

ABSTRACT
Helicobacter pylori causes chronic gastritis and avoids elimination by the immune system of the infected host. The commensal bacterium Lactobacillus acidophilus has been suggested to exert beneficial effects as a supplement during H. pylori eradication therapy. In the present study, we applied whole-genome microarray analysis to compare the immune responses induced in murine bone marrow-derived macrophages (BMDMs) stimulated with L. acidophilus, H. pylori, or both bacteria in combination. While L. acidophilus induced a Th1-polarizing response characterized by high expression of interferon beta (IFN-β) and interleukin 12 (IL-12), H. pylori strongly induced the innate cytokines IL-1β and IL-1α. In BMDMs prestimulated with L. acidophilus, H. pylori blocked the expression of L. acidophilus-induced IFN-β and IL-12 and suppressed the expression of key regulators of the Rho, Rac, and Cdc42 GTPases. The inhibition of L. acidophilus-induced IFN-β was independent of H. pylori viability and the virulence factor CagPAI; however, a vacuolating cytotoxin (vacA) mutant was unable to block IFN-β. Confocal microscopy demonstrated that the addition of H. pylori to L. acidophilus-stimulated BMDMs redirects intracellular processing, leading to an accumulation of L. acidophilus in the endosomal and lysosomal compartments. Thus, our findings indicate that H. pylori inhibits the development of a strong Th1-polarizing response in BMDMs stimulated with L. acidophilus by blocking the production of IFN-β in a VacA-dependent manner. We suggest that this abrogation is caused by a redirection of the endocytotic pathway in the processing of L. acidophilus.
IMPORTANCE
Approximately half of the world’s population is infected with Helicobacter pylori. The factors that allow this pathogen to persist in the stomach and cause chronic infections have not yet been fully elucidated. In particular, how H. pylori avoids killing by macrophages, one of the main types of immune cell underlying the epithelium, remains elusive. Here we have shown that the H. pylori virulence factor VacA plays a key role by blocking the activation of innate cytokines induced by the probiotic Lactobacillus acidophilus in macrophages and suppresses the expression of key regulators required for the organization and dynamics of the intracellular cytoskeleton. Our results identify potential targets for the treatment of H. pylori infection and vaccination, since specific inhibition of the toxin VacA possibly allows the activation of an efficient immune response and thereby eradication of H. pylori in the host.
Introduction
The Gram-negative bacterium Helicobacter pylori colonizes the human stomach and has infected more than half of the world’s population (1, 2). Upon infection, H. pylori activates an inflammatory response which leads to the recruitment of macrophages, neutrophils, and lymphocytes to the gastric tissue (3). Macrophages can efficiently engulf H. pylori; however, this pathogenic bacterium has developed mechanisms to avoid killing upon phagocytosis and prevents the induction of an adaptive immune response (4–7). It is likely that the failure of macrophages to eliminate H. pylori contributes to bacterial persistence in the host and thereby to the development of chronic infection. Indeed, we have previously reported the requirement for macrophages in the development of H. pylori-induced gastritis in vivo (8).
It is currently not fully understood how H. pylori avoids elimination by the immune system and is able to survive and persist in human macrophages. Several different mechanisms have been suggested based on the virulence factors expressed by H. pylori (9). H. pylori retards its own uptake by phagocytes, which has been suggested to be dependent on type IV secretion components encoded by the cytotoxin-associated gene pathogenicity island (CagPAI) (10, 11), one of the main pathogenic factors in H. pylori infection (12). Once it is intracellular, H. pylori actively delays actin polymerization and phagosome formation in addition to disrupting membrane trafficking in macrophages (5).
H. pylori has been indicated to utilize several virulence factors facilitating the escape of intracellular degradation in phagocytes. Upon uptake in macrophages, H. pylori initially localizes in phagosomes that coalesce into “megasomes” containing multiple bacteria (5, 13), which has been proposed to cause resistance to intracellular killing (5). A study by Zheng and coworkers suggested that retention of TACO, a tryptophan aspartate-containing coat protein on phagosomes, impairs fusion of phagosomes and lysosomes in macrophages infected with H. pylori (7).
