distance was congruent with the distance observed within accepted flavivirus species, such as Japanese encephalitis virus (below 9.9%) (29), West Nile virus (below 6.6%) (30), tick-borne encephalitis virus (below 8.7%) (31), or dengue virus types 1, 2, 3, and 4 (each below 5%) (32). It was thus assumed that SLEV-Palenque and all known SLEVs (genotypes I to VIII) form one species.
We tested for possible recombination events by using different substitution models in a single-breakpoint (SBP) analysis and genetic algorithms for recombination detection (GARD). No significant evidence of recombination was found; however, two borderline significant potential breakpoints were detected by GARD at positions 352 and 853 within the NS5 gene. Phylogenetic testing, with the Kishino-Hasegawa test or maximum-likelihood (ML) algorithms, of sequences on either side of the supposed breakpoints did not reveal any significant changes in topology. In addition, Bootscan tests did not yield any evidence of recombination. Phylogenetic analyses of the complete polyprotein, the E gene, the NS3 gene, and the NS5 gene were therefore conducted. Distance- and likelihood-based methods of inference from all data sets (neighbor-joining [NJ] and ML algorithms) placed SLEV-Palenque in a stable sister relationship to all other known SLEV isolates (Fig. 2). Noncoding regions, predicted cleavage sites, and the envelope protein gene were annotated as further described in Table S1B in Data set S2 in the supplemental material.
FIG 2.

Genetic and probabilistic distances of SLEV-Palenque from other viruses of the Japanese encephalitis serocomplex. NJ and ML phylogenies were invested for the NS3 (A), NS5 (B), and E (C) genes, as well as for the complete open reading frame (ORF) (D). ML analyses were performed with the GTR substitution model (NS5, E) and the HKY85 substitution model (NS3, ORF) with invariant sites, four gamma categories, and 1,000 bootstrap replicates with PHYML as implemented in Geneious. Bootstrap values of >60% are shown. NJ analyses were done with the p-distance model in Geneious and are shown on a smaller scale. Genetic lineages are named in accordance with the report by Auguste et al. (28), and genotypes as previously published by Kramer and Chandler (26) are shown in parentheses in panel D. Cosmopolitan strains are blue, sylvatic SLEV strains are red, and representative members of the Japanese encephalitis serocomplex are black.
Virus isolation and growth properties of cosmopolitan SLEV versus SLEV-Palenque.
Palenque-A770 was inoculated into C6/36 and Vero cells. A cytopathic effect (CPE) was observed in C6/36 cells at 4 days postinfection. No CPE was seen in Vero cells. Four further passages in both cell lines were performed. Virus replication was confirmed by real-time RT-PCR in both. To examine virus growth, C6/36 and Vero cells were infected with Palenque-A770 and SLEV reference strain MSI-7 (epidemic strain, high virulence in mice [33]) at multiplicities of infection (MOIs) of 0.1, 0.01, and 0.001. Viral genome copies were measured by real-time RT-PCR at 6-day intervals. Viruses showed only small differences in replication in C6/36 cells (Fig. 3A and B). Compared to MSI-7, Palenque-A770 showed increased infectivity down to an MOI of 0.001 in insect cells. In contrast, Palenque-A770 showed lower and slower replication than MSI-7 in Vero cells (Fig. 3C and D). At an MOI of 0.001, no growth of Palenque-A770 was seen while MSI-7 readily grew to a peak concentration of 1 × 1010 RNA copies per ml within 3 days. Furthermore, in contrast to MSI-7, Palenque-A770 did not induce plaques in Vero cells.
FIG 3.
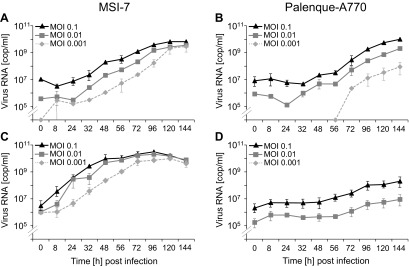
Replication of Palenque-A770 and MSI-7 in insect and primate cells. Numbers of virus genome copies per milliliter (cop/ml) of cell culture supernatant were measured by real-time RT-PCR over 6-day periods. C6/36 and VeroE6/7 cells were infected with MSI-7 (A, C) or Palenque-A770 (B, D) virus at MOIs of 0.1, 0.01, and 0.001, respectively. Each MOI was measured in independent duplicates.
Because SLEV is maintained in a transmission cycle involving birds of the orders Columbiformes and Passeriformes, we tested infectivity in primary cells derived from the mourning dove (Zenaida macroura, order Columbiformes, MD11), the gray catbird (Dumetella carolinensis, order Passeriformes, GC2), and the house sparrow (Passer domesticus, order Passeriformes, HS1). In contrast to MSI-7, no replication of Palenque-A770 was found in any of the bird cells tested (Fig. 4A and B).
FIG 4.
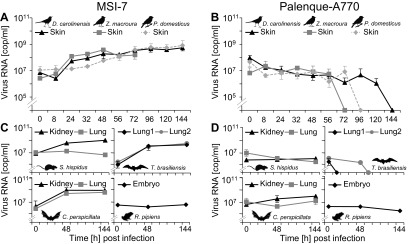
Growth of Palenque-A770 compared to that of MSI-7 in proposed vertebrate host species. Primary fibroblasts derived from a gray catbird (D. carolinensis, GC2), a mourning dove (Z. macroura, MD11), and a house sparrow (P. domesticus, HS1), as well as cells derived from a cotton rat (S. hispidus; S-his-Lu, S-his-Ki cells), a free-tailed bat (T. brasiliensis; Tb1 Lu, Tb2 Lu), a fruit bat (C. perspicillata; C-per-Lu, C-per-Ki), and a grass frog (R. pipiens; ICR2-A) were infected with MSI-7 (A, C) or Palenque-A770 (B, D) virus at an MOI of 0.1. Each MOI was measured in independent duplicates. Numbers of virus genome copies per milliliter (cop/ml) of cell culture supernatant were measured by one-step real-time RT-PCR over 6-day periods. Supernatant of MD11 was removed over 3-day periods. Where no standard deviation is shown, the scale is too small to be visible.
