

ABSTRACT
The pathogenic bacterium Xanthomonas campestris pv. campestris, the causal agent of black rot of Brassicaceae, manipulates the physiology and the innate immunity of its hosts. Association genetic and reverse-genetic analyses of a world panel of 45 X. campestris pv. campestris strains were used to gain understanding of the genetic basis of the bacterium’s pathogenicity to Arabidopsis thaliana. We found that the compositions of the minimal predicted type III secretome varied extensively, with 18 to 28 proteins per strain. There were clear differences in aggressiveness of those X. campestris pv. campestris strains on two Arabidopsis natural accessions. We identified 3 effector genes (xopAC, xopJ5, and xopAL2) and 67 amplified fragment length polymorphism (AFLP) markers that were associated with variations in disease symptoms. The nature and distribution of the AFLP markers remain to be determined, but we observed a low linkage disequilibrium level between predicted effectors and other significant markers, suggesting that additional genetic factors make a meaningful contribution to pathogenicity. Mutagenesis of type III effectors in X. campestris pv. campestris confirmed that xopAC functions as both a virulence and an avirulence gene in Arabidopsis and that xopAM functions as a second avirulence gene on plants of the Col-0 ecotype. However, we did not detect the effect of any other effector in the X. campestris pv. campestris 8004 strain, likely due to other genetic background effects. These results highlight the complex genetic basis of pathogenicity at the pathovar level and encourage us to challenge the agronomical relevance of some virulence determinants identified solely in model strains.
IMPORTANCE
The identification and understanding of the genetic determinants of bacterial virulence are essential to be able to design efficient protection strategies for infected plants. The recent availability of genomic resources for a limited number of pathogen isolates and host genotypes has strongly biased our research toward genotype-specific approaches. Indeed, these do not consider the natural variation in both pathogens and hosts, so their applied relevance should be challenged. In our study, we exploited the genetic diversity of Xanthomonas campestris pv. campestris, the causal agent of black rot on Brassicaceae (e.g., cabbage), to mine for pathogenicity determinants. This work evidenced the contribution of known and unknown loci to pathogenicity relevant at the pathovar level and identified these virulence determinants as prime targets for breeding resistance to X. campestris pv. campestris in Brassicaceae.
Introduction
Phenotypic variation is central for species adaptation and is due to environmental and genetic variation. The latter often arises from complex interactions between multiple loci. Understanding the molecular mechanisms underlying most complex traits thus remains a main challenge in evolutionary biology (1, 2). The first step toward this goal is to perform linkage or association studies. Association studies seek to identify the joint distribution between genotype and phenotype in order to characterize the genetic variants responsible for phenotypic variation. Genome-wide association (GWA) studies have proven to guarantee the detection of significant associations in humans (2) and other model species (3, 4). Such GWA studies begin to emerge in the field of plant-pathogen interactions (4, 5) and are becoming more accessible thanks to the dropping cost of genotyping and sequencing technologies.
Among plant pathogens, the Xanthomonas genus is a complex and large group of gammaproteobacteria that comprises at least 19 species pathogenic to more than 400 host plants (6, 7). Xanthomonas campestris pv. campestris is a seed-borne pathogen distributed worldwide and the causal agent of black rot on leaves of Brassicaceae of economic importance, such as cabbage, mustard, and radish, as well as the model plant Arabidopsis thaliana (8). Currently, there are four complete X. campestris pv. campestris genomes available (9–12). X. campestris pv. campestris strains have been classified in 9 races based on a discriminant set of resistant/susceptible Brassica cultivars (13, 14), but the molecular basis for the race annotation remains elusive.
To infect their host plants, bacteria of the Xanthomonas genus rely on a large arsenal of virulence factors, such as adhesion factors, cell wall-degrading enzymes, extracellular- and lipo-polysaccharides, and a type III secretion (TTS) system and associated type III effector proteins (T3Es) (6, 7). The TTS system is a protein secretion apparatus used by animal and plant pathogens or mutualists to deliver T3E virulence proteins directly into host cells, where they can modulate the host’s physiology and manipulate the host immune system. This TTS system and T3Es are essential for virulence, since mutation in the TTS machinery limits pathogen growth and symptom development, yet the loss of single effector genes has often no or a limited impact on pathogenicity, likely due to functional redundancy.
Plant innate immunity is a multilayer system (15). (i) Plants monitor for the presence of conserved/generic pathogen-associated molecular patterns (PAMPs) and activate PAMP-triggered immunity (PTI) (16). For instance, flagellin is a PAMP perceived by the FLS2 receptor at the Arabidopsis plasma membrane (17). (ii) Plants can detect the modification of host components by strain-specific T3Es, which trigger a rapid and specific immune response (effector-triggered immunity [ETI]) (iii). Importantly, T3Es are known to be able to suppress both PTI and ETI caused by PAMPs and T3Es, respectively (18). Therefore, T3Es are considered prime pathogenicity determinants for plant pathogens. In Xanthomonas, T3Es were called either Xop (Xanthomonas outer protein) (19) or Avr (avirulence) proteins depending on their mode of identification (see http://www.xanthomonas.org/t3e.html). In available genomes, the T3E repertoires (called the type III effectome) can be predicted based on homology to known T3Es, on presence of eukaryotic features and/or of promoter motifs necessary for coexpression with the TTS system. T3Es can also be identified experimentally using screens for TTS system-coexpressed genes, in vitro demonstration of type III-dependent secretion, and in planta demonstration of translocation. Such analyses predict at least 72 T3Es in Ralstonia solanacearum GMI1000 (20), 39 in Pseudomonas syringae pv. tomato DC3000 (21), and 36 in Xanthomonas axonopodis pv. vesicatoria 85-10 (22). So far, only 20 T3Es were reported in X. campestris pv. campestris ATCC 33913 (22). However, a subset of these T3Es are family or species specific; for instance, xopAC (originally called avrAC) is exclusively found in X. campestris species and cannot be found by simple homology searches with effectors from other Xanthomonas species (22, 23). T3Es are distributed heterogeneously in bacterial populations. Thus, sizes of effectomes determined from sequenced strains are underestimates of the species or pathovar T3E repertoires. Individual genomes are shaped by horizontal gene transfer (HGT), a mechanism by which virulence factors are distributed throughout genomes via transposable elements, plasmids, and phages. Because T3Es display characteristics of heterogeneous distribution among strains, participate in host defense suppression, and promote pathogen multiplication and dispersion, T3Es are good candidates for, in part, explaining host specificity in the plant-pathogenic species P. syringae (24) and Xanthomonas axonopodis (25). To date, the genetic basis of X. campestris host specificity remains unknown.
In this study, we report our findings of the natural genetic variation of X. campestris pv. campestris using a panel of 45 strains harvested worldwide on different host cultivars. Our goal was to identify genes or loci with large virulence or avirulence effects relevant at the pathovar level. The aggressiveness of these 45 natural strains was tested on two natural strains (accessions) of Arabidopsis thaliana, and association studies were performed using the presence/absence of T3E genes and a large set of amplified fragment length polymorphism (AFLP) markers. The function of candidate T3Es in pathogenicity was also tested by reverse genetics. This study identified a set of T3E genes and AFLP markers associated with pathogenicity. Our results highlight the complexity of X. campestris pv. campestris pathogenicity and demonstrate the dual virulence or avirulence role of T3Es.
RESULTS
High-resolution AFLP-based phylogeny and molecular evolutionary genetics reveal a high genomic diversity in a world panel of 45 X. campestris pv. campestris strains.
We aimed to investigate the genetic diversity within a collection of 45 X. campestris pv. campestris strains, selected to maximize diversity based on geographic origin, host plant, year of isolation, and race (see Table S1 in the supplemental material; Fig. 1A). Multilocus sequence analysis (MLSA) performed on genes efp and glnA confirmed that the selected strains were genuine X. campestris pv. campestris (Table S1; Fig. S1A) (26), yet this MLSA organized the 45 X. campestris pv. campestris strains in only 5 sequence types and thus had a very low resolution. An AFLP analysis, which generates markers throughout the genomes, was performed to determine more precisely the phylogenic relationships among these 45 X. campestris pv. campestris strains (Fig. 1B). The X. campestris pv. raphani strain CFBP5828 was used as the outgroup. Among the 1,942 neutral markers identified, 929 polymorphic markers had a minor allele frequency (MAF) of >5%. Based upon the AFLP results, placement was supported by high bootstrap values (80%) of 39 strains from our total of 45. The remaining strains could not be assigned unambiguously to any of those clades. Still, AFLP analysis was highly discriminative relative to MLSA, since it relied on a large number of informative polymorphic markers (Fig. S1A). For this reason, AFLP analysis was preferred for subsequent studies. The gene diversity (h) in the panel of the 45 strains is high (estimated h, 0.185), considering that all strains belong to the same pathovar. Irrespective of the distance between markers, a very low averaged pairwise linkage disequilibrium (LD, defined as any nonrandom association between markers) (R2V = 0.015, where R2V is the R2 corrected by the relatedness of genotyped individuals; a noncorrected LD was an R2 of 0.02) was found across the genome, which was close to the expected value under complete equilibrium (1/n = 0.02 [n = 45 strains]). These results are possibly a consequence of the complex evolution of the bacterial genome, where gene duplication, gene gain, and gene loss are common features. Interestingly, the two X. campestris pv. campestris reference strains 8004 and ATCC 33913 (clade C) shared more than 96% marker identity, whereas strain B100 belonged to a distinct clade (A). X. campestris pv. campestris strains harvested in China were present in all clades except clade E, suggestive of a worldwide and human-based expansion of those strains. Finally, clade G strains, though less closely related to the other X. campestris pv. campestris strains, were still classified as X. campestris pv. campestris based on MLSA definition established by Fargier et al. (26) and also because these strains caused typical black rot symptoms on cabbage and Chinese radish (27). Thus, clade G strains might represent a more distant lineage of X. campestris pv. campestris that has not been reported so far in the literature.
