

ABSTRACT
Human cytomegalovirus (HCMV) glycoproteins gB and gH/gL are both necessary and sufficient for cell-cell fusion. However, it is not clear what roles these glycoproteins play in virus entry, whether acting directly in membrane fusion or in binding receptors. With other herpesviruses, it appears that gB is the fusion protein and is triggered by gH/gL, which, in some cases, binds receptors. However, for HCMV, there is published evidence that gB binds cellular ligands necessary to promote virus entry into or signaling of cells. Most mechanistic information on herpesvirus fusion proteins involves cell-cell fusion assays, which do not allow a determination of whether gB or gH/gL in the virion envelope must be oriented toward cellular membranes that contain receptors. Here, we showed that HCMV virions lacking gB were unable to enter normal cells but entered cells that expressed gB. Analyses of gB mutants lacking the cytoplasmic domain or with substitutions in putative “fusion loops” provided evidence that gB fusion activity was required for this “entry in trans.” In gB-mediated entry in trans, gB is oriented toward the virion envelope that apparently lacks receptors, arguing against an essential role for gB in binding receptors or signaling molecules. In contrast, particles lacking gH/gL did not enter cells expressing gH/gL, apparently because gH/gL must be oriented toward cellular membranes (which have receptors). Coupled with our previous interference studies, in which gH/gL expressed in cells blocked HCMV entry, our findings here support the hypothesis that HCMV gH/gL binds cellular receptors before triggering gB, which acts as the fusion protein.
IMPORTANCE
Human cytomegalovirus (HCMV) produces major disease in neonates and immunosuppressed transplant patients. As with other herpesviruses, HCMV requires two membrane glycoproteins, gB and gH/gL, to enter host cells. However, it has not been clear how gB and gH/gL function in two steps of the HCMV entry pathway, i.e., (i) binding of cellular receptors and (ii) fusion of the virion envelope with cellular membranes. There are studies that suggest that HCMV gB is required for receptor binding and other studies suggesting that gH/gL is the receptor binding protein and gB is the fusion protein. Here, we show that HCMV virions lacking gB can enter cells that express gB in cellular membranes. In contrast, virus particles lacking gH/gL could not enter cells expressing gH/gL. Our study supports the hypothesis that gB is the fusion protein and gH/gL acts upstream of gB to bind receptors and then activate gB for fusion.
Introduction
Human cytomegalovirus (HCMV) infects a wide spectrum of cell types by utilizing different forms of glycoprotein gH/gL to enter different cell types by distinct routes of entry (reviewed in reference 1). Entry into human fibroblasts requires at least two glycoproteins, gB and gH/gL, and involves fusion with the plasma membrane (2–4). HCMV entry into epithelial and endothelial cells requires gB and a pentameric form of gH/gL denoted gH/gL/UL128-131 and involves macropinocytosis or endocytosis and low-pH-dependent fusion with endosomes (3, 5, 6). Another glycoprotein, gO, is associated with HCMV gH/gL and is important for incorporation of gH/gL into virions but not for cell surface expression in fibroblasts (7–9). We described interference experiments in which different forms of gH/gL were expressed in cells, apparently saturating or altering intracellular traffic of virus receptors and leading to inhibition of virus entry (9, 10). These studies supported a working model suggesting that HCMV gH/gL binds receptors important for entry into fibroblasts and that gH/gL/UL128-131 binds receptors in epithelial cells. Importantly, another major HCMV glycoprotein, gB, did not cause interference in any cell type.
Studies of the better-characterized herpes simplex virus (HSV) and Epstein-Barr virus (EBV) gB and gH/gL homologues have supported models for how these glycoproteins promote virus entry (reviewed in reference 11). These models are, in part, based on the structures of HSV and EBV gB molecules that suggest that these molecules function as fusion proteins (12, 13). Moreover, HSV and EBV gB molecules contain so-called “fusion loops,” including hydrophobic residues, that promote direct interactions of gB with cellular membranes and liposomes in vitro, decreasing membrane proximity for fusion (14, 15). In contrast, EBV and HSV gH/gL molecules do not have structures similar to any known fusion proteins (reviewed in references 11 and 16), and, importantly, HSV gH/gL does not interact with liposomes unless gB is present (17). For EBV, there is good evidence that different forms of gH/gL (gH/gL or gH/gL/gp42) bind different cellular receptors, and this receptor binding is thought to lead to activation of gB for fusion (reviewed in references 11 and 18). For HSV, a third glycoprotein, gD, binds cellular receptors and apparently activates gH/gL that then triggers gB (reviewed in references 11 and 16).
Currently, we have little or no structural information on HCMV gB and gH/gL, although gB is a trimer (19). In addition, the functional properties of HCMV gB and gH/gL are poorly understood compared to HSV and EBV glycoproteins. Based on analogies with other herpesviruses, HCMV gB may be the fusion protein, although there is no evidence for this currently, except that an HCMV gB-null virus cannot enter cells (4). Our interference data argued that HCMV gB does not bind saturable receptors, while expression of different forms of gH/gL in cells blocks entry, which is consistent with (but by no means proving) the possibility that gH/gL molecules act as receptor binding proteins. In contrast, there have been multiple reports of HCMV gB binding cellular receptors needed for entry. There have been observations that HCMV gB interacts with the epidermal growth factor receptor (EGFR) and that this interaction is important for virus entry (20). Subsequently, conclusions about the importance of EGFR in HCMV entry and whether HCMV activates EGFR were challenged (6, 21). The platelet-derived growth factor receptor (PDGFR) has also been described as an HCMV receptor or entry mediator, and again, gB bound to PDGFR (22). We concluded that PDGFR does not normally act as an important receptor for HCMV entry into fibroblasts and epithelial and endothelial cells (6). There have also been reports that gB binds integrins important for HCMV entry (23, 24). Several of these studies suggested that HCMV gB activates cytoplasmic signaling pathways that are necessary for virus entry.