Because SLEV is suspected to have additional natural hosts in certain mammalian species, including cotton rats (Sigmodon hispidus) and Mexican free-tailed bats (Tadarida brasiliensis) (19, 20, 34), cells from those species were tested as well. MSI-7 replicated in the majority of the cell lines from those species, while SLEV-Palenque did not (Fig. 4C and D). Because of the occurrence of neotropical fruit bats in primary neotropical rainforests such as the study area, several cell lines were also generated from the abundant fruit bat species Carollia perspicillata. Kidney cells were permissive for SLEV-Palenque, albeit to a small extent (Fig. 4C and D). Because Culex nigripalpus mosquitoes carrying SLEV-Palenque are known to feed on amphibians (35), infection experiments were also done with a frog cell line (ICR-2A). However, no growth of either virus was observed.
In order to provide additional support for the genetic evidence of the conspecificity of SLEV and SLEV-Palenque, we investigated serological reactivity. Although there are no clearly defined criteria, Flavivirus species can generally be discriminated in infection neutralization tests in mammalian cells. To implement neutralization testing, the virtual inability of SLEV-Palenque to grow in mammalian cell cultures had to be overcome by exploratory testing of a large number of cell lines commonly used for virus isolation. BHK-J cells, which are deficient in several components of cellular innate immunity and are known to provide exceptional permissibility for several flavivirus species, were found to support SLEV-Palenque growth, albeit to a 10-fold lesser extent than MSI-7 (peak titers, 5 × 106 versus 5 × 107 RNA copies per ml supernatant, respectively). With a standardized, commercial anti-SLEV serum, BHK-J cells were infected at MOIs of 0.1 to 0.0001 in quadruplet assays per dose, with and without anti-SLEV serum at a 1:32 dilution. In each of the parallel assays, the anti-SLEV serum reduced SLEV-Palenque infectivity at least 10-fold. The reduction was even more pronounced than with MSI-7. In contrast, the same assay performed with West Nile virus strain NY showed no reduction of virus infectivity in any of four parallel experiments with the same serum at the same dilution. It was concluded that SLEV-Palenque is serologically related to SLEV but not to the next closest flavivirus, West Nile virus.
Mapping the spread of SLEV.
Knowledge of a conspecific clade in a sister relationship to all previously known SLEVs enabled coalescent-analysis-based inference of phylogeny. To reconstruct the spread of major SLEV stem lineages, global positioning system (GPS) coordinates and times of existence of sequences at ancestral tree nodes were reconstructed along with phylogeny in a Bayesian coalescent analysis with a relaxed random walk model in BEAST (36, 37). Figure 5 shows a maximum clade credibility (MCC) tree that summarizes the 133 taxa contained in the analysis (see Data set S3 in the supplemental material for more information on the composition of the data set, including all virus designations and GenBank accession numbers).
FIG 5.

Dated phylogeny of SLEV, including SLEV-Palenque. The MCC tree shown was calculated in BEAST as indicated. The tree contains the 133 E gene sequences used for phylogeographic reconstruction. Major clades have been collapsed. For a complete representation of this tree with all taxon names and accession numbers, see Data set 3 in the supplemental material. The scale is in years before the present, with the present time set at 2008 AD.
The inferred geographical coordinates of the common ancestor of all previously known SLEVs (genotypes I to VIII) were plotted in Google Maps (Fig. 6). Here, this common ancestor is referred to as the cosmopolitan clade ancestor (CCA). A comparison of Fig. 6A and B suggests that the geographic placement of the CCA was more robust when tree inference based on the Hasegawa, Kishino, and Yano (HKY) substitution model was used than when general time reversible (GTR)-based inference was used (this was systematically observed in parallel runs). To reflect our assumption that SLEV-Palenque-related strains might originally have been distributed more widely than in the region of our study, a parallel analysis was conducted with a slight extension of the hypothetical area of distribution of SLEV-Palenque strains. To implement this, coordinates of the most variant SLEV-Palenque strain, C475, were deliberately placed in the Amazon region at 0°N and 65°W while keeping those of the other two strains unchanged at 17°N and 92°W (original place of SLEV-Palenque isolation). Interestingly, the geographic placement of the CCA obtained better focus with this modification (Fig. 6C). Again, this was systematically observed in parallel runs.
FIG 6.
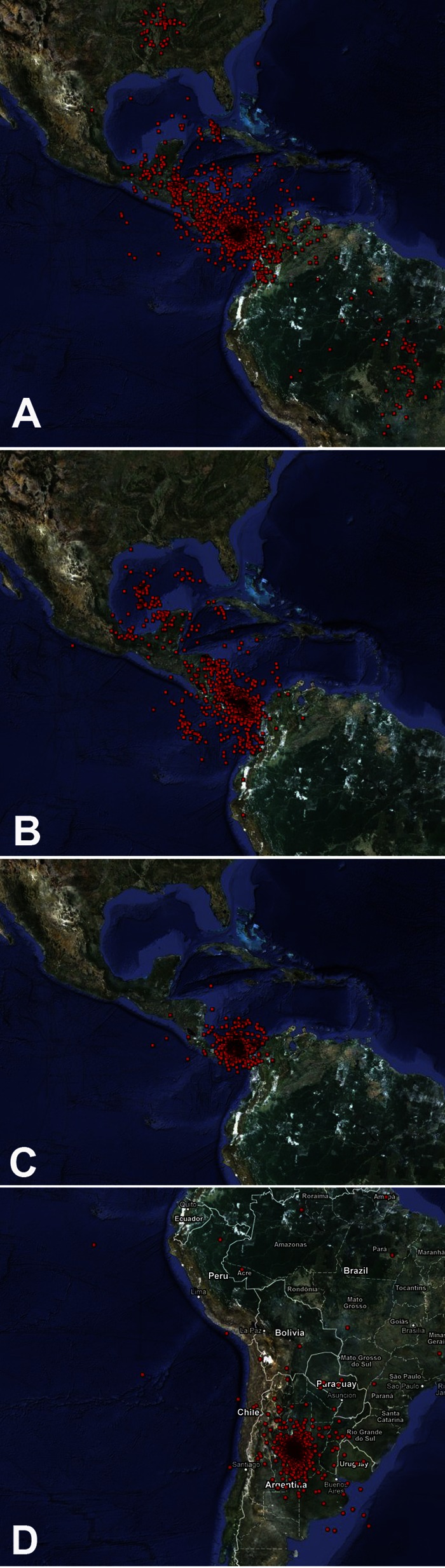
Root point locations under different phylogenetic models with and without knowledge of SLEV-Palenque. Red dots symbolize reconstructed geographic locations of the MRCAs of all cosmopolitan SLEVs (all SLEVs except SLEV-Palenque). Projections for 2,000 trees from the stationary phase of Bayesian phylogeographic analyses are shown. (A) Typical result based on phylogenetic trees constructed under the GTR model. (B) Typical result based on inference and the HKY model. (C) Typical result based on the same approach as in panel B but moving the geographic coordinates of only SLEV-Palenque C475 to 0°N and 65°W. (D) Typical result obtained from a run after the removal of all SLEV-Palenque strains. The image was drawn in GPS Visualizer on the basis of data extracted from marginal tree files by TreeStat (BEAST package). Map data: Google, Sanborn.