FIG 1 .

Origin and neutral phylogeny of a world collection of 45 X. campestris pv. campestris strains. (A) Countries of origin for the different X. campestris pv. campestris strains of the collection are in dark red. (B) A neighbor-joining tree based on Dice similarity indices inferred from 1,942 AFLP markers shows the phylogenetic relationships among X. campestris pv. campestris strains. Bootstrap values higher than 70% are indicated. X. campestris pv. campestris strains with genome sequences available are highlighted in black. On the right, lines indicate AFLP-based clades of X. campestris pv. campestris strains. X. campestris pv. raphani strain CFBP5828 was used to root the tree. XccA to -G, X. campestris pv. campestris of the A to G clades, respectively.
The Spearman rho rank correlation coefficient was calculated to evaluate if the clade distribution might be consistent with life history traits of the bacteria, factors including geographical origin, host of harvest, and race. At the clade level, significant correlations were found with the country of origin (ρ = −0.39, P = 0.01) and with the race distribution (ρ = 0.44, P = 0.004). Because X. campestris pv. campestris races were defined by the host range on different Brassica species, a significant clade/race correlation suggests that the host may shape the evolution of X. campestris pv. campestris.
X. campestris pv. campestris flagellin does not elicit FLS2-dependent innate immunity in Arabidopsis.
The bacterial flagellin FliC and its 22-amino-acid peptide flg22 have been shown to be potent elicitors of Arabidopsis innate immunity. Previous studies have reported that eliciting and noneliciting variants of flg22 are present in X. campestris pv. campestris (28) and might impact the interaction with host plants. Thus, flg22 conservation in 43 X. campestris pv. campestris strains and 1 X. campestris pv. raphani strain was studied by sequencing the 5′ region of fliC. Only two DNA haplotypes could be identified (GenBank accession numbers JX453140 to JX453183). All X. campestris pv. campestris strains but HRI6185 were predicted to produce an flg22 peptide that avoids recognition by the Arabidopsis FLS2 receptor (see Fig. S2 in the supplemental material) (28). The HRI6185 flagellin was identical to that observed in X. campestris pv. raphani strain CFBP5828 and was predicted to elicit FLS2-dependent immune responses. Thus, any variation in pathogenicity that might be observed among those 42 bacterial strains should not be attributed to polymorphisms in the N terminus of X. campestris pv. campestris flagellin, as most strains are predicted to escape recognition by FLS2.
X. campestris pv. campestris strains harbor a highly variable predicted type III secretome.
Because T3Es are globally essential for pathogenicity and are proposed to contribute to host specificity (25, 29), we aimed at identifying and comparing the type III secretomes in our panel of 45 X. campestris pv. campestris strains. The genomes of the 3 sequenced strains, B100, ATCC 33913, and 8004, were first mined by searching by BLAST analysis for homologs of known type III effectors/secreted proteins (22; http://www.xanthomonas.org). In parallel, plant-inducible promoter (PIP) boxes (consensus, TTCG-N16-TTCG), which control the in planta expression of a large hrpX regulon in Xanthomonas, including the TTS system and numerous T3Es, were searched (19, 30). Twenty-nine genes encoding 11 validated T3Es (demonstrated translocation), 15 effector candidates (predicted translocation), and 3 type III secreted proteins (no translocation evidenced) were identified. We preferred to categorize these proteins as type III secretome or type III substrate proteins (T3SPs). The three reference X. campestris pv. campestris strains have at least 25 T3SP-encoding genes in common. In addition, both of the closely related strains ATCC 33913 and 8004 contain xopD2, xopE2, and xopJ5 (which encode at least 28 putative T3SPs), while strain B100 contains xopD1 (which encodes at least 26 putative T3SPs).
To investigate the natural genetic variation in these genes, PCR-based detection of both partial and full-length sequences was performed (Fig. 2). We defined a core type III secretome composed of at least 15 genes that are present in more than 95% of the strains and a variable type III secretome consisting of 14 genes. An accuracy rate estimated from a dot blot analysis of a subset of 10 strains for 51% of the effectome (9 genes in the core effectome and 6 in the variable effectome) was 99.4%, representing 1 conflict out of 150 events; a xopX2 amplicon was present, but no hybridization signal was visualized for strain CFBP5683. Minimal type III secretome sizes varied considerably among the strains, ranging from 18 to 28 genes (average = 23). Genes encoding the variable T3SPs were present on average in 58% of the strains. These results revealed an important and formerly unknown diversity of the X. campestris pv. campestris type III secretome composition at the intrapathovar level. Gene xopX1 was present in only 31% of the strains tested, while xopD was present with two mutually exclusive alleles, which were defined as xopD2 (as found in X. axonopodis pv. vesicatoria 85-10 and X. campestris pv. campestris 8004) (31) and xopD1 (as found in X. campestris pv. campestris B100) (32). We also classified the strains based on their type III secretome composition (see Fig. S1B in the supplemental material). The type III secretome composition is a good descriptor of clades A, B, D, F, and G, suggesting that it results for the most part from vertically inherited genes, as observed in other phytopathogenic bacteria (e.g., Xanthomonas axonopodis [25] and Pseudomonas syringae [21]). Identical insertions of IS1478 in xopAL1 in 5 strains of clade B further illustrate the vertical inheritance of T3SPs, yet some incongruence for clades C and E was observed, indicating that horizontal gene transfer also shaped the variable type III secretome. Furthermore, the GC contents of the variable T3SP genes based on the genes from the three sequenced strains were significantly lower (56.3% ± 7%; P = 1 × 10−5; n = 14) than in the rest of the genes (65% ± 4%) or the core type III secretome (62% ± 4%; P = 0.0054; n = 15). This result suggests the existence of relatively recent horizontal gene transfers within the variable type III secretome. Insertion sequences (IS) were observed in 5 different T3SP genes, with an average frequency per gene of 7%. Insertions were predominant in the variable type III secretome (15 out of 17 insertions). Interestingly, strain CN07 carries four genes interrupted by IS elements, which might impact its host range.
FIG 2 .

Distribution of genes coding for type III secreted proteins among X. campestris pv. campestris strains and X. campestris pv. campestris pathogenicity on Arabidopsis and pepper. A phylogeny of X. campestris pv. campestris strains as determined in Fig. 1B is indicated in the left. The pathogenicity of these X. campestris pv. campestris strains on Arabidopsis natural-accession Col-0 and Kas plants (mean disease index at 7 dpi) is indicated by color coding. For Arabidopsis, 0 to 1 indicates no symptoms, 1 to 2 indicates weak chlorosis, 2 to 3 indicates strong chlorosis, and 3 to 4 indicates necrosis. Strains were also inoculated on nonhost ECW-10R pepper carrying the Bs1 R gene. HR, hypersensitive response; −, no HR. The presence of a flagellin protein eliciting the FLS2 receptor in Arabidopsis was inferred from a fliC partial DNA sequence (Fig. S2) as described previously (28). The presence or absence of a homologous T3SP gene sequence was determinate by PCR either on the 5′ or 3′ part of the gene or on the entire gene (FL [full length]). Dark-gray squares represent the presence of the corresponding genes with both primer combinations at the expected sizes, whereas white squares represent the absence of PCR amplification or PCR with a different amplicon size. Light-gray squares indicate that the expected PCR amplicons were detected only with one of the two primer combinations. The circles indicate that these results are corroborated by dot blot analyses. The core (present in more than 95% of the strains studied) and variable type III secretomes are separated by a vertical dashed line. The dotted frame shows the mutually exclusive presence/absence of xopD1 and xopD2 in X. campestris pv. campestris. A, B, C, and D correspond to the different XopAC protein haplotypes (Fig. 5). Genes detected but interrupted by an insertion sequence are as follows: IS1477, IS1478, and IS1995 in xopAL1, IS1479 in xopAC, IS1404 in xopAL2, IS1404 and IS1481 in xopP, and IS1404 and IS1477 in xopD2. The right column indicates the minimal size of the predicted type III secretome for each strain. Ath, Arabidopsis thaliana; XccA to -G, X. campestris pv. campestris of the A to G clades, respectively; Min, minimum; nd, not determined.
In conclusion, both type III secretomes and genomes of these X. campestris pv. campestris isolates show important natural variation at the intrapathovar level, which could be exploited for genetic approaches.
Genome-wide association study of the pathogenicity of X. campestris pv. campestris on Arabidopsis.
In order to identify natural genetic variants associated with pathogenicity traits on Arabidopsis, we inoculated all 45 strains on both the Columbia-0 (Col-0) and Kashmir (Kas) ecotypes by piercing plant leaves. The aggressiveness among strains varied with each accession, from avirulence (disease index [DI] < 1) to full virulence (DI > 3), and infection outcomes were very different between the two natural accessions (Fig. 2; Fig. S3). A general loss of virulence cannot explain the weak aggressiveness of some strains on Arabidopsis; most strains were virulent on at least one host plant (13, 14, 27), and all strains carrying avrBs1 caused a TTS-dependent hypersensitive response (HR) in resistant pepper ECW-10R, indicating that their TTS system was functional (Fig. 2).