Most of our mechanistic knowledge of how herpesvirus gB and gH/gL promote virus entry into cells comes from the use of a surrogate assay involving cell-cell fusion. In virus entry, the virion envelope fuses with either the plasma membrane or, frequently, endosomes. In contrast, cell-cell fusion involves cell surface membranes studded with viral membrane proteins that fuse with one another. There are frequently major differences between virus entry and cell-cell fusion. For example, HSV entry into nectin-1-expressing CHO cells involves endocytosis and low-pH-dependent fusion with endosomes, yet HSV mediates cell-cell fusion of nectin-expressing CHO cells involving plasma membranes at neutral pH (25). HCMV gB and gH/gL are sufficient for fusion of all cells tested to date, and this fusion is most efficient at neutral pH (26). In contrast, gH/gL/UL128-131 is required for entry into epithelial and endothelial cells, a process that involves endocytosis or macropinocytosis and low-pH-dependent fusion with endosomes (3, 6). These major differences between cell-cell fusion and entry of enveloped virions likely reflect differences in morphologies and compositions of viral and cellular membranes, as well as how virus particles traffic following attachment to cells.
A second problem with relying on cell-cell fusion assays relates to difficulties in determining how individual virus glycoproteins function, whether directly in fusion or by binding receptors. This problem is especially relevant to HCMV gB and gH/gL, since functional studies of these glycoproteins are less extensive. For example, we previously described cell-cell fusion in trans in which cells expressing gB were mixed with other cells expressing gH/gL, producing efficient (50%) cell-cell fusion (26). Related to the question of how gB and gH/gL function, we observed that gH/gL-expressing ARPE-19 epithelial cells fused in trans with gB-expressing HeLa cells, but there was no fusion of gH/gL-expressing HeLa cells mixed with gB-expressing ARPE-19 cells (26). ARPE-19 cells are permissive for HCMV and fuse well in cis, whereas HeLa cells are not infected by HCMV and do not fuse, presumably because these cells lack HCMV receptors (26). We attempted to explain these data by suggesting that gH/gL might interact with receptors in cis, i.e., gH/gL in ARPE-19 membranes interacts with receptors in the same membranes and then reaches across to trigger gB in the opposite HeLa cell membrane. An alternative suggestion was that HCMV gB in the HeLa cells could potentially reach across to permissive ARPE-19 cells, interact with gB receptors, and mediate fusion. Importantly, these cell-cell fusion assays cannot discern between these models and tell us whether gB or gH/gL is necessary to bind receptors.
With the objective of determining whether HCMV gB or gH/gL expressed in the context of the virion envelope must bind cellular ligands, we tested whether HCMV particles lacking either gB or gH/gL could enter cells expressing gB or gH/gL. Different from the situation with cell-cell fusion assays, the virus particles applied to cells were unlikely to possess virus receptors. Thus, when gB or gH/gL was expressed in cells oriented toward the virion envelope, there were unlikely to be receptor interactions. Virions lacking gB, but with gH/gL in their envelopes, were able to enter cells expressing gB in a relatively efficient manner (entry in trans) but could not enter cells that did not express gB. Mutant forms of gB that were unable to mediate cell-cell fusion did not mediate entry in trans. Particles lacking gH/gL could not enter cells expressing gH/gL. These studies demonstrated that HCMV gB can be expressed in cells oriented toward the virion envelope and can mediate entry. This strongly argues that gB is not required to bind gB receptors or signaling molecules on cells that are essential for entry, supporting the hypothesis that gB is the fusion protein. In contrast, gH/gL did not mediate entry in trans, suggesting that gH/gL must be present in the virion envelope oriented toward cells in order to engage cellular receptors.
RESULTS
Construction of an HCMV gH-null mutant.
Previously, an HCMV strain AD169 mutant unable to express gB was constructed by replacing gB (UL55) sequences with the galK gene and complementing gB by using fibroblasts infected with a retrovirus expressing gB (4). We constructed an HCMV strain TR mutant lacking the gH (UL75) gene by using a bacterial artificial chromosome (BAC) copy of the HCMV genome to replace the gH gene with a kanamycin gene cassette. The BAC was transfected into human fibroblasts transduced with a retrovirus expressing gH (27). HCMVΔgB and HCMVΔgH produced plaques on complementing fibroblasts (NHDF+gB, NHDF+gH) that included ~25 to 30% and 70 to 80%, respectively, of the number of infected cells that wild-type HCMV AD169 or strain TR did (Fig. 1A to D). In contrast, when virus preparations from complemented cells were used to infect normal fibroblasts (without gB or gH), viruses entered the cells but did not spread beyond a single infected cell (Fig. 1E and G). The numbers of virus particles elicited in culture supernatants following infection of complementing and noncomplementing cells (infected using 1 PFU/cell) were quantified using quantitative PCR (qPCR) to measure viral DNA, with a comparison to known concentrations of virus genomes in BAC. Both HCMVΔgB and HCMVΔgH produced 3- to 10-fold fewer virus particles following infection of normal fibroblasts than that produced on complementing cells (Fig. 1I), which was apparently related to reduced spread. Western blot analyses were used to evaluate HCMVΔgB and HCMVΔgH particles derived from either complementing (gB- or gH-expressing) or normal fibroblasts and characterized the major capsid protein (MCP), tegument protein pp65, gB, and gH. Figure 1J and K show that the quantities of gB or gH in complementing cells were lower than those from wild-type HCMV infection. This was likely related to the comparatively low copy number of retroviruses in these fibroblasts, despite three to five rounds of reinfection with retroviruses. This incomplete complementation, i.e., lower levels of infectious virus produced in complementing cells, did not compromise our capacity to extend these studies by producing the necessary quantities of particles lacking gB and gH/gL (following infection of normal fibroblasts) for the studies described below.
FIG 1.