It has been reported that probiotic supplementation to H. pylori eradication therapy increases tolerability and decreases side effects (14–16), but the results are contradictory (17). We have previously shown that the probiotic bacterium Lactobacillus acidophilus NCFM is a strong inducer of the Th1-polarizing cytokines interferon beta (IFN-β) and interleukin 12 (IL-12) in dendritic cells (18–20). In this study, we investigated whether L. acidophilus is able to modulate the inflammatory response induced by H. pylori in bone marrow-derived macrophages (BMDMs). We utilized whole-genome microarray analysis to identify the cellular and molecular mechanisms involved in uptake and processing of the bacteria in addition to the immune response generated. BMDMs were stimulated with either L. acidophilus or H. pylori or with both bacteria in combination. Contrary to our expectations, our results demonstrate that H. pylori is able to block the induction of IFN-β and IL-12 by L. acidophilus. Furthermore, Rgs1/2, Fgd2, and Dock8, key regulators of the Rho, Rac, and Cdc42 GTPases, respectively, required for the organization and dynamics of the actin cytoskeleton, were strongly suppressed when H. pylori was added to BMDMs prestimulated with L. acidophilus. This inhibition was independent of the viability of H. pylori or the virulence factor CagPAI but completely dependent on the VacA toxin. Confocal microscopy revealed that the addition of H. pylori to L. acidophilus-treated BMDMs induces alterations in the endocytic pathway that prevent intracellular processing of L. acidophilus and thereby the induction of IFN-β. Thus, H. pylori is able to prevent the activation of innate cytokines that can modulate the adaptive immune response in macrophages via the virulence factor VacA, which possibly allows persistence and limited eradication in the host.
RESULTS
H. pylori modulates the stimulatory profile induced by L. acidophilus in murine bone marrow-derived macrophages.
To compare the gene expression profiles after stimulation with L. acidophilus and H. pylori, murine bone marrow-derived macrophages (BMDMs) were stimulated either with L. acidophilus NCFM or with an H. pylori clinical isolate, strain 251, or with both bacteria in combination. Since we wanted to ensure that L. acidophilus is able to interact with the macrophages prior to their encounter with the motile pathogen H. pylori, we added H. pylori 251 to BMDMs after 1 h of prestimulation with L. acidophilus NCFM (cell-bacterium ratio, 1:1). After 5 h, the BMDMs were harvested and genome-wide microarray analysis was performed. In total, 4,181 upregulated and 3,956 downregulated genes were common in the three groups of stimulatory conditions (Fig. 1A). In the L. acidophilus-stimulated group, 153 genes were exclusively upregulated and 109 genes exclusively downregulated. Upon stimulation with H. pylori alone, 61 genes were exclusively upregulated and 70 genes exclusively downregulated. With both strains in combination, only 2 genes were exclusively upregulated and 22 genes exclusively downregulated. The two groups of BMDMs stimulated with either L. acidophilus or H. pylori alone had 22 upregulated and 2 downregulated genes in common. In contrast, for L. acidophilus stimulation, there were 70 upregulated and 61 downregulated genes, and for H. pylori, there were 109 upregulated and 153 downregulated genes, in common with the BMDMs that were stimulated with both bacteria.
FIG 1 .

H. pylori modulates the stimulatory profile induced by L. acidophilus in murine bone marrow-derived macrophages. Murine BMDMs from three individual mice were stimulated for 5 h with either L. acidophilus NCFM or H. pylori 251 or prestimulated with L. acidophilus NCFM for 1 h prior to the addition of Helicobacter pylori 251. (A) Venn diagram illustrating pairwise overlap of differentially expressed genes (P < 0.01; fold change greater than ±2) by one-way ANOVA with Benjamini-Hochberg multiple testing correction. (B) Heat map of differentially expressed genes (P < 0.01, fold change greater than ±1.5) expressed at a lower or higher rate than with L. acidophilus treatment. Blue designates genes expressed at a lower level than with L. acidophilus alone, while red indicates greater expression. Data were classified using hierarchical clustering by genes with Euclidean distances and centroid linkage rules.