We were interested in investigating the natural genetic variation of the strains for pathogenicity to plants of the Col-0 and Kas ecotypes at 7 days postinoculation (dpi), the time at which symptoms were the strongest. Phenotyping data quality was assessed by calculating the upper bound of the broad-sense heritability (H2) (33). We found suitable values of H2 from our three replicates (H2 = 0.74 for Col-0; H2 = 0.94 for Kas). A genome-wide association study was performed with 29 T3SP markers (15 full-length coding sequences [CDS], 14 5′ CDS) and 929 AFLP markers. Three of the core effectome genes (xopX2, xopP, and xopF) were included in the analysis because polymorphic amplification patterns were observed for one of the PCR markers. To limit the chance of making type I errors, a false-discovery rate (FDR) of 0.05 (P = 0.003 for Kas; P = 0.001 for Col-0) was used. Nonparametric Wilcoxon rank tests with an FDR correction at 5% were also performed, assuming nonnormal distribution. These tests showed 75% congruence of significant markers with the efficient mixed model for Col-0 and Kas data.
With an FDR of 0.05, 28 (2 T3SP markers/26 AFLPs) and 61 (4 T3SP markers/57 AFLPs) significant variants for each trait at a threshold of 7 dpi were detected on Col-0 and Kas ecotypes, respectively (Fig. 3A and C and S4A; Table S2). Pairwise linkage disequilibrium (LD) was estimated among all significant markers (average LD, R2V = 0.08) (Fig. S5). No complete linkage between T3SP and AFLP markers was observed, suggesting that more DNA variants than the currently known T3SPs in the genome were detected. The markers with known physical positions based on their positions in reference strain 8004 presented an average LD value of 0.14, much higher than the average LD observed for all markers (R2V = 0.015) (Fig. S5). This result may suggest some epistatic interactions between the T3SP loci or their simultaneous acquisition by the genome.
FIG 3 .

Association study results 7 days after inoculation on Col-0 (dots) and Kas (diamonds) plants. (A) Negative log10 of the P values from an association test of the effector markers along the chromosome of the X. campestris pv. campestris 8004 reference strain. (B) Average phenotypic differences measured at each significant effector. The significant markers (FDR at 5%) and their respective effects are in red (pathogenicity effect) or blue (avirulence effect). Dashed lines indicate q values (minimum FDR adjusted p-value at which the test may be called). (C) The overlap between the AFLP and T3SP markers associated with gain (red) or loss (blue) of pathogenicity in Col-0 and Kas plants is represented using a Venn diagram. Avr, avirulent; Vir, virulent.
The effectors xopAC (also called avrAC) and xopJ5 on Kas and xopAL2 on Col-0 plants (Fig. 3A and B) were significantly associated with variation in bacterial pathogenicity at 7 dpi. Interestingly, the Wilcoxon nonparametric test revealed significant associations for the following effectors: avrBs1, xopAC, and xopH on Col plants and xopAC, xopJ5, and xopF on Kas plants (Table S3). These T3E markers were fairly common (with a MAF between 26 and 49%). The contribution of these T3E loci to pathogenicity to Arabidopsis was previously unknown except with xopAC (34). However, one has to be cautious in interpreting these results with a sample size of 45 strains. Sufficient statistical power (~80%) can be reached only for quantitative trait loci (QTL) of very large effects that explain more than 50% of the total phenotypic variance (3). When we consider a more stringent nominal significance threshold of 10−5, only 6 markers are significant on Kas plants (including the effectors xopAC and xopJ5) and none are significant on Col-0 plants. The direction and size of the effects estimated from the difference between the mean phenotypic values of the two alleles at each locus are presented in Fig. 3B and S4B. More effects are associated with increased pathogenicity on Col-0 than on Kas plants (χ2 = 5, P = 0.03), and the intensity of virulence effects was higher on Col-0 than on Kas plants (Wilcoxon test, W = 207, where W is the smaller of the rank totals; P = 1.1 × 10−8). Interestingly, the markers significantly associated with X. campestris pv. campestris pathogenicity in Col-0 or Kas plants are essentially different, with only 22% of the markers being common to both responses (Fig. 3C). These 16 markers did not include T3SP markers. Interestingly all showed opposite effects between the two plants: increased pathogenicity to Kas plants but reduced pathogenicity (i.e., avirulence) to Col-0 plants. Thus, these findings illustrate the dual roles of these genetic variants, which depend on host genotype.
A reverse-genetics approach indicates that xopAM confers partial avirulence to X. campestris pv. campestris 8004 on Arabidopsis plants of ecotype Col-0.
We assessed the contribution to pathogenicity of the predicted T3SP genes of X. campestris pv. campestris 8004 by constructing single or double T3SP gene deletions; we studied all 28 genes except xopAL2, xopA, hpaA, avrBs2, and xopAG from the core type III secretome. Col-0 and Kas plants were inoculated with each mutant (Fig. 4 and S6). No significant effect was observed on Kas plants (Fig. S6A). Only the ∆xopAC and ∆xopAM mutants showed a significantly increased aggressiveness on Col-0 plants compared to that of the wild-type strain (wt) and all other mutants (P < 10−3 for all significant comparisons) (Fig. 4A). Both mutant phenotypes could be complemented (Fig. 4B and 5C). These results indicate that xopAC and xopAM contribute to the avirulence of X. campestris pv. campestris 8004 on Col-0 plants, yet xopAM seems to confer a limited avirulence to X. campestris pv. campestris on Col-0 plants (average mean difference between the ∆xopAM mutant and the wt, −1.25) compared to that conferred by xopAC (between the ∆xopAC mutant and the wt, −2.25) (Fig. 4B). Interestingly, the ∆xopAC ∆xopAM double mutant behaved like the xopAM mutant, so that the ∆xopAM mutation was epistatic to the xopAC mutation in terms of symptom development. However, the bacterial growth of the xopAM deletion mutant in planta was not significantly increased in Col-0 leaves, unlike that of the ∆xopAC and ∆xopAC ∆xopAM mutants (Fig. 4C). In conclusion, the conserved effector XopAM is a novel determinant of X. campestris pv. campestris 8004 avirulence on Col-0 plants that does not affect bacterial growth under the conditions tested.
FIG 4 .

Pathogenicity of T3SP mutants in X. campestris pv. campestris strain 8004 inoculated on Arabidopsis plants of the Col-0 ecotype. (A, B) Bacteria were inoculated by piercing in the central vein, and infection symptoms were scored over 10 days. The disease index was as follows: 0 to 1, no symptoms; 1 to 2, weak chlorosis; 2 to 3, strong chlorosis; and 3 to 4, necrosis. Box plot representations of symptoms are as follows: middle bar, median; box limit, upper and lower quartiles; and extremes, minimum and maximum values. (A) Inoculation of deletion mutants. (B) Complementation of the xopAM mutant (∆AM) by pFAJ1700-xopAM (∆AM+AM) and phenotypes of the xopAM xopAC double-deletion mutant (∆AM∆AC) and xopAC mutant (∆AC). EV, empty vector. Statistical groups were determined using a Kruskal-Wallis test (P < 0.001) and are indicated by different letters. (C) Wild-type X. campestris pv. campestris strain 8004 and its deletion mutant derivatives in the xopAM (∆AM), xopAC (∆AC), and xopAM xopAC (∆AM∆AC) mutants were inoculated by piercing leaves of Col-0 plants and inoculating them at a bacterial density of 108 CFU/ml. In planta bacterial populations around the inoculated areas were determined 0 and 5 days postinoculation and are expressed as the log of the number of CFU per square cm. Standard deviations were calculated in three independent experiments with three samples of two leaf discs from different plants for each strain. Statistical groups identified using a Wilcoxon test (P < 0.01) are indicated by different letters.
Natural genetic variation at the xopAC locus.
Our association data based on the presence or absence of xopAC suggest that all xopAC allelic variants are able to confer avirulence to X. campestris pv. campestris on Col-0 plants. We thus sequenced the xopAC locus of the 29 xopAC-containing X. campestris pv. campestris strains (Fig. 5A) (GenBank accession numbers JX453111 to JX453139). Sequencing data covered 473 bp upstream of the annotated start site to 53 bp after the stop codon. Seven bi-allelic single-nucleotide polymorphisms (SNPs) were identified: two in the cis-regulatory regions and five nonsynonymous substitutions in the xopAC coding sequence. The XopAC protein is composed of an N-terminal leucine-rich repeat (LRR) domain and a C-terminal Fic (Filamentation induced by cyclic AMP) domain. Four out of these five substitutions occur in the Fic domain and none in the LRRs (Fig. 5A and B). However, core residues of the Fic domain were absolutely conserved, suggesting that these polymorphisms should not abolish enzymatic functions of the four XopAC haplotypes (A to D) (Fig. 5). Haplotype B (20 out of 29 occurrences) is present throughout the phylogenetic tree (Fig. 2). Haplotype D was exclusively found in 4 Chinese strains, but Chinese strains also expressed XopAC of the B and C haplotypes. Haplotype C was also found in X. campestris pv. raphani strain CFBP5828. In order to test the functionality of all four haplotypes, complementation tests were performed with X. campestris pv. campestris 8004 ∆xopAC. Strains were inoculated onto Col-0 plants (Fig. 5C). While xopAC deletion strains were virulent, all four complemented mutants of X. campestris pv. campestris 8004 ∆xopAC were avirulent to similar extents (P < 0.001). The same complementation tests on Kas plants did not reveal any significant variation in pathogenicity (Fig. 5E). These experiments indicate that strains of all four XopAC haplotypes are functionally equivalent in avirulence on ecotype Col-0 plants.