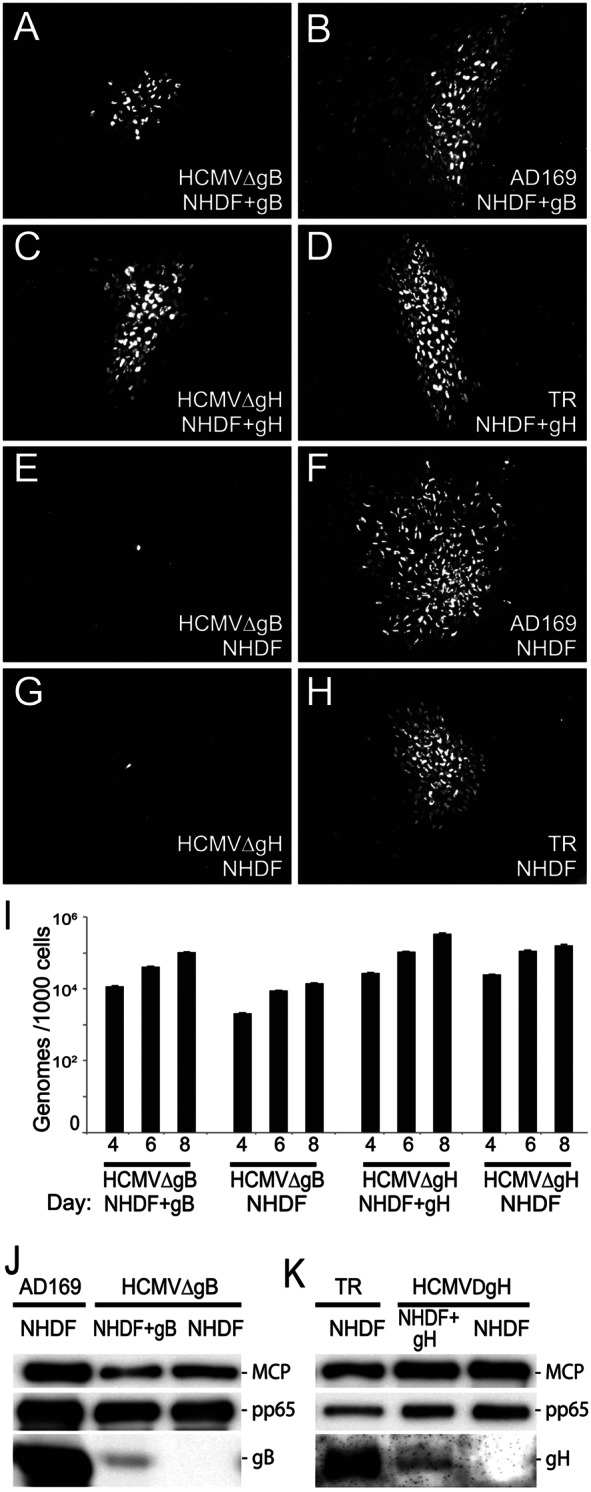
Analyses of HCMV mutants lacking gB or gH. (A to H) Normal fibroblasts (NHDF) or fibroblasts transduced with retroviruses expressing gB (NHDF+gB) or gH (NHDF+gH) were infected with wild-type HCMV strain AD169 or TR or with HCMVΔgB or HCMVΔgH (mutant viruses derived from complementing cells). After 10 days, the cells were fixed, permeabilized, and stained for HCMV IE86. (I) Similar quantities of cell culture supernatants derived from HCMV-infected NHDF, NHDF+gB, or NHDF+gH were subjected to qPCR analyses for HCMV DNA and compared to virus genomes in HCMV BAC. (J and K) Similar quantities of virus particles were subjected to immunoblotting to detect gB, gH, the major capsid protein (MCP), or pp65.
HCMV mutants lacking gB or gH do not enter fibroblasts but can enter following PEG treatment.
Virus particles lacking either gB or gH were produced by infecting normal fibroblasts with HCMVΔgB or HCMVΔgH derived from complementing cells. Cell culture supernatants were harvested, and virus was concentrated and partially purified using centrifugation through a 20% sorbitol cushion. Initially, these particles were enumerated by qPCR as shown in Fig. 1. We anticipated that these particles were noninfectious. When HCMV mutants unable to enter cells are treated with polyethylene glycol (PEG), this chemical fusogen mediates entry (3). Thus, we characterized and enumerated HCMVΔgB or HCMVΔgH particles lacking gB or gH by using PEG treatment. Sufficient HCMV particles were incubated with fibroblasts, followed by PEG, so that 48% of the cells expressed HCMV IE86 after 24 h (Fig. 2B and D). When PEG was omitted, there was no entry into the cells (Fig. 2A and C). It is important to point out that the background here was zero, i.e., no cells expressed IE86 even after 48 h without PEG. Moreover, the PEG experiment proved that these particles could adsorb onto cell surface heparan sulfate glycosaminoglycans in sufficient numbers in order to enter the cells with PEG treatment. In other experiments, binding of [35S]methionine-labeled HCMV particles lacking gB or gH onto cells was similar to that with wild-type HCMV (data not shown).
FIG 2.
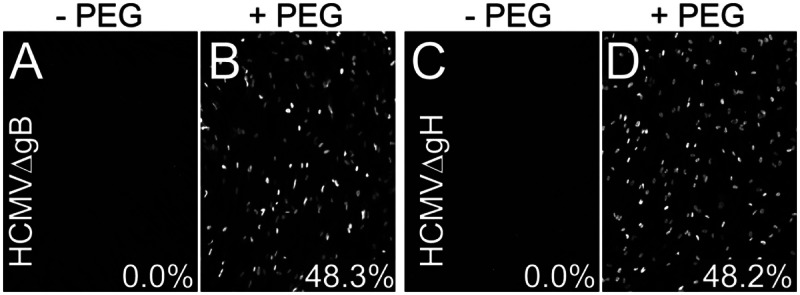
NHDF were infected with preparations of HCMVΔgB (A and B) or HCMVΔgH (C and D) derived from NHDF (lacking gB or gH) by incubating virus with cells and then immediately centrifuging the cells at 800 × g for 30 min at 10°C. Other cells were incubated with virus, centrifuged, treated with 44% polyethylene glycol (PEG) for 30 s at 37°C, and then extensively washed. The cells were incubated for 24 h at 37°C and then stained for IE86. Numbers indicate the average number of IE86-positive cells derived from triplicate wells.
HCMV particles lacking gB can enter cells that express gB in trans.
To try to determine whether HCMV particles lacking gB or gH could enter cells expressing these glycoproteins, we used nonreplicating (with E1 deleted) adenovirus (Ad) vectors to express HCMV gB or both gH and gL in neonatal human dermal fibroblasts (NHDF) for 24 to 48 h and then the cells were incubated with HCMV particles lacking gB or gH. The same quantities of HCMV particles lacking gB or gH were used to infect these cells as shown in Fig. 2, and PEG treatment promoted entry so that 45 to 50% of fibroblasts were infected (data not shown). Fibroblasts transduced with a control Ad vector, Ad-green fluorescent protein (AdGFP), and incubated with particles lacking either gB or gH showed no IE86-positive cells (Fig. 3A and B). Particles lacking gB entered 14% of fibroblasts expressing gB, approximately 2,800 cells in each well (Fig. 3C). In numerous other experiments, entry of particles lacking gB into cells expressing gB was always observed at levels of 6% to 20%.