To further challenge the outcome of the analysis, the coordinates of all three SLEV-Palenque strains were modified simultaneously so as to simulate SLEV-Palenque placements further north and south, up to northern Mexico and down to the southern Amazon region. The collective results of these analyses are presented in Data set S4 in the supplemental material. The CCA projection for an analysis completely omitting SLEV-Palenque is shown in Fig. 6D. These simulations, in summary, suggest that the inclusion of SLEV-Palenque was a necessary prerequisite for the inference of the proposed CCA location.
In order to test whether knowledge of SLEV-Palenque might also have an influence on the spatial placement of hierarchically higher tree nodes, the projected locations of the most recent common ancestors (MRCAs) of three major clades were extracted from marginal tree densities with and without the inclusion of SLEV-Palenque (Fig. 7). Latitude traces were analyzed, as they were generally less stable than longitude traces (not shown). The root point marked A in Fig. 7 represents the ancestors of the 1964 Houston epidemic. Point B is the MRCA of all United States strains. The latitudes of these high- and intermediate-level nodes were hardly affected by the inclusion of SLEV-Palenque (compare black data traces for the model with SLEV-Palenque and green for the model without). A major loss of stability was seen in the inference of the root point latitude of the major lineages occurring in Brazil (root point C). While the common ancestor of these viruses was stably placed in the Amazon delta region on the basis of the full data set, it could not be inferred from the data set lacking the SLEV-Palenque strains because of a failure of the latitude trace to converge. This failure affected the latitude placement of the roots of all deeper branches leading up to this node (compare latitude-dependent coloring of branches leading to node C between trees in Fig. 7).
FIG 7.

Effect conferred by the knowledge of SLEV-Palenque on geographic reconstruction. Shown are MCC trees extracted by Bayesian phylogeographic analyses without (right, top tree) and with (right, bottom tree) SLEV-Palenque. The circle in each tree symbolizes the root point of the cosmopolitan tree. Tree branches are colored according to the latitude of the nodes they lead to; the darker they are, the further south the latitude location. Three deep nodes are identified in both trees correspondingly, i.e., the MRCA of the 1964 SLEV outbreak in St. Louis, MO (A); the MRCA of all of the viruses isolated in the United States (B); and the MRCA of all of the viruses derived from an early singular introduction of SLEV in the Amazon delta region (C). At the left are the latitude traces extracted from the same runs for root points A, B, and C. In each of the panels, the black trace represents node-specific latitude inferences collected during the whole run of the data set with SLEV-Palenque while the green trace represents results obtained with the data set without SLEV-Palenque.
Plotting of MCC trees in SPREAD suggested an emergence of the CCA from its original location in the Panama region by the year 1687, followed by short-range transmission movements and a split into two major stem lineages that reached the Mississippi region in the area of New Orleans between 1722 and 1744 and the Amazon delta region between 1736 and 1810, respectively (Fig. 8A to D). Argentinian strains were suggested to have resulted from an independent introduction from Middle America. Major lineages of North American SLEV strains (including the introduction of novel major lineages into geographic regions where other SLEV lineages were previously present) reached St. Louis around 1875, Texas around 1902, California around 1944, Florida around 1950, central Mississippi around 1955, Texas around 1963, and Florida around 1989 (see Data set S5 in the supplemental material, which is provided separately as a .kml file at http://www.virology-bonn.de/index.php?id=28 [this file requires Google Earth to be installed for viewing; it can be imported into Google Earth to obtain a fully animated spatiotemporal reconstruction of SLEV spread]). The major influence of SLEV-Palenque on deep tree node placements became evident in the deviating reconstruction of spatiotemporal spread of SLEV, as shown in Fig. 8E to H. The analysis ignoring SLEV-Palenque suggested a CCA emergence from Argentina with parallel transmissions to the Amazon region, southern Brazil, and the Mississippi delta, from which the Central American strains would have been secondarily derived.
FIG 8.
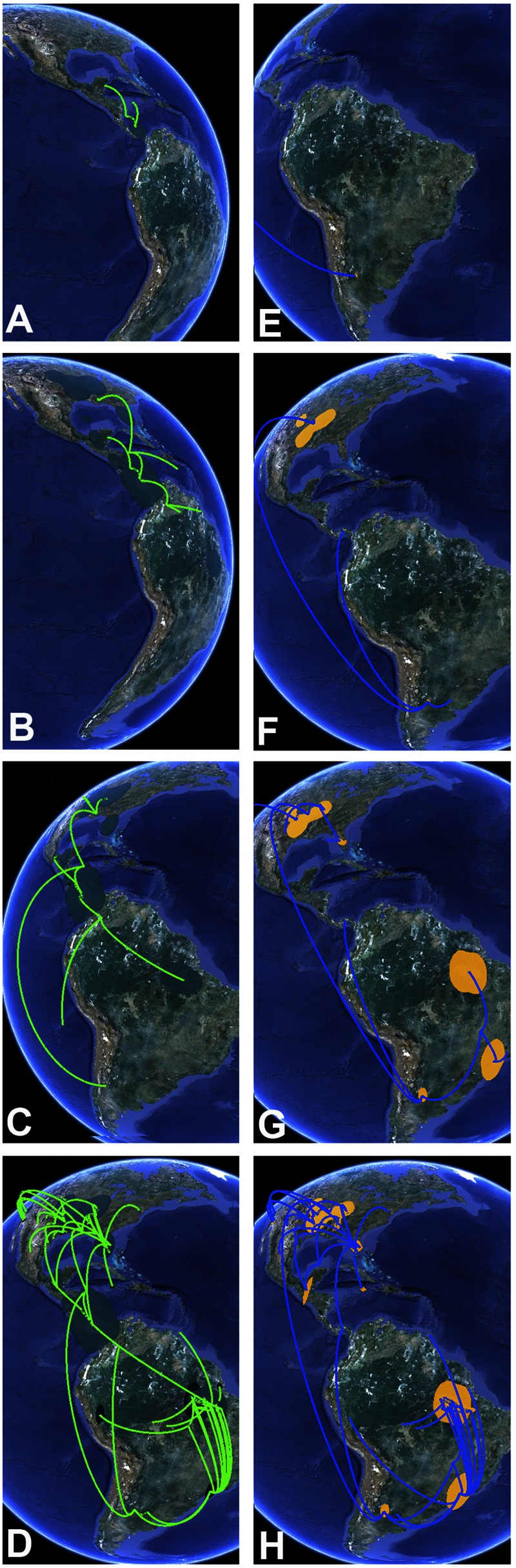
Excerpt from a continuous projection of SLEV phylogeography with and without knowledge of SLEV-Palenque. Shown is the projected spread of the geographic range of root point locations (colored polygons on the map), as well as hypothetical positions of major tree branches (lines) over time. A to D, snapshots from a reconstruction, including SLEV-Palenque; E to H, snapshots from a reconstruction after the removal of SLEV-Palenque. Panel elements represent snapshots taken in Google Earth by using different time ruler positions and appropriate globe rotation angles. Time projections in each panel element roughly correspond to the years 1710 (A), 1740 (B), 1870 (C), 2008 (D), 1750 (E), 1780 (F), 1890 (G), and 2008 (H). Map data: Google, Sanborn.