FIG 5 .
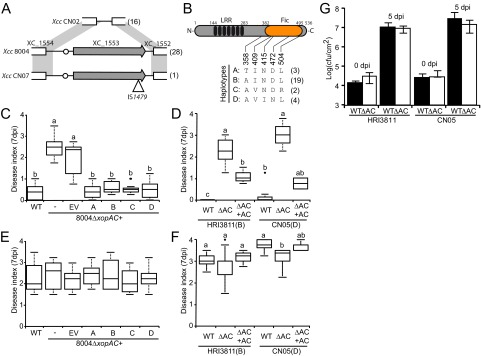
xopAC sequence diversity and functionality of the different XopAC haplotypes. (A) Schematic representation of the XC_1552/XC_1554 locus in X. campestris pv. campestris strain 8004 and orthologous regions in strains CN02 and CN07. Arrows indicate xopAC, and the circles indicate the PIP box promoter element. IS1479 (triangle) is inserted at the 3′ end of the xopAC coding region in strain CN07. Haplotype occurrence is indicated in brackets. (B) Schematic illustration of XopAC functional domains, namely, the N-terminal leucine-rich repeats (LRRs) and C-terminal Fic domain (filamentation induced by cAMP). Numbers indicate domain borders. The different polymorphisms shared by at least two XopAC haplotypes are represented and their positions within XopAC indicated. Four haplotypes (A to D) can be distinguished in X. campestris pv. campestris, and their occurrence in the 29 xopAC-containing strains is indicated in brackets. (C to F) The functionality of the different XopAC haplotypes was tested by inoculations on Col-0 (C, D) and Kas (E, F) natural accessions of Arabidopsis. Disease symptoms were scored at 7 dpi. Each inoculated leaf was individually scored as follows: no symptoms, 0; weak chlorosis surrounding the wound sites, 1; strong V-shaped chlorosis, 2; developing necrosis, 3; and leaf death, 4. The represented average disease scored and the standard deviations were calculated from the values from four plants with four inoculated leaves per plant. Experiments were reproduced at least 3 times. (C, E) Complementation of the 8004 ∆xopAC strain with the pCZ917 empty vector (EV) containing the respective haplotypes of xopAC as defined for panel B. (D, F) Deletion of xopAC was obtained in Rifr derivatives of HRI3811 and CN05 strains (∆AC) and complemented with pCZ917 carrying haplotype A of xopAC (∆AC+AC). X. campestris pv. campestris genotypes are indicated below the graph, and the haplotype of the endogenous xopAC is indicated in brackets. Statistical groups were determined using a Kruskal-Wallis test (P < 0.001) and are indicated by different letters. (G) The wild types and xopAC deletion mutants (∆AC) of X. campestris pv. campestris strains HRI3811 and CN05 were inoculated by piercing the leaves of Col-0 plants and inoculating them at a bacterial density of 108 CFU/ml. In planta bacterial populations around the inoculated areas were determined 0 and 5 days postinoculation and are expressed as the log number of CFU per square cm. Standard deviations were calculated from three independent experiments with three samples of two leaf discs from different plants for each strain. Statistical groups identified using a Wilcoxon test (P < 0.01) are indicated by different letters.
These sequences were then used to perform association tests on haplotype variants of the xopAC sequence (28 strains). A significant haplotype effect was evidenced on both ecotypes at 7 dpi (P = 4.17 × 10−9 on Col-0 and P = 1.02 × 10−8 on Kas plants). An association test for the SNP effect showed one significant association with Kas plants for the nonsynonymous I409V SNP present in the Fic domain (P = 0.02 by the nonparametric test). In Kas plants, the substitution of I for V (minor allele) was associated with a decrease of DI. The pairwise linkage disequilibrium at the locus is intermediate (average R2V of 0.26) (Table S4).
Since the four haplotypes seem functionally equivalent in conferring avirulence to X. campestris pv. campestris 8004, either the detected association is not causal or there are other interacting haplotype-specific genetic factors that are absent/present from the X. campestris pv. campestris 8004 genetic background.
Visualization of xopAC virulence functions in Arabidopsis depends on X. campestris pv. campestris strain genotype.
To test these hypotheses, xopAC was also deleted from two other X. campestris pv. campestris strains (HRI3811 and CN05), which differ in the composition of their variable type III secretomes. Again, the deletion event rendered the strains virulent to Col-0 plants with the same phenotypic differences as previously observed (Fig. 5D), demonstrating that haplotypes B and D in their respective backgrounds are functional (P = 1.2 × 10−12). The insertion of haplotype A in both deletion strains rescued the avirulent phenotype (Fig. 5D). On Kas plants, CN05 ∆xopAC (but not HRI3811 ∆xopAC) was weakly but significantly less aggressive than its wild type (P = 0.0003) (Fig. 5F), suggesting that in this particular genetic background, we may observe a significant contribution of xopAC to pathogenicity. Interestingly, CN05 is a very virulent strain with only 5 predicted variable T3SPs. To study this phenotype further, in planta bacterial populations of the xopAC mutants were measured in Kas leaves inoculated by piercing. At 5 dpi, the ∆xopAC mutation did not significantly affect the growth of strains HRI3811 and CN05 (Fig. 5G). Thus, the virulence role of xopAC on Kas leaves could be visualized by forward genetics in one but not two other X. campestris pv. campestris strains and for symptom development but not in planta growth. This suggests that xopAC’s contribution to pathogenicity might be masked by some genetic background effects in several strains due to functional redundancy or slightly different virulence strategies.
DISCUSSION
X. campestris pv. campestris genetic diversity was unsuspected based on the available reference genomes.
In this study, we observed substantial genetic and phenotypic variation in our collection of X. campestris pv. campestris strains, likely illustrating the potential of adaptation of the species to infect different hosts and to adjust to environmental heterogeneity. A world collection of 45 X. campestris pv. campestris strains collected over 55 years mostly on diseased Brassica oleracea and Brassica rapa was gathered. Their identification as X. campestris pv. campestris was confirmed by MLSA. However, robust phylogenetic relationships could be inferred only from detailed AFLP analysis, which generates markers throughout the genome. This approach clearly identified one coherent genomic group encompassing all X. campestris pv. campestris strains. It also unraveled a significant level of genetic diversity and a low LD among markers, suggesting that recombination may be common in X. campestris pv. campestris populations. Another explanation for a low LD could be a high level of HGT and insertion/deletion markers (indels). Clade G strains seem quite distinct at the genomic level from other X. campestris pv. campestris clades, yet those strains were unambiguously identified as X. campestris pv. campestris by MLSA and caused typical black rot symptoms on Chinese radish (27). Thus, clade G might represent an uncharacterized group of X. campestris pv. campestris strains for which genome sequence data would be very informative. Interestingly, X. campestris pv. campestris phylogeny was only partially accounted for by the geographic origins of the strains. For instance, Chinese strains (CN) were distributed in all X. campestris pv. campestris clades but one, which suggests an efficient introduction into China of strains from worldwide origins or vice versa. Finally, we also observed that the two X. campestris pv. campestris reference strains 8004 and ATCC 33913 are very closely related and that both belong to clade C, while X. campestris pv. campestris B100 belongs to clade A. Thus, these three X. campestris pv. campestris reference genomes are not representative of the natural diversity of this pathogen. Whether this diversity originates from large indels, gene gain, or gene loss, SNPs or the presence of a plasmid would have to be determined by whole-genome sequencing.
High diversity of the type III secretome composition is observed within X. campestris pv. campestris.
The type III secretome predictions for X. campestris pv. campestris can provide only an estimate of the effectome’s minimal size. It varied from 18 type III substrates for strain CFBP12824 to 28 for strains 8004, ATCC 33913, and HRI3851A. We found a significant negative correlation between the type III secretome size and aggressivity on Kas plants (P = 0.007, ρ = −0.39). Genetic diversity observed among X. campestris pv. campestris strains suggests that genome sequencing might unravel other T3SPs that are absent from the genomes of the 3 X. campestris pv. campestris reference strains or other Xanthomonas pathovars (12). Furthermore, no systematic experimental T3SP mining has been reported for X. campestris pv. campestris yet. Thus, the size and composition of these type III secretomes are likely underestimated; X. campestris pv. campestris type III secretome sizes are increasing compared to earlier estimates (20 T3SPs in X. campestris pv. campestris ATCC 33913) (22) and are now in the range of those of many other Xanthomonas strains (20 to 34 non-transcription activator-like T3Es) (22, 35) and P. syringae pathovars (9 to 39 Hrp-dependent outer proteins [Hops]) (21). Only functional screens and analyses of the complete genomes would fully uncover the X. campestris pv. campestris type III secretome, as was done for X. axonopodis pv. manihotis (35).