FIG 3.
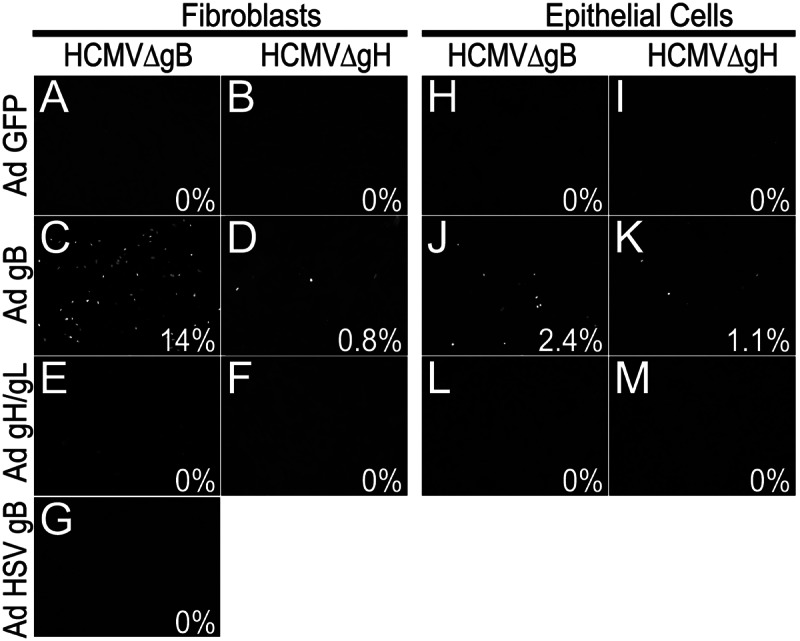
Entry in trans of HCMV particles. NHDF (fibroblasts) (A to G) or ARPE-19 epithelial cells (H to M) were transduced with nonreplicating Ad vectors to express GFP (A, B, H, and I), HCMV gB (C, D, J, and K), HCMV gH and gL (E, F, L, and M), or HSV gB (G) for 24 h. The cells were then incubated with HCMVΔgB particles lacking gB or HCMVΔgH particles lacking gH (in both cases particles derived from NHDF) and immediately centrifuged at 800 × g for 30 min at 10°C and then incubated for 4 h at 37°C. Virus was removed, and cells were incubated for an additional 20 h before being stained for HCMV IE86. Assays were done in triplicate, and the average percentages of IE86-positive cells are indicated.
In contrast to the gB-mediated entry in trans, particles lacking gH were unable to enter gH/gL-expressing fibroblasts, i.e., not a single IE86-positive cell was observed (Fig. 3F), as with AdGFP-transduced fibroblasts (Fig. 3B). Increasing or decreasing the expression of gH/gL (by using different input doses of Ad vectors) and coexpression of gO with gH/gL did not alter the entry of particles lacking gH into fibroblasts (data not shown). Moreover, there was no entry of particles lacking gB into gH/gL-expressing cells (Fig. 3E). Interestingly, HCMV particles lacking gH entered 0.8% of gB-expressing cells (range, 0.1% to 1%) (Fig. 3D), which was 20-fold less efficient than with particles lacking gB entering gB-expressing cells (Fig. 3C). Expression of HSV gB in fibroblasts did not promote entry of HCMV particles lacking gB (Fig. 3G). Entry in trans of HCMV particles lacking gB into gB-expressing cells was observed when the centrifugal enhancement step was omitted, but infection was lower by 1 to 2% (200 to 400 cells/well) (data not shown).
ARPE-19 epithelial cells can be infected by HCMV, although much less efficiently than fibroblasts; i.e., when 3 to 5 PFU/cell of HCMV TR are used, ~15 to 30% of ARPE-19 cells express IE86 (26). Incubation of ARPE-19 cells expressing gB with HCMVΔgB particles lacking gB (the same levels as shown in Fig. 3C) produced 2.4% infection (Fig. 3J) compared with no cells infected when ARPE-19 cells were transduced with AdGFP (Fig. 3H). There was 1.1% entry of HCMV particles lacking gH into gB-expressing ARPE-19 cells (Fig. 3K). No entry of particles lacking either gB or gH into gH/gL-expressing cells was observed (Fig. 3L and M). Coexpression of UL128-131 with gH/gL did not affect these results (data not shown). In comparing entry of particles lacking gB with that lacking gH into gB-expressing cells, two important points must be considered. HCMVΔgB is derived from AD169 and lacks gH/gL/UL128-131, which is important for entry into epithelial cells. That said, HCMV AD169 can enter epithelial cells inefficiently, e.g., into 0.5 to 1% of cells (3, 6). In contrast, HCMVΔgH was derived from the TR stain that expresses gH/gL/UL128-131 and can more efficiently enter epithelial cells (3, 6). Thus, it is not accurate to directly compare the efficiencies of entry in trans of HCMVΔgB and HCMVΔgH into epithelial cells. What is more important is that gB expressed in epithelial cells can significantly increase entry into cells from 0 (no gB) to 2.4% and there was no entry of particles lacking gH into gH-expressing ARPE-19 cells. However, given the strain differences and different entry pathways, some caution must be used in interpreting these results.
Cytoplasmic domain and fusion loop mutations in HCMV gB block entry in trans.