DISCUSSION
Here we have identified a novel clade of SLEV from a tropical rainforest region in southern Mexico, highlighting tropical forests as ecosystems with high pathogen diversity. SLEV-Palenque is a phylogenetic outgroup conspecific with SLEV genotypes I to VIII. The idea that SLEV-Palenque might be a putatively sylvatic virus was supported by its very low detection rate of ca. 0.1% (3/3,491), matching the concept that the prevalence of viruses in sylvatic (enzootic) amplification cycles should be lower than in epizootic cycles because of a lower host density (38). The low replication level of the virus in cell culture also matches this idea, as sylvatic viruses are generally believed to show lower virulence than epidemic strains (39). SLEV-Palenque reached a 100-fold-lower concentration in Vero cells than MSI-7 did and took three times as long to reach its peak concentration. Notably, in contrast to MSI-7, SLEV-Palenque did not replicate in primary fibroblasts derived from birds of the orders Columbiformes and Passeriformes (D. carolinensis, P. domesticus, Z. macroura). These birds are assumed to be major amplificatory hosts of cosmopolitan SLEV, and skin fibroblasts should be among the cells primarily exposed to the virus by mosquito bites. It should be noted, however, that primary dendritic cells could constitute another primary target, which has not been assessed here. Still, the overall contrast between SLEV-Palenque and MSI-7 was striking. In cells from presumed mammalian SLEV hosts (19, 20, 40), no or low replication of SLEV-Palenque was seen while MSI-7 replicated well. This is interesting because it suggests that SLEV-Palenque would have to be maintained in different transmission cycles than epidemic SLEV.
For epidemic SLEV, it is unclear how the virus is maintained in mosquito populations and what range of vertebrate species participate in amplification cycles (5). Passeriform and columbiform birds seem to be the principal summer vertebrate hosts of epizootic SLEV in North America. However, antibodies against SLEV have been detected in rodents (9, 41), bats (19, 20, 40), rats (34), and armadillos (10, 42), suggesting that mammals participate in SLEV transmission. Interestingly, SLEV-Palenque was isolated from Culex nigripalpus mosquitoes, a common vector of SLEV in Florida and Central and South America (43–46). Culex nigripalpus feeds primarily on birds but also on mammals, including humans, horses, cattle, dogs, rabbits, rodents, and armadillos, as well as lower vertebrates, including lizards and frogs. Notably, Culex nigripalpus shows a seasonal shift from avian hosts in the spring to mammalian hosts in the summer (47). Culex nigripalpus mosquitoes thus may be able to act as bridging vectors between enzootic and epidemic amplification cycles (48). A shift in their feeding behavior because of habitat invasion may have facilitated the escape of SLEV from its original putative sylvatic amplification cycle. Knowledge of the transmission of SLEV-Palenque in highly promiscuous Culex nigripalpus mosquitoes may uncover unknown vertebrate hosts in South America and could unravel how the virus overwinters in North America.
The advent of improved probabilistic approaches in phylogeny creates new opportunities to reconstruct viral phylogeography (49, 50). In the present study, we have used templates from seminal studies on other viruses and applied them to the case of SLEV (36, 49). Their application was based on the fact that we could trust SLEV-Palenque and all other strains of SLEV to form one viral species. By definition, coalescent-analysis-based phylogenies, including their inherent population size assumptions, should be applied only to viral species fulfilling, at least theoretically, the criterion of panmixia at the hypothetical starting point (in time) of the observed population.
The application of novel methodology should involve comparisons with established approaches. The SLEV E gene is already a well-studied data set on grounds of the hitherto-used approach of parsimony analysis (28). We could therefore validate our approach of continuous trait reconstruction by comparison with these existing results.
The parsimony-based study by Auguste et al. suggested a region between 15 and 30°N latitude to be the major source of SLEV gene flow in North America, which is in very good agreement with our results (28). Another study not employing any specific ancestral reconstruction algorithms suggested that SLEV might alternatively have emerged from South America and spread to North America because of the high virus diversity observed in Brazil (25). However, all of the data from that study are already included in our data set, leading to a different conclusion that is in better agreement with that of Auguste et al. It was nevertheless interesting that our analysis yielded an origin in South America when excluding the knowledge of SLEV-Palenque. The projected origin region in northern Argentina would be suitable as a place of virus emergence, as warm summer temperatures allow efficient extrinsic incubation of virus in insects and intermittent rainfall events synchronize oviposition and blood feeding, facilitating transmission (48, 51). However, the placement of this origin was contributed simply by the existence of Argentinian strains in both basal sister clades of the South American stem lineage. This placement was overruled by the inclusion of the Palenque viruses, leading to a root point placement in the area of Panama. Interestingly, this overruling effect was maintained only if the isolation place of the Palenque strains was not dislocated too far north or south of its true location. Since there was considerable variation in these analyses in the distance between the isolation place and the root point projection in Panama, we can reject the idea that the placement of the root simply follows the positioning of the outgroup locality.