Interestingly, the X. campestris pv. campestris type III secretome is highly polymorphic; the X. campestris pv. campestris core type III secretome is essentially identical to the Xanthomonas core type III secretome (22). Within the variable type III secretome, XopD is present in two variants, XopD1 and XopD2, whose N-terminal extensions differ and whose distributions are mutually exclusive. Importantly, it was recently reported that both isoforms are not functionally equivalent (36). These observations of high type III secretome diversity at the pathovar level are in agreement with other recent observations of X. axonopodis pv. manihotis or P. syringae pv. avellanae (35, 37). These observations contrast with earlier work performed with X. axonopodis where little variation in type III secretome composition was observed at the pathovar level (25). Both vertical and horizontal inheritances of T3SPs in X. campestris pv. campestris are suggested by our analysis. On the one hand, IS1478 insertion in xopAL1 throughout clade B indicates that vertical inheritance of T3SPs plays a role in X. campestris pv. campestris. On the other hand, the low GC content of the variable type III secretome of X. campestris pv. campestris 8004 and the variability of the type III secretome within genomic clades (e.g., X. campestris pv. campestris E) suggest the occurrence of horizontal gene transfers (Fig. 2).
A genome-wide approach to identify pathovar-relevant rather than strain-specific pathogenicity determinants in X. campestris pv. campestris.
The observed genomic diversity among these 45 X. campestris pv. campestris strains indicates that the three reference genomes are not representative of the natural X. campestris pv. campestris diversity. Thus, some of the knowledge acquired from those three strains might not be transferrable to many other X. campestris pv. campestris strains and might be of limited interest with regard to our understanding of X. campestris pv. campestris biology and pathogenicity evolution. Similarly, several biologically relevant processes might be ignored because they are absent from those three strains.
GWA was performed on candidate genes and AFLP markers, leading to the identification of 73 significant markers with a low LD. These results suggest that X. campestris pv. campestris pathogenicity is controlled by many genetic determinants rather than by a few factors. However, these findings are not contradictory to the hypothesis that a proportion of these genetic determinants may be located within genomic islands, as was observed in P. syringae and Pseudomonas viridiflava (5, 38). Genomic islands can indeed harbor many loci acquired independently via HGT. The exact coverage of our study is unfortunately unknown. Assuming a random distribution of the 929 AFLP markers in the X. campestris pv. campestris genome (average LD = 0.015), we would have a marker every 5.5 kb (the X. campestris pv. campestris 8004 genome is ~5.15 Mb). However, this estimate is highly unlikely because of the poor correspondence between the LD and chromosomal distance in bacterial genomes. With only 45 strains and a low LD among markers across the genome, our GWA certainly lacks statistical power, so true associations are likely being missed. In best-case scenarios, only QTL with large effects can be identified (3, 39). In future studies, using a larger sample size and genome data should greatly improve this GWA approach and should allow the detection of variants associated with smaller phenotypic effects. Besides, our current approach precluded the possibility that T3SPs detected by PCR would be expressed and functional. Though this was verified for XopAC, studying polymorphisms within T3SP sequences and whole-genome data should allow the detection of subtler haplotypic differences which affect gene function or expression. For instance, a significant haplotype effect at the xopAC locus was detected and could be pinpointed to one SNP corresponding to amino acid position 409 located in the Fic domain. This polymorphism was associated with a change in pathogenicity on Arabidopsis of ecotype Kas. It might be worth testing experimentally by site-directed mutagenesis in several X. campestris pv. campestris strains whether this polymorphism impacts xopAC virulence and avirulence functions.
Functional validation of these associations between the presence of T3SPs and increased aggressiveness was tested experimentally with single- or double-deletion mutants of X. campestris pv. campestris 8004. Except for the xopAC strain, none could be confirmed because either the effect is too small to be detected with the current patho-assay or the T3SP gene is not the causal determinant of the association but a linked polymorphism. Alternatively, the X. campestris pv. campestris 8004 genetic background might mask these subtle virulence effects. In support of this hypothesis, xopAC virulence functions on Arabidopsis could be observed only for strains CN05 (ecotype Kas) and B186 (Col-0) (34), not strains HRI3811 and 8004. Both strains B186 and CN05 are highly aggressive on Arabidopsis, and strain CN05 has one of the smallest predicted effectomes of the 45 X. campestris pv. campestris strains studied here, yet xopAC virulence functions on cabbage were easily detected in X. campestris pv. campestris 8004 (34), suggesting that these genetic background effects are both strain and host specific. Detecting avirulence functions can also be problematic, since many effectors suppress ETI themselves, as was observed in P. syringae (40) or X. axonopodis (41). The effect of such suppressors may explain why we did not validate xopJ5’s avirulence function in strain 8004 on Arabidopsis of ecotype Kas. Finally, our results show differences in intensity and direction of effects of markers, depending on the plant genetic background, suggesting that these bacterial genes meet different proteins/networks in the plant. All these observations suggest that complex genetic interactions will complicate the validation of the GWA results and that mutational approaches should be performed with multiple strains to draw solid conclusions.
The avirulence of X. campestris pv. campestris 8004 on Col-0 plants is mediated by both xopAC and xopAM.
The XopAC avirulence role in X. campestris pv. campestris strain 8004 was well known from previous studies (23). Our association studies confirmed that xopAC is a major avirulence gene in natural X. campestris pv. campestris isolates and suggests that the 4 haplotypes are functionally equivalent with respect to their avirulence properties. Protein sequence alignments showed that the LRRs necessary for interaction with potential substrates, such as RPM1-induced protein kinase (RIPK) and Botrytis-induced kinase 1 (BIK1) (34), are highly conserved. Only a few polymorphisms were identified around the Fic region without affecting the Fic consensus motif important for uridylylation of substrate proteins. The avirulence function of these four haplotypes was confirmed on Col-0 plants by deletion and complementation approaches. Importantly, XopAH (also called AvrXccC), a T3E of the fido family (like XopAC) (42), did not confer avirulence in our assays, in contrast to published results with the same strain (43), and it was not associated with avirulence on Col-0 plants in GWA studies at the population level. Thus, this GWA analysis suggests that other effectors, such as xopAC, avrBs1, or xopH, are determinants of avirulence on Arabidopsis of the Col-0 ecotype for X. campestris pv. campestris.
Still, conserved invariable genes are not amendable to GWA studies, and conserved T3SPs were studied by reverse genetics in X. campestris pv. campestris 8004. Besides the known pathogenicity function of the conserved effector gene xopAM on Chinese radish (10), the avirulence function of xopAM on Col-0 plants was revealed by our study. Thus, xopAM is a prime target for breeding resistance into Brassicaceae. XopAM is a 2,049-amino-acid protein highly conserved in X. campestris pv. campestris with homologies to HopR1 from P. syringae. This T3E belongs to the AvrE DspA/E HopR family of effectors, which is widely distributed in type III-containing phytopathogenic bacteria (44). dspA/E was shown to be essential for Erwinia amylovora virulence (45). In Nicotiana benthamiana, hopR1 was also shown to be important for suppression of callose deposition and growth of a P. syringae pv. tomato DC3000 mutant with compromised pathogenicity (44). Virulence targets for members of the AvrE DspA/E HopR family are yet unknown. Interestingly, the xopAM mutation was epistatic to the xopAC mutation for virulence but not bacterial growth. Several scenarios could be proposed to explain such observations. For instance, both xopAM and xopAC may have avirulence activity, and epistasis could explain the reduced virulence of the double mutant. However, such virulence functions are not visible in the susceptible Kas ecotype (Fig. S6), and other Arabidopsis genotypes should be tested to exclude genotype-specific susceptibility in Kas versus Col-0 plants. Alternatively, xopAM and xopAC might suppress recognition of a yet-unknown avirulence gene on Col-0 plants in a manner similar to the suppression of avrBs1-dependent HR on pepper by avrBsT in X. axonopodis pv. vesicatoria (41). Obviously, prime candidates are the T3E genes identified in our GWA analysis. However, many other hypotheses could be envisaged for the moment, but only detailed genetic and biochemical analyses will resolve these questions in the future.
This genome-wide association study of a bacterial plant pathogen evidences the relevance of using complementary multidisciplinary approaches; GWA has its advantages but also its own limitations, which have to be supplemented with other classical strain-specific genetic approaches. With the decreasing cost of whole-genome sequencing and genotyping, such generic GWA studies may allow us to dissect our favorite biological questions with new perspectives and broader relevance.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
X. campestris pv. campestris strains and plasmids used in this study are listed in Table S1 in the supplemental material. X. campestris pv. campestris cells were grown at 28°C in MOKA medium (46). Escherichia coli cells were grown on Luria-Bertani medium at 37°C. For solid media, agar was added at a final concentration of 1.5% (wt/vol). Antibiotics were used at the following concentrations: for X. campestris pv. campestris, 50 µg/ml rifampin, 50 µg/ml kanamycin, and 5 µg/ml tetracycline, and for E. coli, 50 µg/ml ampicillin, 25 µg/ml kanamycin, 40 µg/ml spectinomycin, and 10 µg/ml tetracycline. Spontaneous rifampin-resistant derivatives of X. campestris pv. campestris HRI3811 and CN05 were selected on MOKA-rifampin.
Plant material, growth conditions, and infection tests.