In order to characterize how gB mediates entry in trans, we mutated gB to produce molecules deficient in membrane fusion. The cytoplasmic domain of HSV gB is required for cell-cell fusion and entry, although mutations in the more C-terminal (CT) region of HSV gB can produce the syncytial phenotype (28). We previously described Ad vectors that express truncated forms of HCMV gB: gBΔCT lacks all but 3 residues of the gB cytoplasmic domain, and syncytial gB (sgB) lacks both the CT and transmembrane domains (29). Both gBΔCT and sgB were found on cell surfaces (see Fig. S1 in the supplemental material; Fig. 4A) (sgB likely binds to heparan sulfate) and did not mediate cell-cell fusion in either ARPE-19 epithelial cells or MRC-5 fibroblasts (Fig. S2; Fig. 4A). Neither gBΔCT nor sgB mediated entry in trans involving HCMV particles lacking gB (Fig. 4A). Based on observations that an HSV gB mutant truncated to remove 41 C-terminal residues produced enhanced cell-cell fusion (28), we constructed HCMV gB882stop, with residue 882 replaced with a stop codon (removing 25 C-terminal residues). gB882stop was present on cell surfaces (Fig. S1) and produced extensive cell-cell fusion that appeared much earlier (16 h) after gB expression than with wild-type gB, which required 24 to 28 h for the same level of fusion (Fig. S2). In contrast, gB882stop produced less entry in trans, i.e., 50 to 60% compared with wild-type gB (Fig. 4B). Thus, gB882stop functions more efficiently or quickly in cell-cell fusion but performs less efficiently in entry in trans.
FIG 4.

Mutant forms of HCMV gB that cannot cause fusion do not promote entry in trans. NHDF were transduced for 48 h with Ad vectors that express the following wild-type and mutant forms of gB: (A) wild-type AD169 gB, sgB, and gBΔCT; (B) L241R and 882stop mutant gB; or (C) wild-type TR gB or Y157S, A154D, A154W, W240A, YIY157/GHR, or GSTW240/AAAA mutant gB, or no gB (same multiplicity of infection of AdtetTrans). The cells were then incubated with HCMVΔgB particles lacking gB, centrifuged at 800 × g for 30 min, and incubated for 4 additional hours with virus, and then the virus was removed and the cells were incubated for 20 h before being stained for HCMV IE86. Results of cell surface expression assays (Fig. S1) and cell-cell fusion assays (Fig. S2) are summarized.
HCMV gB contains two putative fusion loops (Y153-T158, G237-E244) based on homology to HSV and EBV gB fusion loops (19). We made a panel of HCMV gB fusion loop mutants. In early studies, one mutant, the L241R mutant, constructed in AD169 gB, produced no fusion in either ARPE-19 cells or MRC-5 cells. However, in later experiments, we observed a low level of fusion in ARPE-19 cells but still none in MRC-5 cells (Fig. S2). Interestingly, the L241R mutant produced wild-type levels of entry in trans (Fig. 4B). Later, other gB fusion loop mutants were constructed in the TR strain gB. TR gB was less efficient in promoting entry in trans involving particles lacking gB than was AD169 gB (Fig. 4A and C). In this case, TR gB must interact with AD169 gH/gL (in particles lacking gB), which might explain the lower levels of entry in trans. However, keeping in mind that background entry in trans was zero, we could readily characterize TR gB mutants. The gB Y157S mutant mediated efficient cell-cell fusion and entry in trans (Fig. 4C). The A154D mutant produced low levels of cell-cell fusion in ARPE-19 cells and none in MRC-5 cells (Fig. S2) yet mediated entry in trans similar to that of TR gB (Fig. 4C), like the L241R mutant. Therefore, low levels of gB cell-cell fusion activity can suffice for wild-type levels of entry in trans. We note that gB882stop produced more rapid cell-cell fusion and less entry in trans, probably because the cytoplasmic domain of gB has other effects on the entry process. However, this result does not compromise the conclusion that low levels of cell-cell fusion activity can suffice for entry in trans. The A154W, W240A, YIY157/GHR (3 residues altered), and GSTW240/AAAA (four residues altered) mutants were all present on cell surfaces (Fig. S1), did not cause any detectable cell-cell fusion (Fig. S2), and produced no detectable entry in trans (Fig. 4C). We concluded that gB molecules with no fusion activity cannot function in HCMV entry in trans.
Entry of HSV particles lacking gB into cells expressing gB.
Given that a great deal more is known about the how HSV gB and gH/gL function than how HCMV gB and gH/gL function, we attempted to extend these studies to HSV glycoproteins. Ad vectors were used to express HSV gB or gH/gL in human fibroblasts, and the glycoproteins were observed on cell surfaces (Fig. S3A). HSV particles lacking gB (30) could not enter fibroblasts expressing gB, and there was no increased entry of these particles compared to entry into normal fibroblasts lacking gB (Fig. S3B). There was some background in these studies related to contamination with repaired HSV (with gB) derived from complementing cells (30). Expression of gH/gL in fibroblasts did not promote entry of HSV particles lacking gH (Fig. S3B). In a demonstration that there were many particles adsorbed onto the surfaces of these cells, PEG treatment promoted entry, and over 50% of cells expressed ICP4 (Fig. S3B). Similar experiments were repeated using C10 mouse melanoma cells, which express relatively high levels of HSV receptor nectin-1 (cells used to study cell-cell fusion) (31), and Vero, U373, Hep2, ARPE-19, HeLa, and HaCat cells. In no case did we observe any HSV entry in trans, not even a low level of entry above background, whether or not HSV was centrifuged onto cells (data not shown). One might argue that these are negative results. However, given all that is known about HSV entry and the need for three glycoproteins (gB, gD, and gH/gL) for entry, it was important to report the results.
DISCUSSION
How HCMV glycoproteins gB and gH/gL participate in virus entry is not well established. Our interference studies supported models in which different forms of gH/gL bind saturable receptors in cells (reviewed in reference 1). Importantly, HCMV gB did not interfere with virus entry into any cell type tested. Similar conclusions were drawn from interference studies with HSV gD, and these were later proven correct (reviewed in reference 16). Given the interference studies and evidence that that HSV and EBV gB molecules are fusion proteins, we proposed that gH/gL molecules bind receptors and function upstream of gB, which is not required for binding ligands, and instead acts as the fusion protein. However, arguing against that model are numerous reports that HCMV gB binds cellular receptors, i.e., platelet-derived growth factor receptor (22), epidermal growth factor receptor (20), and integrins (24), which are necessary for virus entry. Certain of these conclusions have been challenged (6, 21). Several of these studies also suggested that gB interacts with cellular signaling molecules to produce signals inside cells that are necessary for entry or early stages of infection. It should be kept in mind that gB could act both as the fusion protein and to bind receptors or signaling molecules.