The top 20 most strongly upregulated genes in BMDMs stimulated with L. acidophilus or H. pylori are listed in Table S1 and Table S2 in the supplemental material, respectively. Analysis of the genes expressed revealed that L. acidophilus strongly induced innate response genes that promote the development of an adaptive immune response, characterized by IL-12 and IFN-β and the chemokines Cxcl10 and Cxcl11, known to be involved in recruitment of activated T cells to the site of infection (Table S1). In contrast, H. pylori induced primarily an innate immune response upregulating the cytokines IL-1β (Il1b) and IL-1α (Il1a) and the neutrophil-recruiting chemokines (Cxcl3, Cxcl1, and Cxcl2) (see Table S2). Notably, H. pylori strongly induced serum amyloid A 3 (Saa3), an acute-phase reactant elevated in chronic low-grade inflammation (21), and Socs3 (suppressor of cytokine signaling 3), a negative regulator of cytokine signaling (22). Figure S1 illustrates a heat map of the genes differentially expressed upon addition of H. pylori, L. acidophilus, or both strains in combination compared to expression in nonstimulated cells (P < 0.01; fold change of greater than ±2). Figure 1B displays the downregulated genes (blue color) and upregulated genes (red color) induced by L. acidophilus, H. pylori, or both strains in combination compared to L. acidophilus alone (P < 0.01; fold change of greater than ±2). Blue designates genes expressed at a lower level than with L. acidophilus alone, while red indicates greater expression.
Table 1 and Fig. 1B present genes that were either strongly induced or suppressed upon the addition of both L. acidophilus and H. pylori to BMDMs relative to results with L. acidophilus alone. Only two genes were strongly induced: P2rx2 (purinergic receptor P2X, ligand-gated ion channel 2) and Pdxp (pyridoxal [pyridoxine, vitamin B6] phosphatase), both regulators of actin cytoskeleton dynamics, were upregulated 7.7-fold and 3.9-fold, respectively. P2rx2 belongs to the family of ligand-gated ion channels that open in response to ATP (23). Activation of P2X receptors induces actin cytoskeleton alterations via Rho activation in macrophages (24, 25). Pdxp is located in the cytosol, colocalizes with the actin cytoskeleton, and acts as a positive regulator of actin filament depolymerization (26). In total, 12 genes were strongly suppressed (Table 1). The gene encoding IFN-β (Ifnb) was the most significantly inhibited (18.5-fold) over 41,000 genes and transcripts analyzed. Interleukin 12b (encoded by Il12b) was likewise significantly suppressed (4.3-fold). Other strongly suppressed genes identified were Rgs2 and Rgs1 (regulator of G-protein signalling 2 and 1) (27), Dock8 (dedicator of cytokinesis 8) (28), and Fgd2 (FYVE, RhoGEF, and PH domain-containing 2) (29), all involved in signaling of Rho, Rac, or Cdc42 GTPase, which are key regulators of the actin cytoskeleton (P < 0.01) (30).
TABLE 1.
Strongest induced/suppressed genes in BMDMs stimulated with L. acidophilus and H. pylori in combination
| Probe name | Annotation | Fold change |
Description | |
|---|---|---|---|---|
| Induced | Suppressed | |||
| A_51_P498526 | P2rx2 | 7.68 | Purinergic receptor P2X, ligand-gated ion channel 2 | |
| A_52_P609120 | Pdxp | 3.90 | Pyridoxal (pyridoxine, vitamin B6) phosphatase | |
| A_51_P144180 | Ifnb | 18.47 | Interferon beta 1 | |
| A_51_P419768 | Rgs2 | 11.13 | Regulator of G-protein signalling 2 | |
| A_51_P478556 | Cited2 | 10.42 | Cbp/p300-interacting transactivator 2 | |
| A_51_P306017 | Dll1 | 8.73 | Delta-like 1 | |
| A_51_P455326 | Sele | 7.97 | Selectin, endothelial cell | |
| A_51_P315904 | Gadd45g | 4.46 | Growth arrest and DNA-damage-inducible 45 gamma | |
| A_51_P260683 | Rgsl | 4.43 | Regulator of G-protein signaling 1 | |
| A_51_P385812 | 1112b | 4.29 | lnterleukin 12b | |
| A_51_P367866 | Egrl | 4.23 | Early growth response 1 | |
| A_51_P149349 | Nfxl1 | 3.72 | Nuclear transcription factor, X-box binding-like 1 | |