Arabidopsis plants were grown on Jiffy pots in a growth chamber at 22°C, with a 9-h light period and a light intensity of 192 µmol m−2 s−1. Natural variation in X. campestris pv. campestris pathogenicity was assayed on the A. thaliana natural accessions Columbia and Kashmir by piercing inoculation of a bacterial suspension at 108 CFU/ml as described previously (47). Each of the 45 strains was tested on 4 plants per ecotype and 4 leaves per plant. Three independent repetitions were done in 16 blocks. After inoculation, plants were covered by a plastic film and kept at nearly 100% relative humidity. Disease development was scored from 3 to 7 dpi using a disease index ranging from 0 (no symptom) to 4 (full leaf necrosis) as described previously (47). Single-deletion mutants and complemented strains of the X. campestris pv. campestris 8004, HRI3811, and CN05 backgrounds were tested on Col-0 and Kas plants using 4 plants and 16 leaves in 3 independent replicates. Annotations were done at 3 to 10 dpi (Fig. 4C and D). Nonhost ECW-10R pepper plants were grown and inoculated at an optical density at 600 nm (OD600) of 0.4 as previously described (48). The HR was scored 36 h postinfiltration.
Detection and analysis of endogenous X. campestris pv. campestris plasmids.
Plasmids were isolated from overnight cultures in liquid MOKA medium as described previously (49) and resolved by electrophoresis on a 0.7% agarose gel. Plasmids pXCV2, pXCV19, pXCV38, and pXCV183 isolated from X. axonopodis pv. vesicatoria 85-10 were used as size makers to estimate the size of X. campestris pv. campestris plasmids.
PCR-based detection of T3E genes in X. campestris pv. campestris strains and dot blotting.
Two genomic DNA (gDNA) extractions were prepared independently for each strain as described by the manufacturer (50) and used either for dot blot or PCR analyses. The presence of T3E genes was determined using 2 pairs of gene-specific primers designed from the X. campestris pv. campestris 8004-orthologous sequence but lacking small genes, such as xopA, xopG, and xopH. For each gene, one of the primer pairs amplified the full-length T3SP DNA sequence, while the other one amplified a shorter sequence of ca. 300 bp usually in the 5′ coding region. All oligonucleotide sequences are available upon request. A reaction was considered positive (the gene was present) if a single clear band with the expected size was observed after separation on 1% agarose gel. Dot blot hybridizations were performed on a subset of genes (n = 15) for 10 strains with the probe set and hybridization conditions described in reference 25. The estimated accuracy rate with regard to consistency over replicated experiments and internal controls is ca. 99.4%.
Determination of in planta bacterial populations.
Six leaves from different plants were inoculated by piercing the leaves with an X. campestris pv. campestris suspension of 108 CFU/ml. Three pools of two leaf discs encompassing the inoculated zones were sampled using a cork borer (area = 0.33 cm2) at 0 or 5 days after inoculation. Fresh tissues were homogenized in 200 µl sterile water. Serial dilutions of the homogenates were performed, and a 5-µl drop was spotted for each dilution on plates supplemented with appropriate antibiotics. The plates were incubated at 28°C for 48 h, and colonies were counted in spots containing 1 to 30 colonies. Experiments were performed at least three times.
Sequencing of T3SP genes and fliC fragment and sequence data analysis.
The xopAC locus and fliC 5′ region were PCR amplified from genomic DNA using primers LN191/LN193 and LN625/LN626, respectively (sequences are available upon request). During PCR-based detection of T3E genes, single amplicons with an unexpectedly large size were excised from the gel and revealed the presence of IS elements in several T3E genes. After purification (Wizard SV gel and PCR cleanup purification kit; Promega), each amplicon was sequenced and analyzed using Geneious software (Biomatters, New Zealand).
The average GC contents of the core and conserved type III secretome were calculated from the GC content of each T3SP CDS as inferred from Geneious and compared to the whole-genome GC content.
X. campestris pv. campestris genotyping.
In order to confirm that all strains from our working collection were indeed X. campestris pv. campestris, we performed an MLSA with the efp and glnA amplicons as described previously (26).
Amplified fragment length polymorphism (AFLP) analysis with the SacI and MspI restriction enzymes of X. campestris pv. campestris gDNA was performed as previously described (51). One selective nucleotide was used on each adapter-specific primer. All 16 possible primer combinations but Sac-T/Msp-C were used. The presence/absence of DNA fragments was determined using GeneMapper (Applied Biosystems, CA) with the following criteria: a size between 60 and 500 bp, a peak area of >1,000, a peak high of >800 relative fluorescence units, and no signal in negative controls.
Phylogeny and molecular evolutionary genetics.
Phylogenetic distances among strains were estimated from AFLP marker data using Dice similarity indices with 5,000 bootstraps. The Dice coefficient of similarity among strains was used to construct a weighted neighbor-joining tree using the Darwin software package (version 5.0.158; http://darwin.cirad.fr/Home.php). The robustness of the tree was assessed by bootstrapping (5,000 resamplings).
The allelic frequency of nonnull alleles was set equal to the frequency of the AFLP fragment in the sample. The Nei gene diversity index h was estimated using the sample allele frequencies from all AFLP markers (52). The estimate of the linkage disequilibrium between all pairs of polymorphic markers with a minor allele frequency of >5% was calculated using an extension of the usual R2 measure, which accounts for the relatedness between the genotypes per the R2V in R package LDcorSV (53). The kinship matrix used to estimate R2V was calculated from the dissimilarity matrix produced by neighbor-joining analysis.
Mutagenesis of X. campestris pv. campestris.
Deletion mutants in X. campestris pv. campestris were obtained using the SacB system with a pK18 suicide vector (54) modified for GoldenGate cloning (55). To this end, the pDONR207 ccdB-Cmr cassette (Invitrogen) was amplified using primers LN431/LN432 (sequences are available upon request) and cloned into pK18 at the HindIII and XbaI sites, giving p∆13. Primers used for amplification of sequences flanking the deleted region are available upon request. Plasmids were introduced into E. coli by electroporation and into X. campestris pv. campestris by triparental mating as described previously (56, 57). Deletion events were selected as previously described (54) and verified by PCR (primer details are available upon request).
Complementation of xopAC and xopAM deletion mutants.
For complementation studies, the xopAC promoter and coding sequence were amplified from X. campestris pv. campestris 8004 (haplotype A), X. campestris pv. campestris B100 (haplotype B), X. campestris pv. campestris CN01 (haplotype C), and X. campestris pv. campestris CN05 (haplotype D). Amplicons were cloned into pCZ917 (58), giving pCZ917-xopACA, pCZ917-xopACB, pCZ917-xopACC, and pCZ917-xopACD, respectively. pCZ917 is a plasmid derived from pFAJ1700 (59) with the lacI gene, Ptac promoter, and T7 terminator. The xopAM promoter and coding sequences were amplified from X. campestris pv. campestris 8004, cloned into the pCRII-Blunt vector (Invitrogen), and used for cointegration into pFAJ1700, giving pFAJ1700-xopAM. All primer sequences used for PCR amplification are available upon request. Cloning details are available upon request. DNA manipulations were performed with standard protocols as described previously (60). Plasmids were introduced into E. coli by electroporation and into X. campestris pv. campestris using pRK2073 as a helper plasmid in triparental matings (56, 57).
Statistical analyses.
The upper bound of the broad-sense heritability (H2) of the raw phenotypic data was calculated using the mean squares method (33). The model coefficients for each bacterial strain were calculated using an ordinal logistic regression model (function “lmr” in package “rms” in R). The strain, plant, and experiment effects were included in the model. Association studies were carried out using the coefficients at 7 dpi.
An efficient mixed-model analysis with a likelihood ratio test from the “emma” package in R (function “emma.ML.LRT”) was used to test for an association between trait and genetic marker (3). All AFLP markers with a minor allele frequency of >5% were tested. For T3SP markers, singletons were excluded. A total of 960 markers were tested, marker by marker. To control for confounding effects of genetic relatedness in the sample, a similarity matrix, which is 1 minus the dissimilarity matrix constructed from the neighbor-joining tree, was used. We obtained a much greater statistical power using the phylogeny-based kinship matrix than the kinship matrix calculated in the emma package.
A nonparametric Wilcoxon test (R function “wilcox.test”) was performed to see association results without correction for genetic relatedness. For the association tests using SNP data at the xopAC locus with only 28 strains, a Wilcoxon test was performed to compare the phenotypic values among the four haplotypes, and we used the emma and Wilcoxon tests to search for an association with SNPs.
To correct for false positives when hundreds of association tests were performed, significance thresholds were adjusted using a false discovery rate set at 0.05 (61). The function “mt.rawp2adjp” of the “multtest” package in Bioconductor was used. At each significant marker, effect size was defined as the difference between the mean phenotypic values of the two alleles.
The effects of the single-gene deletions and complementations were assessed from a nonparametric Kruskal-Wallis test, using the “kruskal” function in the package “agricolae” in R software. The significance threshold for multiple pairwise comparisons was set at 0.001.
All analyses were performed using R software version 2.14.1 (http://www.r-project.org).
Nucleotide sequence accession numbers.
GenBank accession numbers for the 29 xopAC loci and the 44 5′ sequences of fliC from X. campestris are JX453111 to JX453139 and JX453140 to JX453183, respectively.