Here we showed that HCMV particles lacking gB can enter fibroblasts that express gB. This entry of particles lacking gB was not an inefficient process, i.e., 6 to 20% of AD169 gB-expressing cells were infected, compared with PEG-mediated entry of 45 to 50% of these particles. A comparison to PEG-mediated entry of the same particles is more accurate than comparisons to entry of wild-type HCMV because the numbers of mutant particles were low. Importantly, the background numbers were extremely low or zero; e.g., dishes of cells with no gB typically showed not a single IE86-positive cell, yet there were thousands of IE86-positive cells in dishes containing gB-expressing cells. Therefore, gB expressed in trans, with the glycoprotein oriented toward the virion envelope, can promote relatively efficient entry of virus particles (Fig. 5A). It is a reasonable assumption that virions do not contain substantial quantities of cellular ligands that might serve as receptors or entry mediators. In that case, these results argue that gB can mediate entry of HCMV virions without binding cellular receptors, entry mediators, or signaling molecules. This is not to say that gB does not, in some instances, interact with receptors or signaling molecules, but our results demonstrate that these interactions are not essential for relatively efficient virus entry and IE86 expression in fibroblasts. Instead, our model suggests that gH/gL in the virion envelope interacts with cellular receptors and then interacts with gB in trans, triggering gB for fusion (Fig. 5A).
FIG 5.

Cartoon depicting HCMV particles lacking gB or gH/gL interacting with cells expressing these glycoproteins. (A) An HCMV particle lacking gB but containing gH/gL in the virion envelope can interact with cell surface receptors (green) and then activate gB (lightning bolt) that then causes fusion of the virion envelope with cell surface membranes. (B) An HCMV virion containing gB but lacking gH/gL fails to enter cells because gH/gL is oriented toward the virion envelope, which lacks gH/gL receptors, so that gH/gL does not trigger gB for fusion.
Support for this model came from observations that HCMV particles lacking gH/gL (but containing gB) were unable to enter fibroblasts and epithelial cells expressing gH/gL. In this case, gH/gL in cell membranes is oriented toward the virion envelope, which is unlikely to contain receptors (see Fig. 5B). In the model shown in Fig. 5A, gH/gL is present in the virion envelope oriented toward cells and cellular receptors and functions in entry. One might argue that our observations involving virions lacking gH represent negative results. A number of points are relevant here. First, the gH/gL results do not stand alone; the comparison is to gB-expressing cells that mediated entry in trans of particles lacking gB. Those particles lacking gB do contain gH/gL that is oriented toward the gB in cells and is obviously functioning in entry, since particles lacking gH entered these cells inefficiently. Second, there is good evidence that gH/gL is present on cell surfaces at relatively high concentrations (9, 32), compared to HCMV gB, which was expressed at much lower levels on cell surfaces (19). It seems unlikely that gH/gL is simply less efficient in promoting entry in trans, because fibroblasts made to express a broad range of gH/gL did not exhibit any entry. Third, interference (gH/gL downregulation of fibroblast receptors) did not explain these results. There was only 20% interference when gH/gL was expressed in fibroblasts (9), not sufficient to account for the zero entry of particles lacking gH. Coexpression of gO with gH/gL increased interference, although there was still 30% entry, and gO did not increase surface expression in fibroblasts (9) and did not alter entry in trans. Therefore, the inability of particles lacking gH to enter gH/gL-expressing cells was not related to low cell surface gH/gL or interference. We stress that these results do not prove that gH/gL in the virion envelope must engage receptors in cell membranes. However, this possibility is highly consistent with our interference studies.
The important conclusion that HCMV gB is not required to bind cellular ligands is based on the assumption that there are not sufficient quantities of certain cellular proteins (acting as gB receptors) incorporated into virions. Cellular proteins are present in purified HCMV particles, mainly derived from trans-Golgi membranes and at low concentrations (33). One could argue that even low concentrations of cellular ligands in the virion might promote entry in trans. However, there are several arguments against this notion. If there were gB or gH/gL receptors in virions, wild-type HCMV particles might be prematurely triggered for fusion in cis rather than able to fuse only once the virus engages cellular membranes. It makes little sense for HCMV to package functionally important receptors in the virion if those receptors are the trigger for fusion. Related to this, if there were gB receptors in virions, it seems reasonable that there should also be gH/gL receptors, yet particles lacking gH did not enter gH/gL-expressing cells.
Additional evidence for the model that HCMV gB acts as the fusion protein came from studies of a panel of gB mutants. gB molecules lacking most of the cytoplasmic domain (gBΔCT) or both the cytoplasmic and transmembrane domains (sgB) were unable to cause cell-cell fusion and entry in trans. Mutant gB882stop lacks the 25 C-terminal residues of gB and exhibited enhanced cell-cell fusion, extensive cell fusion occurred earlier after expression than with wild-type gB, and yet entry in trans was reduced. This fit with analyses of a similar HSV gB mutant, gBΔ41, that displayed 7-fold more cell-cell fusion and yet was markedly reduced (~15%) in its capacity to mediate entry. Four HCMV gB mutants in presumed fusion loops, the A154W, W240A, YIY157/GHR and GSTW240/AAAA mutants, exhibited no cell-cell fusion and no entry in trans. Therefore, combined with gBΔCT and sgB, there were a total of six mutant gB molecules that did not function either in cell-cell fusion or entry in trans. However, two gB mutants, A154D and L241R, produced much less or no fusion and the cell-cell fusion produced was delayed but mediated entry in trans similar to the wild-type gB. This suggested that the threshold for gB-mediated entry in trans is lower than that for cell-cell fusion, again highlighting important differences.
Our previous cell-cell fusion assays did not allow a determination of whether HCMV gH/gL or gB must be present in the virion envelope oriented toward cellular membranes that contain receptors, entry mediators, or signaling molecules. Permissive ARPE-19 epithelial cells expressing gH/gL fused with gB-expressing, nonpermissive HeLa cells, but there was no fusion of gH/gL-expressing HeLa cells mixed with gB-expressing ARPE-19 cells (26). We previously explained this by suggesting that gH/gL might be able to interact in cis with epithelial cell receptors, i.e., receptors in the same cell, and then with gB in trans, i.e., in the cell opposite. Our results here establish that gH/gL in cells cannot interact with receptors in cis for virus entry but instead that gH/gL in the virion envelope can clearly interact with cellular receptors in trans. We would argue that the studies of entry in trans shown here more accurately represent how gH/gL functions in nature because these experiments involve virus particles rather than cells fusing with one another. An alternative explanation for our previous cell-cell fusion results suggested that HCMV gB in the HeLa cells could reach across to permissive epithelial cells and interact with gB receptors in trans. Again, the data here involving entry of virus particles argue against the possibility that gB must bind essential gB receptors or ligands for virus to enter cells. It is not clear how the entry in trans results should be interpreted. However, these data further emphasize that cell-cell fusion and entry assays provide different pictures of the HCMV fusion machinery, and we would argue that the cell-cell fusion assays are less accurate in their portrayal of some aspects of the entry pathway. Virion envelopes are studded with membrane proteins, with fewer cellular proteins, and differ substantially in curvature, and particles can be internalized by cells and subjected to low pH or endosomal contents. These differences further underscore the need to study virus entry where possible.