The phylogeographic algorithm employed in our study reconstructed the times of origin of MRCAs together with their geographic locations. Earlier studies suggested the emergence of the cosmopolitan strains from their MRCA to have occurred around 214 (28) or 107 (25) years ago. Again, our result was closer to that achieved by Auguste et al. (28). In our analysis, the MRCA of the cosmopolitan clade was projected to have existed until ca. 330 years ago.
Projection of the MRCA in an area including Nicaragua, Costa Rica, Panama, and northern Colombia seemed to be congruent with biological and historical considerations. At that time, this area was at the center of the Spanish colonial empire in Middle America (52). It was already a place with colonial settlements from which intensive exploration of forested regions began. The Gulf of Mexico was an area of busy trade and transport by sea, with ships landing and departing from harbors in the whole Caribbean and Gulf region, including the southeastern colonies in today’s United States. The reconstruction projected the cosmopolitan clade to have undergone several initial local transmissions in this area and to have departed from there to the southeastern United States, including locations that could have been reached by sea transport. Displacement of viruses released from sylvatic habitats could also have occurred via migratory birds visiting the region as part of their annual migrations along the Mississippi flyway (53). The Gulf region, moreover, is connected to the Amazon delta by a separate migratory route, the Atlantic flyway (53). Strict separation of the Mississippi and Atlantic flyways can explain why the viruses were separated into two basal sister clades after leaving their hypothetical common origin. Interestingly, the model projects a third basal movement of viruses from northern Colombia to Argentina that would be congruent with the Pacific flyway (53). The origin in Middle America is thus highly concordant with a hypothesis under which human invasion during the post-Columbian era triggered an escape of virus from sylvatic habitats into epizootic cycles accessible to migrating birds. Their migration routes blend in the predicted area of virus emergence. In contrast, the alternative model ignoring SLEV-Palenque suggested a direct initial distribution from northern Argentina to the southeastern United States. While this way of dispersal would be conceivable along the extremes of the Atlantic flyway, it would be highly unlikely that no virus lineages were founded along the way. Long-distance transport requires prolonged viremia in migrating birds without any disadvantage conferred by virus infection.
Finally, it was highly interesting that even recent epidemiological events were reflected in the phylogeographic reconstruction. For instance, a new virus lineage was projected to have arrived in Houston, TX, in 1963, 1 year before the actual Houston SLEV epidemic with over 500 human cases (5, 54). Another new introduction was projected in Tampa, FL, in 1990, 1 year before the Tampa outbreak with over 200 cases (5, 54). Moreover, the largest ever recorded SLEV epidemics occurred between 1975 and 1980 in the central eastern United States, causing around 2,000 human cases (5, 54). In our reconstruction, a virus lineage arriving by 1955 in Mississippi was projected to have spread further north by 1968, and it is conceivable that this lineage was involved in those outbreaks.
In summary, the knowledge of a conspecific and sylvatic sister clade of SLEV has enabled the deployment of advanced phylogeographic methodology, linking the emergence of a major epidemic arbovirus with human ecosystem invasion during colonial times. It will be highly relevant to apply this methodology to other virus models in the future. In particular, the phylogeographic investigation of sylvatic relatives of other arboviruses might enhance our understanding of the timing and localization of virus emergence in general.
MATERIALS AND METHODS
Mosquito collection and species identification.
Adult mosquitoes were collected with BG-Sentinel traps (Biogents, Regensburg, Germany) in the area of Palenque National Park in the Mexican state of Chiapas between July and September 2008. Permits were approved by the Mexican environment agency Semarnat and by the Mexican National Autonomous Institute of Biology. For species identification, see Data set S1 in the supplemental material.
Cell culture.
Primary fibroblast cells from D. carolinensis, P. domesticus, and Z. macroura were generated by J.H. and R.M. and cultivated as described previously (55) but without oxygen regulation. For establishment of lung and kidney cell lines from cotton rats (S. hispidus, S-his-Lu and S-his-Ki) and lung cells from short-tailed bats (C. perspicillata, C-per-Lu), one single adult animal of each species bred in breeding colonies was euthanized and dissected. Tissues fragments were placed in a six-well cell culture plate (PAA) and submerged in 37°C serum-free cell culture medium (PromoCell, Heidelberg, Germany). All primary cell culture media were also supplemented with penicillin-streptomycin (PAA), ofloxacin (Tarivid; Sanofi-Aventis), and amphotericin B (PAA). When cells had nearly reached confluence, they were immortalized by lentiviral transduction of the large T antigen of simian virus 40 as described previously (56). After four or five passages, cells were adapted to standard conditions (Dulbecco’s modified Eagle’s medium [DMEM] containing 10% fetal calf serum [FCS], 1% glutamine, 1% nonessential amino acids, 1% sodium pyruvate, and 1% penicillin-streptomycin at 37°C with 5% CO2). Primary kidney cells from inbred C. perspicillata (C-per-Ki) were generated as described previously (56) and cultivated under the standard conditions described above. Lung cells derived from free-tailed bats (T. brasiliensis), Tb1 Lu and Tb2 Lu, were obtained from the American Type Culture Collection (catalogue number CCL-88) and the Interlab Cell Line Collection, Genoa, Italy (catalogue number ATL96010), respectively, and cultivated with DMEM containing 5% FCS at 37°C with 5% CO2. ICR-2A cells derived from grass frog (Rana pipiens) embryos were obtained from the European Collection of Cell Cultures, Porton Down, Salisbury, United Kingdom (catalogue number 89072615), and cultivated at 28°C with medium containing L15 (50%), distilled water (40%), FCS (10%), and glutamine. SLEV strain MSI-7 (accession number AY289618) was obtained from the National Collection of Pathogenic Viruses, Porton Down, Salisbury, United Kingdom.
Screening PCRs and genome sequencing.
Pools were generated by using 100 µl of supernatant of 10 individually homogenized mosquito suspensions. RNA was extracted with the QIAamp viral RNA minikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. cDNA synthesis was performed with the SuperScript III RT system (Life Technologies GmbH, Darmstadt, Germany) and random hexamers. Mosquito pools were pretested for flavivirus infection by a genus-specific RT-PCR (57). SLEV-specific RT-PCRs used primer pairs A1-F (5′-GCTGGCGTTGGTGTGATG) and A1-R (5′-CTCTTGAATGATGCCACTAACG) for the NS3 gene, B1-F (5′-ACGAAGACCATTGGATGAGC) and B1-R (5′-CACTCACTGCCATCCTGCT) for the NS5 gene, and C1-F (5′-CATGCAGGAACCAGGATGAG) and C1-R (5′-ATGTTCTCAGCCCAAGTTGC) for the NS5 gene, respectively. PCR products were used for seminested PCRs with A1-F and A2-R (5′-CTCTCCTTTCCTGTTGATGATTG), B2-F (5′-AAGATGTACGCAGACGACACAG) and B1-R, and C1-F and C2-R (5′-CTATCAAGCTTCCGCACCA), respectively. Full-genome sequencing was achieved by applying primer-walking techniques with viral RNA extracted from individual mosquitoes or infectious cell culture supernatant of C6/36 cells. The 5′ and 3′ untranslated regions were determined by rapid amplification of cDNA ends (RACE)-PCR with the 5′ RACE System (Life Technologies GmbH, Darmstadt, Germany). All of the primers used are available from S.J. on request.