SUPPLEMENTAL MATERIAL
Relationships between X. campestris pv. campestris strains analyzed by MLSA or based on the diversity of type III secretomes. Download
FliC amino acids 14 to 92 were deduced by partial fliC sequencing in the 45 X. campestris pv. campestris strains and 1 X. campestris pv. raphani strain studied (Table S1). Download
Box plot representation of the pathogenicities of 45 X. campestris pv. campestris strains on Arabidopsis thaliana plants of the Col-0 (A) and Kas (B) ecotypes, which were assayed by piercing of the central vein, and infection symptoms scored 7 days postinoculation. Download
Association study results 7 days after inoculation on plants of the Col-0 (dots) and Kas (diamonds) ecotypes. Download
Pairwise-linkage disequilibrium among all significant markers (n = 73) after emma analysis at 7 dpi with the T3SP PCR markers demarcated from AFLP markers by blue lines (R2 under the diagonal, R2V above the diagonal). Download
Pathogenicity preferred of T3SP mutants of X. campestris pv. campestris strain 8004 10 days postinoculation of Arabidopsis plants of the Kas ecotype. Download
Genotype and characteristics of Xanthomonas campestris strains used in this study.
The emma results on all markers at 7 dpi (see Materials and Methods).
Wilcoxon nonparametric test results on all markers at 7 dpi (see Materials and Methods)
Pairwise linkage disequilibrium measures (R2 and R2V) using the LDcorSV package in R (53) among SNPs present at the xopAC locus.
ACKNOWLEDGMENTS
We are thankful to Brigitte Mangin for statistical advice, to Lionel Gagnevin for help with AFLP analysis, and to Joana Vicente and Eric Holub (Warwick University) for contributing Xanthomonas strains. We also thank the reviewers for their constructive comments to improve the manuscript.
A.H. was supported by grants from the Tunisian Government, from the Conseil Général du Maine-et-Loire, and from INRA. This work was supported by a Ph.D. grant from the French Ministry of National Education and Research and French Guyana to E.G., an INRA-SPE postdoctoral fellowship to M.C., an INRA-SPE grant to S.P. and L.D.N., the LabEx TULIP project (grant ANR-10-LABX-41), and an Agence Nationale de la Recherche-Jeunes Chercheurs grant (Xopaque grant ANR-10-JCJC-1703-01) to L.D.N.
Footnotes
Citation Guy E, Genissel A, Hajri A, Chabannes M, David P, Carrere S, Lautier M, Roux B, Boureau T, Arlat M, Poussier S, Noël LD. 2013. Natural genetic variation of Xanthomonas campestris pv. campestris pathogenicity on Arabidopsis revealed by association and reverse genetics. mBio 4(3):e00538-12. doi:10.1128/mBio.00538-12.
REFERENCES
- 1. Glazier AM, Nadeau JH, Aitman TJ. 2002. Finding genes that underlie complex traits. Science 298:2345–2349 [DOI] [PubMed] [Google Scholar]
- 2. Flint J, Mackay TF. 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 19:723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. 2008. Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JD, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M. 2010. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Araki H, Tian D, Goss EM, Jakob K, Halldorsdottir SS, Kreitman M, Bergelson J. 2006. Presence/absence polymorphism for alternative pathogenicity islands in Pseudomonas viridiflava, a pathogen of Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 103:5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryan RP, Vorhölter FJ, Potnis N, Jones JB, Van Sluys MA, Bogdanove AJ, Dow JM. 2011. Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nat. Rev. Microbiol. 9:344–355 [DOI] [PubMed] [Google Scholar]
- 7. Büttner D, Bonas U. 2010. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 34:107–133 [DOI] [PubMed] [Google Scholar]
- 8. Alvarez AM. 2000. Black rot of crucifers, p 21–52 In Slusarenka AJ, Fraser RSS, van Loon LC, Mechanisms of resistance to plant diseases. Kluwer Publishers Academic, Dordrecht, Netherlands [Google Scholar]
- 9. da Silva AC, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, Monteiro-Vitorello CB, Van Sluys MA, Almeida NF, Alves LM, do Amaral AM, Bertolini MC, Camargo LE, Camarotte G, Cannavan F, Cardozo J, Chambergo F, Ciapina LP, Cicarelli RM, Coutinho LL, Cursino-Santos JR, El-Dorry H, Faria JB, Ferreira AJ, Ferreira RC, Ferro MI, Formighieri EF, Franco MC, Greggio CC, Gruber A, Katsuyama AM, Kishi LT, Leite RP, Lemos EG, Lemos MV, Locali EC, Machado MA, Madeira AM, Martinez-Rossi NM, Martins EC, Meidanis J, Menck CF, Miyaki CY, Moon DH, Moreira LM, Novo MT, Okura VK, Oliveira MC, Oliveira VR, Pereira HA, Rossi A, Sena JA, Silva C, de Souza RF, Spinola LA, Takita MA, Tamura RE, Teixeira EC, Tezza RI, Trindade dos Santos M, Truffi D, Tsai SM, White FF, Setubal JC, Kitajima JP. 2002. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417:459–463 [DOI] [PubMed] [Google Scholar]
- 10. Qian W, Jia Y, Ren SX, He YQ, Feng JX, Lu LF, Sun Q, Ying G, Tang DJ, Tang H, Wu W, Hao P, Wang L, Jiang BL, Zeng S, Gu WY, Lu G, Rong L, Tian Y, Yao Z, Fu G, Chen B, Fang R, Qiang B, Chen Z, Zhao GP, Tang JL, He C. 2005. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15:757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vorhölter FJ, Schneiker S, Goesmann A, Krause L, Bekel T, Kaiser O, Linke B, Patschkowski T, Rückert C, Schmid J, Sidhu VK, Sieber V, Tauch A, Watt SA, Weisshaar B, Becker A, Niehaus K, Pühler A. 2008. The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J. Biotechnol. 134:33–45 [DOI] [PubMed] [Google Scholar]
- 12. Bolot S, Guy E, Carrere S, Barbe V, Arlat M, Noël LD. 2013. Genome sequence of Xanthomonas campestris pv. campestris strain Xca5. Genome Announc. 1(1):e00032-00012 http://dx.doi.org/10.1128/genomeA.00032-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vicente JG, Conway J, Roberts SJ, Taylor JD. 2001. Identification and origin of Xanthomonas campestris pv. campestris races and related pathovars. Phytopathology 91:492–499 [DOI] [PubMed] [Google Scholar]
- 14. Fargier E, Manceau C. 2007. Pathogenicity assays restrict the species Xanthomonas campestris into three pathovars and reveal nine races within X. campestris pv. campestris. Plant Pathol. 56:805–818 [Google Scholar]
- 15. Jones JD, Dangl JL. 2006. The plant immune system. Nature 444:323–329 [DOI] [PubMed] [Google Scholar]
- 16. Boller T, Felix G. 2009. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 60:379–406 [DOI] [PubMed] [Google Scholar]
- 17. Gómez-Gómez L, Boller T. 2000. FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 5:1003–1011 [DOI] [PubMed] [Google Scholar]
- 18. Deslandes L, Rivas S. 2012. Catch me if you can: bacterial effectors and plant targets. Trends Plant Sci. 17:644–655 [DOI] [PubMed] [Google Scholar]
- 19. Noël L, Thieme F, Nennstiel D, Bonas U. 2001. cDNA-AFLP analysis unravels a genome-wide hrpG-regulon in the plant pathogen Xanthomonas campestris pv. vesicatoria. Mol. Microbiol. 41:1271–1281 [DOI] [PubMed] [Google Scholar]
- 20. Poueymiro M, Genin S. 2009. Secreted proteins from Ralstonia solanacearum: a hundred tricks to kill a plant. Curr. Opin. Microbiol. 12:44–52 [DOI] [PubMed] [Google Scholar]
- 21. Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, Cherkis K, Roach J, Grant SR, Jones CD, Dangl JL. 2011. Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7(7):e1002132 http://dx.doi.org/10.1371/journal.ppat.1002132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. White FF, Potnis N, Jones JB, Koebnik R. 2009. The type III effectors of Xanthomonas. Mol. Plant Pathol. 10:749–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu RQ, Blanvillain S, Feng JX, Jiang BL, Li XZ, Wei HY, Kroj T, Lauber E, Roby D, Chen B, He YQ, Lu GT, Tang DJ, Vasse J, Arlat M, Tang JL. 2008. AvrAC(Xcc8004), a type III effector with a leucine-rich repeat domain from Xanthomonas campestris pathovar campestris confers avirulence in vascular tissues of Arabidopsis thaliana ecotype Col-0. J. Bacteriol. 190:343–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarkar SF, Gordon JS, Martin GB, Guttman DS. 2006. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics 174:1041–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hajri A, Brin C, Hunault G, Lardeux F, Lemaire C, Manceau C, Boureau T, Poussier S. 2009. A “repertoire for repertoire” hypothesis: repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS One 4(8):e6632 http://dx.doi.org/10.1371/journal.pone.0006632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fargier E, Fischer-Le Saux M, Manceau C. 2011. A multilocus sequence analysis of Xanthomonas campestris reveals a complex structure within crucifer-attacking pathovars of this species. Syst. Appl. Microbiol. 34:156–165 [DOI] [PubMed] [Google Scholar]