| A_51_P484718 | Dock8 | 3.71 | Dedicator of cytokinesis 8 | |
| A_52_P260994 | Fgd2 | 3.66 | FYVE, RhoGEF and PH domain containing 2 | |
H. pylori blocks production of L. acidophilus-induced IFN-β and IL-12 in murine bone marrow-derived macrophages.
Our microanalysis data revealed that genes encoding the cytokines IFN-β and IL-12 were strongly inhibited upon the addition of H. pylori to BMDMs prestimulated with L. acidophilus (Table 1), hence altering the effect of L. acidophilus. To confirm this, BMDMs were stimulated with either L. acidophilus NCFM or H. pylori 251 or pretreated with L. acidophilus NCFM for 1 h prior to the addition of H. pylori 251. The levels of the IFN-β and IL-12 proteins were determined in the supernatants after 10 h of stimulation (Fig. 2A). L. acidophilus induced high expression of IFN-β (840 pg/ml) and IL-12 (10,700 pg/ml), whereas H. pylori 251 induced small amounts of IFN-β and IL-12 (20 pg/ml and 850 pg/ml, respectively). As expected, the addition of H. pylori to BMDMs prestimulated with L. acidophilus significantly suppressed the release of IFN-β by 90% and that of IL-12 by 66% (*, P < 0.01).
FIG 2 .

H. pylori 251 blocks the induction of IFN-β and IL-12 in murine bone marrow-derived macrophages stimulated with L. acidophilus. BMDMs were stimulated with either L. acidophilus NCFM or H. pylori 251 or prestimulated with L. acidophilus NCFM for 1 h prior to addition of Helicobacter pylori 251. (A) IFN-β and IL-12 were measured by ELISA in supernatants after 10 h of stimulation (L. acidophilus NCFM plus H. pylori 251 versus L. acidophilus NCFM alone; *, P < 0.01). Unstim., unstimulated. (B) RNA was extracted after 2, 4, 6, or 8 h of stimulation, cDNA was synthesized, and the expression of Ifn-β and Il-12 was analyzed by RT-PCR (L. acidophilus NCFM plus H. pylori 251 versus L. acidophilus NCFM alone; *, P < 0.01). (C) H. pylori 251 was added at various MOI (1:1, 1:2, 1:4, 1:8, and 1:16, H. pylori/BMDM ratio) to BMDMs prestimulated with L. acidophilus (1 h). RNA was extracted after 4 h, cDNA was synthesized, and the expression of Ifn-β and Il-12 was analyzed by RT-PCR. (D) BMDMs were prestimulated with L. acidophilus (L. acid. pre-incub.) for 1 h, 1.5 h, 2 h, 2.5 h, and 3.5 h prior to addition of H. pylori 251, RNA was extracted after 4 h, cDNA was synthesized, and the expression of Ifn-β and Il-12 was analyzed by RT-PCR (L. acidophilus NCFM plus H. pylori 251 versus L. acidophilus NCFM alone; *, P < 0.01). Data are means and SD for triplicate cultures and are representative of three independent experiments.
To determine whether this inhibition is time dependent, the gene expression of Ifn-β and Il-12 was analyzed by reverse transcription-PCR (RT-PCR) after 2, 4, 6, and 8 h of stimulation (Fig. 2B). After 4 h, the expression of Ifn-β in the L. acidophilus-stimulated BMDMs reached a maximum of 400-fold, whereas H. pylori induced Ifn-β only 15-fold compared to results for controls. However, when H. pylori was added to BMDMs prestimulated with L. acidophilus, the expression of Ifn-β was completely blocked, and the expression of Il-12 was strongly reduced (by 94%) at all time points compared to results for BMDMs stimulated with L. acidophilus alone (*, P < 0.01), indicating that the inhibitory effect is exerted at a very early stage.
In all of our previous experiments, we added both L. acidophilus and H. pylori at a multiplicity of infection (MOI) of 1:1 to the BMDMs. We next sought to investigate the potency of H. pylori to block the expression of Ifn-β and added H. pylori to L. acidophilus-stimulated cells at MOIs of 1:2, 1:4, 1:8, and 1:16 (H. pylori-cell ratio) (Fig. 2C). Even at an MOI as low as 1:8, H. pylori was still able to significantly downregulate the L. acidophilus-induced Ifn-β expression by 51% (*, P < 0.01). Only at an MOI of 1:16 the inhibition of Ifn-β was no longer significant. A similar effect was observed when the expression of Il-12 was measured. Since H. pylori is able to suppress the induction of Ifn-β and Il-12 at a very low MOI, it is indicated that this inhibitory effect is not restricted to the action of an individual bacterium but rather to a virulence determinant secreted by H. pylori.