Phylogenetic analyses.
Testing for recombination was done by SBP analysis and GARD on the Datamonkey server (58). Phylogeographic investigations involved Bayesian phylogenetic analysis with simultaneous reconstruction of the geographic coordinates and times of existence of phylogenetic root points by a continuous relaxed random walk approach in BEAST v. 1.6 (37). Initial runs used the parameters proposed in reference 36. In extensive optimizations, the substitution model (GTR versus HKY), trait randomization parameters, and chain operators were tuned to lose and regain convergence in order to determine how each of the parameters affects the efficiency of chain convergence. GTR-based substitution models were compared to HKY-based models. Efficiency of mixing was controlled by observation of chains in TRACER. Efficiency of convergence was tested by defining grossly misplaced root priors and then confirming departure from these priors after short runs. For all analyses, parallel chains were observed and results were accepted only if chains converged on equivalent root point locations. Selected latitude and longitude traces were extracted from tree files with TREESTAT (BEAST package), stripped to ca. 10,000 trees by removal of data from the burn-in period in Excel, and plotted in Google Maps in GPS Visualizer by using 1,200 waypoint values.
Nucleotide sequence accession numbers.
The sequences determined in this study have been deposited in GenBank under accession numbers JQ957868 to JQ957871 and JQ957876.
SUPPLEMENTAL MATERIAL
Mosquito species identification. Download
Annotations of the viral polyprotein, noncoding genome regions, and envelope protein. Download
Assembly of the data set for phylogeographic reconstruction. Download
Experimental modifications of geographic outgroup placement and influence of topological uncertainty in deep tree nodes. Download
ACKNOWLEDGMENTS
We thank Juan Francisco and Juan Montejo for field assistance, as well as the Mexican authorities and the directory of the Palenque National Park for long-term support. We thank Sabine Specht (Institute of Medical Microbiology, Parasitology, and Immunology, University of Bonn) for providing S. hispidus from a breeding colony and Karl-Heinz Esser (Institute of Zoology, University of Veterinary Medicine, Hannover, Germany) for providing C. perspicillata from a breeding colony. We are grateful to Biogents, Regensburg, Germany, for support with insect traps and Jan Felix Drexler and René Kallies (Institute of Virology, University of Bonn) for advice and technical assistance. We thank Jonas Schmidt-Chanasit (Bernhard Nocht Institute, Hamburg, Germany) for kindly providing West Nile virus.
This project was supported by European Union DG Research through the programs EMPERIE (grant agreement no. 223498) and EVA (grant agreement no. 228292) to C.D., as well as intramural grants from the University of Bonn Medical Center to S.J. (grant agreement no. O-156.0006), the Robert Koch Institute to F.H.L. and Emory University to T.R.G. F.H.L. acknowledges financial support from the Global Viral Forecasting Initiative. The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
Footnotes
Citation Kopp A, Gillespie TR, Hobelsberger D, Estrada A, Harper JM, Miller RA, Eckerle I, Müller MA, Podsiadlowski L, Leendertz FH, Drosten C, Junglen S. 2013. Provenance and geographic spread of St. Louis encephalitis virus. mBio 4(3):e00322-13. doi:10.1128/mBio.00322-13.
REFERENCES
- 1. Chamberlain RW. 1980. History of St. Louis encephalitis, p 3–61 In Monath TP, St. Louis encephalitis. American Public Health Association, Washington, DC [Google Scholar]
- 2. Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Crise B, Volpe KE, Crabtree MB, Scherret JH, Hall RA, MacKenzie JS, Cropp CB, Panigrahy B, Ostlund E, Schmitt B, Malkinson M, Banet C, Weissman J, Komar N, Savage HM, Stone W, McNamara T, Gubler DJ. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286:2333–2337 [DOI] [PubMed] [Google Scholar]
- 3. Lumsden LL. 1958. St. Louis encephalitis in 1933; observations on epidemiological features. Public Health Rep. 73:340–353 [PMC free article] [PubMed] [Google Scholar]
- 4. Monath TP, Tsai TF. 1987. St. Louis encephalitis: lessons from the last decade. Am. J. Trop. Med. Hyg. 37(3 Suppl):40S–59S [DOI] [PubMed] [Google Scholar]
- 5. Reisen WK. 2003. Epidemiology of St. Louis encephalitis virus. Adv. Virus Res. 61:139–183 [DOI] [PubMed] [Google Scholar]
- 6. Tsai TF, Mitchell CJ. 1988. St. Louis encephalitis, p 431–458 In Monath TP. (ed), The arboviruses: epidemiology and ecology. CRC Press, Boca Raton, FL [Google Scholar]
- 7. Mettler NE, Casals J. 1971. Isolation of St. Louis encephalitis virus from man in Argentina. Acta Virol. 15:148–154 [PubMed] [Google Scholar]
- 8. Rocco IM, Santos CL, Bisordi I, Petrella SM, Pereira LE, Souza RP, Coimbra TL, Bessa TA, Oshiro FM, Lima LB, Cerroni MP, Marti AT, Barbosa VM, Katz G, Suzuki A. 2005. St. Louis encephalitis virus: first isolation from a human in São Paulo State, Brazil. Rev. Inst. Med. Trop. Sao Paulo 47:281–285 [DOI] [PubMed] [Google Scholar]
- 9. Sabattini MS, Monath TP, Mitchell CJ, Daffner JF, Bowen GS, Pauli R, Contigiani MS. 1985. Arbovirus investigations in Argentina, 1977–1980. I. Historical aspects and description of study sites. Am. J. Trop. Med. Hyg. 34:937–944 [PubMed] [Google Scholar]