- 27. He YQ, Zhang L, Jiang BL, Zhang ZC, Xu RQ, Tang DJ, Qin J, Jiang W, Zhang X, Liao J, Cao JR, Zhang SS, Wei ML, Liang XX, Lu GT, Feng JX, Chen B, Cheng J, Tang JL. 2007. Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. campestris. Genome Biol. 8:R218 http://dx.doi.org/10.1186/gb-2007-8-10-r218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun W, Dunning FM, Pfund C, Weingarten R, Bent AF. 2006. Within-species flagellin polymorphism in Xanthomonas campestris pv campestris and its impact on elicitation of Arabidopsis flagellin SENSING2-dependent defenses. Plant Cell 18:764–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White FF, Yang B. 2009. Host and pathogen factors controlling the rice-Xanthomonas oryzae interaction. Plant Physiol. 150:1677–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang W, Jiang BL, Xu RQ, Huang JD, Wei HY, Jiang GF, Cen WJ, Liu J, Ge YY, Li GH, Su LL, Hang XH, Tang DJ, Lu GT, Feng JX, He YQ, Tang JL. 2009. Identification of six type III effector genes with the PIP box in Xanthomonas campestris pv. campestris and five of them contribute individually to full pathogenicity. Mol. Plant Microbe Interact. 22:1401–1411 [DOI] [PubMed] [Google Scholar]
- 31. Noël L, Thieme F, Nennstiel D, Bonas U. 2002. Two novel type III-secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. J. Bacteriol. 184:1340–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Canonne J, Marino D, Noël LD, Arechaga I, Pichereaux C, Rossignol M, Roby D, Rivas S. 2010. Detection and functional characterization of a 215 amino acid N-terminal extension in the Xanthomonas type III effector XopD. PLoS One 5(12):e15773 http://dx.doi.org/10.1371/journal.pone.0015773 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Falconer DS, Mackay TFC. 1996. Introduction to quantitative genetics. Longman, New York, NY. [Google Scholar]
- 34. Feng F, Yang F, Rong W, Wu X, Zhang J, Chen S, He C, Zhou JM. 2012. A Xanthomonas uridine 5′-monophosphate transferase inhibits plant immune kinases. Nature 485:114–118 [DOI] [PubMed] [Google Scholar]
- 35. Bart R, Cohn M, Kassen A, McCallum EJ, Shybut M, Petriello A, Krasileva K, Dahlbeck D, Medina C, Alicai T, Kumar L, Moreira LM, Rodrigues Neto J, Verdier V, Santana MA, Kositcharoenkul N, Vanderschuren H, Gruissem W, Bernal A, Staskawicz BJ. 2012. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc. Natl. Acad. Sci. U. S. A. 109:E1972–E1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Canonne J, Marino D, Jauneau A, Pouzet C, Brière C, Roby D, Rivas S. 2011. The Xanthomonas type III effector XopD targets the Arabidopsis transcription factor MYB30 to suppress plant defense. Plant Cell 23:3498–3511 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37. O’Brien HE, Thakur S, Gong Y, Fung P, Zhang J, Yuan L, Wang PW, Yong C, Scortichini M, Guttman DS. 2012. Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol. 12:141 http://dx.doi.org/10.1186/1471-2180-12-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, van Dijk K, Collmer A. 2000. The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc. Natl. Acad. Sci. U. S. A. 97:4856–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Long AD, Langley CH. 1999. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 9:720–731 [PMC free article] [PubMed] [Google Scholar]
- 40. Guo M, Tian F, Wamboldt Y, Alfano JR. 2009. The majority of the type III effector inventory of Pseudomonas syringae pv. tomato DC3000 can suppress plant immunity. Mol. Plant Microbe Interact. 22:1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szczesny R, Büttner D, Escolar L, Schulze S, Seiferth A, Bonas U. 2010. Suppression of the AvrBs1-specific hypersensitive response by the YopJ effector homolog AvrBsT from Xanthomonas depends on a SNF1-related kinase. New Phytol. 187:1058–1074 [DOI] [PubMed] [Google Scholar]
- 42. Kinch LN, Yarbrough ML, Orth K, Grishin NV. 2009. Fido, a novel AMPylation domain common to fic, doc, and AvrB. PLoS One 4(6):e5818 http://dx/doi.org/10.1371/journal.pone.0005818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rong W, Feng F, Zhou J, He C. 2010. Effector-triggered innate immunity contributes Arabidopsis resistance to Xanthomonas campestris. Mol. Plant Pathol. 11:783–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kvitko BH, Park DH, Velásquez AC, Wei CF, Russell AB, Martin GB, Schneider DJ, Collmer A. 2009. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5(4):e1000388 http://dx.doi.org/10.1371/journal.ppat.1000388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gaudriault S, Malandrin L, Paulin JP, Barny MA. 1997. DspA, an essential pathogenicity factor of Erwinia amylovora showing homology with AvrE of Pseudomonas syringae, is secreted via the Hrp secretion pathway in a DspB-dependent way. Mol. Microbiol. 26:1057–1069 [DOI] [PubMed] [Google Scholar]
- 46. Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, Vasse J, Lauber E, Arlat M. 2007. Plant carbohydrate scavenging through TonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One 2(2):e224 http://dx.doi.org/10.1371/journal.ppat.1000970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meyer D, Lauber E, Roby D, Arlat M, Kroj T. 2005. Optimization of pathogenicity assays to study the Arabidopsis thaliana-Xanthomonas campestris pv. campestris pathosystem. Mol. Plant Pathol. 6:327–333 [DOI] [PubMed] [Google Scholar]
- 48. Bonas U, Schulte R, Fenselau S, Minsavage GV, Staskawicz BJ, Stall RE. 1991. Isolation of a gene cluster from Xanthomonas campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol. Plant Microbe Interact. 4:81–88 [Google Scholar]
- 49. Kado CI, Liu ST. 1981. Rapid procedure for detection and isolation of large and small plasmids. J. Bacteriol. 145:1365–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boucher CA, Van Gijsegem F, Barberis PA, Arlat M, Zischek C. 1987. Pseudomonas solanacearum genes controlling both pathogenicity on tomato and hypersensitivity on tobacco are clustered. J. Bacteriol. 169:5626–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ah-You N, Gagnevin L, Chiroleu F, Jouen E, Neto JR, Pruvost O. 2007. Pathological variations within Xanthomonas campestris pv. mangiferaeindicae support its separation into three distinct pathovars that can be distinguished by amplified fragment length polymorphism. Phytopathology 97:1568–1577 [DOI] [PubMed] [Google Scholar]
- 52. Nei M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY. [Google Scholar]
- 53. Mangin B, Siberchicot A, Nicolas S, Doligez A, This P, Cierco-Ayrolles C. 2012. Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity (Edinb.) 108:285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73 [DOI] [PubMed] [Google Scholar]
- 55. Engler C, Kandzia R, Marillonnet S. 2008. A one pot, one step, precision cloning method with high throughput capability. PLoS One 3(11):e3647 http://dx.doi.org/10.1371/journal.pone.0003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U. S. A. 76:1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ditta G, Stanfield S, Corbin D, Helinski DR. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 77:7347–7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boulanger A, Déjean G, Lautier M, Glories M, Zischek C, Arlat M, Lauber E. 2010. Identification and regulation of the N-acetylglucosamine utilization pathway of the plant pathogenic bacterium Xanthomonas campestris pv. campestris. J. Bacteriol. 192:1487–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dombrecht B, Vanderleyden J, Michiels J. 2001. Stable RK2-derived cloning vectors for the analysis of gene expression and gene function in gram-negative bacteria. Mol. Plant Microbe Interact. 14:426–430 [DOI] [PubMed] [Google Scholar]
- 60. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1996. Current protocols in molecular biology. John Wiley & Sons, New York, NY. [Google Scholar]
- 61. Benjamini Y, Hochberg Y. 2000. The adaptive control of the false discovery rate in multiple hypotheses testing. J. Behav. Educ. Statist. 25:60–83 [Google Scholar]
- 62. Daniels MJ, Barber CE, Turner PC, Sawczyc MK, Byrde RJ, Fielding AH. 1984. Cloning of genes involved in pathogenicity of Xanthomonas campestris pv. campestris using the broad host range cosmid pLAFR1. EMBO J. 3:3323–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relationships between X. campestris pv. campestris strains analyzed by MLSA or based on the diversity of type III secretomes. Download
FliC amino acids 14 to 92 were deduced by partial fliC sequencing in the 45 X. campestris pv. campestris strains and 1 X. campestris pv. raphani strain studied (Table S1). Download
Box plot representation of the pathogenicities of 45 X. campestris pv. campestris strains on Arabidopsis thaliana plants of the Col-0 (A) and Kas (B) ecotypes, which were assayed by piercing of the central vein, and infection symptoms scored 7 days postinoculation. Download
Association study results 7 days after inoculation on plants of the Col-0 (dots) and Kas (diamonds) ecotypes. Download
Pairwise-linkage disequilibrium among all significant markers (n = 73) after emma analysis at 7 dpi with the T3SP PCR markers demarcated from AFLP markers by blue lines (R2 under the diagonal, R2V above the diagonal). Download
Pathogenicity preferred of T3SP mutants of X. campestris pv. campestris strain 8004 10 days postinoculation of Arabidopsis plants of the Kas ecotype. Download
Genotype and characteristics of Xanthomonas campestris strains used in this study.
The emma results on all markers at 7 dpi (see Materials and Methods).
Wilcoxon nonparametric test results on all markers at 7 dpi (see Materials and Methods)
Pairwise linkage disequilibrium measures (R2 and R2V) using the LDcorSV package in R (53) among SNPs present at the xopAC locus.