Next, we aimed to determine the time point at which H. pylori is no longer able to suppress the induction of Ifn-β and Il-12 by L. acidophilus (Fig. 2D). At a preincubation time of 1.5 h with L. acidophilus, H. pylori 251 significantly inhibited the expression of Ifn-β by 92%, at 2 h by 83%, and at 2.5 h by 63%, compared to results for BMDMs stimulated with L. acidophilus alone (*, P < 0.01). However, a downregulation of Ifn-β was no longer evident after 3.5 h of stimulation with L. acidophilus prior to addition of H. pylori 251. A similar but less-pronounced inhibitory effect was observed when the expression of Il-12 was measured, since significant suppression was evident only until the L. acidophilus preincubation time of 2 h prior to addition of H. pylori 251 (*, P < 0.01). Thus, our results suggest that H. pylori-mediated inhibition of Ifn-β and Il-12 responses takes place after endocytosis and that cellular uptake and processing of L. acidophilus may be required for an induction of IFN-β and IL-12.
We then aimed to confirm this inhibitory effect on other cytokines and chemokines using RT-PCR. Expression levels of the T cell- and macrophage-recruiting chemokine gene Cxcl10, as well as those of the proinflammatory cytokine genes Tnf-α and IL-1b, were analyzed after 4 h (see Fig. S2 in the supplemental material). Our results show that the addition of H. pylori 251 to BMDMs preincubated with L. acidophilus is able to significantly downregulate the expression of Cxcl10 and Tnf-α (*, P < 0.01). However, the expression of IL-1β was significantly upregulated by the addition of H. pylori 251 to BMDMs preincubated with L. acidophilus compared to results with L. acidophilus alone (*, P < 0.01), which is in accordance with our microarray results. Thus, we hereby demonstrate that H. pylori is able to inhibit the activation of cytokines that modulate an adaptive immune response but has an additive effect on IL-1β when added to BMDMs prestimulated with L. acidophilus.
Inhibition of IFN-β by H. pylori is independent of viability and the virulence factor CagPAI but dependent on the toxin VacA.
To establish the role of the virulence factor CagPAI in blocking the expression of IFN-β, we added isogenic H. pylori 251 CagPAI mutant bacteria to BMDMs preincubated with L. acidophilus and measured the gene expression of Ifn-β after 4 h (Fig. 3A). The H. pylori 251 CagPAI mutant inhibited Ifn-β expression in BMDMs preincubated with L. acidophilus to a level equivalent to that of the wild-type H. pylori 251 strain, indicating that the inhibitory effect is independent of CagPAI. To determine whether bacterial viability is essential, we added heat-killed (HK) H. pylori 251 and HK 251 CagPAI mutant to BMDMs prestimulated with L. acidophilus and measured the level of IFN-β in the supernatants (Fig. 3B). Both HK H. pylori strains were capable of significantly blocking L. acidophilus-induced IFN-β (*, P < 0.01). Hence, our findings suggest that the inhibitory effect of H. pylori on IFN-β expression in L. acidophilus-stimulated BMDMs is dependent neither on bacterial viability nor on the presence of CagPAI.
FIG 3 .