- 10. Vasconcelos PF, Da Rosa JF, Da Rosa AP, Dégallier N, Pinheiro Fde P, Sá Filho GC. 1991. Epidemiology of encephalitis caused by arbovirus in the Brazilian Amazonia. Rev. Inst. Med. Trop. Sao Paulo 33:465–476 [PubMed] [Google Scholar]
- 11. Diaz LA, Ré V, Almirón WR, Farías A, Vázquez A, Sanchez-Seco MP, Aguilar J, Spinsanti L, Konigheim B, Visintin A, Garciá J, Morales MA, Tenorio A, Contigiani M. 2006. Genotype III Saint Louis encephalitis virus outbreak, Argentina, 2005. Emerg. Infect. Dis. 12:1752–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mondini A, Cardeal IL, Lázaro E, Nunes SH, Moreira CC, Rahal P, Maia IL, Franco C, Góngora DV, Góngora-Rubio F, Cabrera EM, Figueiredo LT, da Fonseca FG, Bronzoni RV, Chiaravalloti-Neto F, Nogueira ML. 2007. Saint Louis encephalitis virus, Brazil. Emerg. Infect. Dis. 13:176–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spinsanti LI, Díaz LA, Glatstein N, Arselán S, Morales MA, Farías AA, Fabbri C, Aguilar JJ, Ré V, Frías M, Almirón WR, Hunsperger E, Siirin M, Da Rosa AT, Tesh RB, Enría D, Contigiani M. 2008. Human outbreak of St. Louis encephalitis detected in Argentina, 2005. J. Clin. Virol. 42:27–33 [DOI] [PubMed] [Google Scholar]
- 14. Haddow AJ. 1968. The natural history of yellow fever in Africa. Proc. R. Soc. Edinb. Biol. 70:191–227 [Google Scholar]
- 15. Rudnick A, Lim TW. 1986. Dengue fever studies in Malaysia. Inst. Med. Res. Malays. Bull. 23:51–152 [Google Scholar]
- 16. Hervé JP, Dégallier N, Travassos da Rosa APA, Pinheiro FP, Sá Filho GC. 1986. Aspectos ecológicos das arboviroses, p. 409–437 In Instituto Evandro Chagas: 50 anos de Contribuicao às Ciências Biológicas e à Medicina Tropical. Fundação Serviços de Saúde Pública, Belém, Brazil [Google Scholar]
- 17. McLean RG, Bowen GS. 1980. Vertebrate hosts, p 381–450 In Monath TP, St. Louis encephalitis. American Public Health Association, Washington, DC [Google Scholar]
- 18. Sabattini MS, Avilés G, Monath TP. 1998. Historical, epidemiological and ecological aspects of arboviruses in Argentina: Flaviviridae, Bunyaviridae and Rhabdoviridae, p 113–134 In Travassos da Rosa APA, Vasconcelos PF, Travassos da Rosa JFS, An overview of Arbovirology in Brazil and neighboring countries. Instituto Evandro Chagas, Belém, Brazil [Google Scholar]
- 19. Sulkin SE, Sims RA, Allen R. 1966. Isolation of St. Louis encephalitis virus from bats (Tadarida b. mexicana) in Texas. Science 152:223–225 [DOI] [PubMed] [Google Scholar]
- 20. Allen R, Taylor SK, Sulkin SE. 1970. Studies of arthropod-borne virus infections in Chiroptera. 8. Evidence of natural St. Louis encephalitis virus infection in bats. Am. J. Trop. Med. Hyg. 19:851–859 [DOI] [PubMed] [Google Scholar]
- 21. Díaz LA, Albrieu Llinás G, Vázquez A, Tenorio A, Contigiani MS. 2012. Silent circulation of St. Louis encephalitis virus prior to an encephalitis outbreak in Cordoba, Argentina (2005). PLoS Negl. Trop. Dis. 6:e1489 http://dx.doi.10.1371/journal.pntd.0001489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitchell CJ, Francy DB, Monath TP. 1980. Arthropod vectors, pp. 313–380 In Monath TP, St Louis encephalitis. American Public Health Association, Washington, DC [Google Scholar]
- 23. Mitchell CJ, Gubler DJ, Monath TP. 1983. Variation in infectivity of Saint Louis encephalitis viral strains for Culex pipiens quinquefasciatus (Diptera: Culicidae). J. Med. Entomol. 20:526–533 [DOI] [PubMed] [Google Scholar]
- 24. Mitchell CJ, Monath TP, Sabattini MS. 1980. Transmission of St. Louis encephalitis virus from Argentina by mosquitoes of the Culex pipiens (Diptera: Culicidae) complex. J. Med. Entomol. 17:282–285 [DOI] [PubMed] [Google Scholar]
- 25. Rodrigues SG, Nunes MR, Casseb SM, Prazeres AS, Rodrigues DS, Silva MO, Cruz AC, Tavares-Neto JC, Vasconcelos PF. 2010. Molecular epidemiology of Saint Louis encephalitis virus in the Brazilian Amazon: genetic divergence and dispersal. J. Gen. Virol. 91(Pt 10):2420–2427 [DOI] [PubMed] [Google Scholar]
- 26. Kramer LD, Chandler LJ. 2001. Phylogenetic analysis of the envelope gene of St. Louis encephalitis virus. Arch. Virol. 146:2341–2355 [DOI] [PubMed] [Google Scholar]
- 27. Ottendorfer CL, Ambrose JH, White GS, Unnasch TR, Stark LM. 2009. Isolation of genotype V St. Louis encephalitis virus in Florida. Emerg. Infect. Dis. 15:604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Auguste AJ, Pybus OG, Carrington CV. 2009. Evolution and dispersal of St. Louis encephalitis virus in the Americas. Infect. Genet. Evol. 9:709–715 [DOI] [PubMed] [Google Scholar]
- 29. Mohammed MA, Galbraith SE, Radford AD, Dove W, Takasaki T, Kurane I, Solomon T. 2011. Molecular phylogenetic and evolutionary analyses of Muar strain of Japanese encephalitis virus reveal it is the missing fifth genotype. Infect. Genet. Evol. 11:855–862 [DOI] [PubMed] [Google Scholar]
- 30. Charrel RN, Brault AC, Gallian P, Lemasson JJ, Murgue B, Murri S, Pastorino B, Zeller H, de Chesse R, de Micco P, de Lamballerie X. 2003. Evolutionary relationship between Old World West Nile virus strains. Evidence for viral gene flow between Africa, the Middle East, and Europe. Virology 315:381–388 [DOI] [PubMed] [Google Scholar]
- 31. Grard G, Moureau G, Charrel RN, Lemasson JJ, Gonzalez JP, Gallian P, Gritsun TS, Holmes EC, Gould EA, de Lamballerie X. 2007. Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology 361:80–92 [DOI] [PubMed] [Google Scholar]