There were much lower levels of entry of particles lacking gH into gB-expressing cells (~1/20th that observed with particles lacking gB). We do not understand this in terms of simple versions of our model, as there was no gH/gL present and gH/gL is essential for entry. This apparently involves a much less efficient gB-mediated fusion in trans, triggered by virions lacking gH/gL. Perhaps HCMV gB normally present in the virion envelope is held in check by gH/gL until gH/gL interacts with receptors and unleashes gB for fusion. Fusion between virus particles and the outer nuclear envelope can occur with gB alone in cells infected with an HSV gH-null mutant (30). Another caveat was that the gB-null mutant was derived from laboratory strain AD169 and the gH-null mutant was derived from clinical strain TR. One might argue that strain differences could potentially affect our conclusions. However, both AD169 gB and TR gB mediated entry in trans of AD169 particles lacking gB (Fig. 4), and thus, the main conclusion of the paper is well supported. In terms of gH/gL, we found the following: (i) the sequences of TR gH and AD169 gH are very similar (98% amino acid identity); (ii) HCMVΔgH was propagated on cells transduced with AD169 gH and gL, showing that these proteins are interchangeable; and (iii) entry in trans of TR particles lacking gH was tested using cells expressing TR gH and gL. As explained above, there may be more subtle differences, e.g., TR gH/gL might interact with AD169 gB less than with TR gB. However, given the zero background levels, the data supported our conclusions that gB can mediate entry in trans, yet gH/gL cannot.
In summary, these results for entry in trans provide strong evidence against the hypothesis that HCMV gB must bind receptors or signaling molecules that are essential for virus entry or early gene expression. Instead, gH/gL appears to fulfill this function of binding receptors and then activating gB for fusion. This is the first example in which herpesvirus fusion proteins have been separated, one expressed in cells and the other in the virion envelope, to function in a relatively efficient manner for virus entry.
MATERIALS AND METHODS
Cells and viruses.
Neonatal human dermal fibroblasts (NHDF, passages 8 to 15; Cascade Biologics), human MRC-5 embryonic lung fibroblasts, and ARPE-19 retinal pigmented epithelial cells (American Type Culture Collection) were maintained in medium with 10% fetal bovine serum (FBS). HCMV laboratory strain AD169 and HCMV strain TR were propagated using NHDF. An HCMV AD169 strain lacking gB (HCMVΔgB) denoted pAD/CreΔUL55 was propagated using NHDF transduced with a retrovirus expressing gB as described previously (4). HCMVΔgH was derived from the TR BAC by using λ phage linear recombination to replace the gH gene with a kanamycin resistance cassette, by methods described previously (8). This BAC was transfected into NHDF infected with a retrovirus expressing gH and GFP (27). Virus particles lacking gB or gH were produced by infecting NHDF with complemented HCMVΔgB or HCMVΔgH using 2 to 5 PFU/cell for 2 h, followed by washing the cells with citrate buffer (40 mM citric acid, 10 mM KCl, 135 mM NaCl, pH 3.0) for 30 s to neutralize virus that did not enter. Virus particles were collected from cell culture supernatants after 7 to 10 days and concentrated by centrifugation through a 20% sorbitol cushion (26). HSV mutants lacking gB (F-BAC gB−) or gH (F-BAC gH−) were described previously (30). Nonreplicating (E1−) adenovirus (Ad) vectors expressing HCMV TR gB, gH, and gL and AD169 gB were described previously (26), as were vectors expressing sgB and gBΔCT (29) and HSV gB and gH/gL (34). Construction of all other mutant forms of HCMV gB involved overlapping PCR mutagenesis, followed by subcloning PCR fragments into plasmid pAdtet7 and rescue into nonreplicating (E1−) Ad vectors as described previously (29). All Ad vectors were used in conjunction with Adtet-Trans, which expresses a transactivator protein that upregulates the herpesvirus genes (29).
qPCR of HCMV DNA.
HCMV capsids were pelleted from culture supernatants and then resuspended and treated with DNase I. Capsids were disrupted using the QIAamp MinElute virus kit (Qiagen), and DNA was quantified by real-time qPCR using 26-nucleotide primers and proven specific for the AD169 UL115 gene and by comparing it to HCMV TR and AD169 BAC DNA.
Antibodies, immunofluorescence, and immunoblotting.
A rabbit polyclonal serum (R6658) specific for HCMV immediate early protein IE86 was used to measure early gene expression in fixed, permeabilized cells 24 h after infection (26). A mouse monoclonal antibody (MAb), 58S, specific for HSV IE protein ICP4 and rabbit sera specific for HSV gB (R68) or gH/gL (R137) were used in immunofluorescence experiments involving HSV-infected cells, with staining for gB and gH/gL at 12 h and ICP4 at 6 h postinfection as described previously (26). HCMV particles were characterized by Western blotting using MAbs specific for the HCMV major capsid protein (284), tegument protein pp65 (2819), and glycoproteins gB (27-156) and gH (AP86-SA4) provided by Bill Britt (University of Alabama).
Entry in trans assays.
Cells in 96-well dishes were transduced with Ad vectors (100 PFU/cell) for 24 to 48 h and then incubated with HCMV particles, followed by immediate centrifugation at 800 × g for 30 min at 10°C. In some cases, virus entry was promoted by treating cells with 44% PEG at 37°C for 30 s (3). The cells were then incubated with virus for 4 h, the virus was removed, and cells were incubated for an additional 20 h before being stained for HCMV IE86. For HSV entry in trans, cells were not centrifuged and stained for ICP4 6 h after the addition of HSV particles.
Cell surface biotinylation.