- 32. Wang E, Ni H, Xu R, Barrett AD, Watowich SJ, Gubler DJ, Weaver SC. 2000. Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J. Virol. 74:3227–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Monath TP, Cropp CB, Bowen GS, Kemp GE, Mitchell CJ, Gardner JJ. 1980. Variation in virulence for mice and rhesus monkeys among St. Louis encephalitis virus strains of different origin. Am. J. Trop. Med. Hyg. 29:948–962 [DOI] [PubMed] [Google Scholar]
- 34. Day JF, Stark LM, Zhang JT, Ramsey AM, Scott TW. 1996. Antibodies to arthropod-borne encephalitis viruses in small mammals from southern Florida. J. Wildl. Dis. 32:431–436 [DOI] [PubMed] [Google Scholar]
- 35. Christensen HA, de Vasquez AM, Boreham MM. 1996. Host-feeding patterns of mosquitoes (Diptera: Culicidae) from central Panama. Am. J. Trop. Med. Hyg. 55:202–208 [DOI] [PubMed] [Google Scholar]
- 36. Lemey P, Rambaut A, Welch JJ, Suchard MA. 2010. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 27:1877–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214 http://dx.doi.10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weaver SC, Barrett AD. 2004. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Diallo M, Ba Y, Sall y, Diop OM, Ndione JA, Mondo M, Girault L, Mathiot C. 2003. Amplification of the sylvatic cycle of dengue virus type 2, Senegal, 1999–2000: entomologic findings and epidemiologic considerations. Emerg. Infect. Dis. 9:362–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ubico SR, McLean RG. 1995. Serologic survey of neotropical bats in Guatemala for virus antibodies. J. Wildl. Dis. 31:1–9 [DOI] [PubMed] [Google Scholar]
- 41. de Souza Lopes O, de Abreu Sacchetta L, Coimbra TL, Pereira LE. 1979. Isolation of St. Louis encephalitis virus in South Brazil. Am. J. Trop. Med. Hyg. 28:583–585 [DOI] [PubMed] [Google Scholar]
- 42. Day JF, Storrs EE, Stark LM, Lewis AL, Williams S. 1995. Antibodies to St. Louis encephalitis virus in armadillos from southern Florida. J. Wildl. Dis. 31:10–14 [DOI] [PubMed] [Google Scholar]
- 43. Aitken TH, Downs WG, Spence L, Jonkers AH. 1964. St. Louis encephalitis virus isolations in Trinidad, West Indies, 1953–1962. Am. J. Trop. Med. Hyg. 13:450–451 [DOI] [PubMed] [Google Scholar]
- 44. Belle EA, Grant LS, Page WA. 1964. The isolation of St. Louis encephalitis virus from Culex nigrlpalpus mosquitoes in Jamaica. Am. J. Trop. Med. Hyg. 13:452–454 [DOI] [PubMed] [Google Scholar]
- 45. Dow RP, Coleman PH, Meadows KE, Work TH. 1964. Isolation of St. Louis encephalitis viruses from mosquitoes in the Tampa Bay area of Florida during the epidemic of 1962. Am. J. Trop. Med. Hyg. 13:462–468 [DOI] [PubMed] [Google Scholar]
- 46. Shroyer DA. 1991. The 1990 Florida epidemic of St. Louis encephalitis: virus infection rates in Culex nigripalpus. J. Fla. Mosq. Control Assoc. 62:69–71 [Google Scholar]
- 47. Edman JD, Taylor DJ. 1968. Culex nigripalpus: seasonal shift in the bird-mammal feeding ratio in a mosquito vector of human encephalitis. Science 161:67–68 [DOI] [PubMed] [Google Scholar]
- 48. Day JF, Curtis GA. 1993. Annual emergence patterns of Culex nigripalpus females before, during and after a widespread St. Louis encephalitis epidemic in South Florida. J. Am. Mosq. Control Assoc. 9:249–255 [PubMed] [Google Scholar]
- 49. Faria NR, Suchard MA, Rambaut A, Lemey P. 2011. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 1:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lemey P, Rambaut A, Drummond AJ, Suchard MA. 2009. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5:e1000520 http://dx.doi.10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hurlbut HS. 1973. The effect of environmental temperature upon the transmission of St. Louis encephalitis virus by Culex pipiens quinquefasciatus. J. Med. Entomol. 10:1–12 [DOI] [PubMed] [Google Scholar]
- 52. Mahoney J. 2010. Colonialism and postcolonial development: Spanish America in comparative perspective. Cambridge University Press, New York, NY. [Google Scholar]
- 53. Berthold P. 2001. Bird migration: a general survey. Oxford University Press, New York, NY. [Google Scholar]
- 54. Day JF. 2001. Predicting St. Louis encephalitis virus epidemics: lessons from recent, and not so recent, outbreaks. Annu. Rev. Entomol. 46:111–138 [DOI] [PubMed] [Google Scholar]
- 55. Harper JM, Wang M, Galecki AT, Ro J, Williams JB, Miller RA. 2011. Fibroblasts from long-lived bird species are resistant to multiple forms of stress. J. Exp. Biol. 214(Pt 11):1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Biesold SE, Ritz D, Gloza-Rausch F, Wollny R, Drexler JF, Corman VM, Kalko EK, Oppong S, Drosten C, Müller MA. 2011. Type I interferon reaction to viral infection in interferon-competent, immortalized cell lines from the African fruit bat Eidolon helvum. PLoS One 6:e28131 http://dx.doi.10.1371/journal.pone.0028131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Crochu S, Cook S, Attoui H, Charrel RN, De Chesse R, Belhouchet M, Lemasson JJ, de Micco P, de Lamballerie X. 2004. Sequences of flavivirus-related RNA viruses persist in DNA form integrated in the genome of Aedes spp. mosquitoes. J. Gen. Virol. 85(Pt 7):1971–1980 [DOI] [PubMed] [Google Scholar]
- 58. Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mosquito species identification. Download
Annotations of the viral polyprotein, noncoding genome regions, and envelope protein. Download
Assembly of the data set for phylogeographic reconstruction. Download
Experimental modifications of geographic outgroup placement and influence of topological uncertainty in deep tree nodes. Download