Cells were labeled with sulfo-HHS-SS biotin then washed and lysed with buffer containing 1% Triton X-100. Biotinylated proteins were precipitated using NeutraAvidin beads (Thermo Scientific) as described previously (35). Precipitated proteins were then subjected to immunoblotting with HCMV gB-specific MAb 27-156.
SUPPLEMENTAL MATERIAL
Cell surface expression of mutant gB molecules. MRC-5 fibroblasts were infected with Ad vectors expressing wild-type HCMV AD169 gB or mutant forms of gB derived from AD169 gB (A) or wild-type HCMV TR gB or mutant forms of gB derived from TR gB (B) for 24 h. Cells were subjected to cell surface biotinylation and then lysed in buffer containing 1% Triton X-100. Most of the extract (95%) was incubated with NeutrAvidin beads, and cell surface proteins were precipitated. Total cell extracts (5% of extract) and surface-biotinylated proteins were subjected to electrophoresis, followed by immunoblotting with anti-gB MAb 27-156. Download
Cell-cell fusion caused by mutant gB. MRC-5 (A) or ARPE-19 (B) cells were infected with Ad vectors expressing wild-type AD169 gB or TR gB or mutant forms of gB for 24 to 48 h, and then the cells were fixed, permeabilized, and stained with SYTO 13 green fluorescent nucleic acid stain. Download
HSV entry in trans. (A) NHDF were transduced with Ad vectors expressing either HSV gB or gH and gL, incubated for 24 h, then: (A) fixed, permeabilized and stained with gB- or gH-specific antibodies (Total) or not fixed or permeabilized then stained with gB or gH-specific antibodies on ice before being fixed (Cell surface). (B) NHDF were either not transduced with Ad vectors (no Ad) or transduced with Ad vectors expressing gB or gH and gL for 24 h then infected with HSV particles lacking gB or gH (gB-null or gH-null). In some cases these cells were treated with 44% polyethylene glycol (+PEG) for 30 s then washed. Six hours after addition of HSV the cells were fixed, permeabilized and stained for HSV ICP4. Download
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health, R01AI081517, to D.C.J.
We are very grateful to Teresa Compton and Marisa Isaacson for providing HCMVΔgB and retroviruses and to Rob Kalejta for digging these out of freezers. We thank Catherine Wright for performing aspects of the HSV experiments. We are most grateful to Bill Britt, Jay Nelson, Roselyn Eisenberg, and Gary Cohen for providing important anti-HCMV and anti-HSV antibodies.
Footnotes
CitationWille PT, Wisner TW, Ryckman B, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4(3):e00332-13. doi:10.1128/mBio.00332-13.
REFERENCES
- 1. Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2:37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395 [DOI] [PubMed] [Google Scholar]
- 3. Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Isaacson MK, Compton T. 2009. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J. Virol. 83:3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. 2012. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog. 8:e1002905 http://dx.doi.org/10.1371/journal.ppat.1002905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 72:8191–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 84:2585–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 85:11638–11645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 13. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Backovic M, Leser GP, Lamb RA, Longnecker R, Jardetzky TS. 2007. Characterization of EBV gB indicates properties of both class I and class II viral fusion proteins. Virology 368:102–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J. Virol. 85:6175–6184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutt-Fletcher LM, Chesnokova LS. 2010. Integrins as triggers of Epstein-Barr virus fusion and epithelial cell infection. Virulence 1:395–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma S, Wisner TW, Johnson DC, Heldwein EE. 2013. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435:239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461 [DOI] [PubMed] [Google Scholar]
- 21. Isaacson MK, Feire AL, Compton T. 2007. Epidermal growth factor receptor is not required for human cytomegalovirus entry or signaling. J. Virol. 81:6241–6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395 [DOI] [PubMed] [Google Scholar]
- 23. Feire AL, Koss H, Compton T. 2004. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. U. S. A. 101:15470–15475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feire AL, Roy RM, Manley K, Compton T. 2010. The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J. Virol. 84:10026–10037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kinzler ER, Compton T. 2005. Characterization of human cytomegalovirus glycoprotein-induced cell-cell fusion. J. Virol. 79:7827–7837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruel N, Zago A, Spear PG. 2006. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology 346:229–237 [DOI] [PubMed] [Google Scholar]
- 29. Hegde NR, Dunn C, Lewinsohn DM, Jarvis MA, Nelson JA, Johnson DC. 2005. Endogenous human cytomegalovirus gB is presented efficiently by MHC class II molecules to CD4+ CTL. J. Exp. Med. 202:1109–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 104:10187–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 83:3115–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wisner TW, Johnson DC. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-Golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519–11535 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell surface expression of mutant gB molecules. MRC-5 fibroblasts were infected with Ad vectors expressing wild-type HCMV AD169 gB or mutant forms of gB derived from AD169 gB (A) or wild-type HCMV TR gB or mutant forms of gB derived from TR gB (B) for 24 h. Cells were subjected to cell surface biotinylation and then lysed in buffer containing 1% Triton X-100. Most of the extract (95%) was incubated with NeutrAvidin beads, and cell surface proteins were precipitated. Total cell extracts (5% of extract) and surface-biotinylated proteins were subjected to electrophoresis, followed by immunoblotting with anti-gB MAb 27-156. Download
Cell-cell fusion caused by mutant gB. MRC-5 (A) or ARPE-19 (B) cells were infected with Ad vectors expressing wild-type AD169 gB or TR gB or mutant forms of gB for 24 to 48 h, and then the cells were fixed, permeabilized, and stained with SYTO 13 green fluorescent nucleic acid stain. Download
HSV entry in trans. (A) NHDF were transduced with Ad vectors expressing either HSV gB or gH and gL, incubated for 24 h, then: (A) fixed, permeabilized and stained with gB- or gH-specific antibodies (Total) or not fixed or permeabilized then stained with gB or gH-specific antibodies on ice before being fixed (Cell surface). (B) NHDF were either not transduced with Ad vectors (no Ad) or transduced with Ad vectors expressing gB or gH and gL for 24 h then infected with HSV particles lacking gB or gH (gB-null or gH-null). In some cases these cells were treated with 44% polyethylene glycol (+PEG) for 30 s then washed. Six hours after addition of HSV the cells were fixed, permeabilized and stained for HSV ICP4. Download