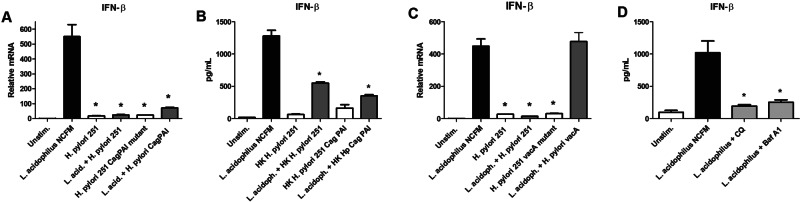
Inhibition of IFN-β by H. pylori is independent of viability and the virulence factor CagPAI but dependent on the toxin VacA. (A) BMDMs were stimulated with L. acidophilus NCFM, H. pylori 251, or the H. pylori 251 CagPAI mutant alone or prestimulated with L. acidophilus NCFM (L. acid.) for 1 h prior to addition of H. pylori strains. RNA was extracted after 4 h, cDNA was synthesized, and the expression of Ifn-β was measured by RT-PCR (L. acidophilus NCFM plus H. pylori 251 or plus the H. pylori 251 CagPAI mutant versus L. acidophilus NCFM alone; *, P < 0.01). (B) BMDMs were prestimulated with L. acidophilus NCFM for 1 h prior to addition of heat-killed (HK) H. pylori 251 and heat-killed H. pylori 251 CagPAI mutant. IFN-β was measured in the supernatants 10 h after stimulation by ELISA (L. acidophilus NCFM [L. acidoph.] plus HK H. pylori 251 or plus the H. pylori 251 CagPAI mutant versus L. acidophilus NCFM alone; *, P < 0.01). (C) BMDMs were prestimulated with L. acidophilus NCFM for 1 h prior to addition of H. pylori 251 or the H. pylori 251 vacA mutant. RNA was extracted after 4 h, cDNA was synthesized, and the expression of Ifn-β was measured by RT-PCR (L. acidophilus NCFM plus the H. pylori 251 vacA mutant versus L. acidophilus NCFM alone; *, P < 0.01). (D) BMDMs were preincubated for 30 min with the endosomal acidification blockers chloroquine (CQ) (final concentration, 20 µM) and bafilomycin A1 (Baf A1) (final concentration, 25 µM) prior to stimulation with L. acidophilus NCFM. IFN-β was measured in the supernatants 10 h after stimulation by ELISA (L. acidophilus NCFM plus inhibitors versus L. acidophilus NCFM alone; *, P < 0.01). Represented are the means ± SEM for triplicate macrophage cultures, which are representative of three independent experiments.
Our microarray analysis revealed that key regulators of the Rho, Rac, and Cdc42 GTPases are strongly suppressed upon stimulation of BMDMs with L. acidophilus and H. pylori in combination. Entry of the H. pylori VacA toxin into epithelial cells is dependent on pinocytosis and controlled by the Cdc42 GTPase (31). Furthermore, in epithelial cells, VacA utilizes the Rac1 GTPase to induce cell vacuolation in host cells (32). We therefore speculated that the H. pylori VacA toxin may also play a role in macrophages and may be implicated in the inhibition of Ifn-β (Fig. 3C). Our hypothesis was confirmed, since an isogenic H. pylori 251 vacA mutant, in contrast to the wild-type strain, was unable to block Ifn-β when added to BMDMs prestimulated with L. acidophilus (P < 0.01). To recapitulate that VacA is essential for the immunosuppression of L. acidophilus-induced IFN-β, we added bacterial-free supernatants (SN) from wild-type H. pylori 251 in various dilutions to BMDMs prestimulated with L. acidophilus (see Fig. S3A in the supplemental material). Our finding was confirmed, since the VacA-containing supernatant from wild-type H. pylori 251 strongly inhibited the IFN-β response induced by L. acidophilus. In contrast, bacterial-free supernatant from the H. pylori 251 vacA mutant was not capable of suppressing IFN-β produced by L. acidophilus-stimulated macrophages (see Fig. S3B).
In epithelial cells, VacA is known to disrupt vesicular trafficking due to formation of anion-selective channels in the membranes of late endocytic compartments (33). We therefore sought to determine whether a similar mechanism is employed in macrophages and whether phagosomal processing of L. acidophilus is necessary for the induction of IFN-β. BMDMs were preincubated for 30 min with the endosomal acidification blockers chloroquine and bafilomycin A1 prior to stimulation with L. acidophilus (Fig. 3D). Viability of the cells was verified via trypan blue staining (data not shown). Both inhibitors completely blocked the release of IFN-β, indicating that phagosomal maturation and processing of L. acidophilus are required for the production of IFN-β (*, P < 0.01).
H. pylori redirects the endocytic pathway of L. acidophilus.
Since our previous experiment revealed that phagosomal processing of L. acidophilus is required to trigger the release of IFN-β in macrophages, we hypothesized that H. pylori redirects the intracellular processing and degradation of L. acidophilus. To determine the intracellular location of H. pylori and L. acidophilus, we applied deconvolution microscopy and analyzed the uptake of the bacteria with the early endosome marker EEA1 (34) and the late endosome and lysosome marker LAMP-1 (35). Since we have shown that H. pylori is able to block the induction of Ifn-β at a very early stage (Fig. 2B), we added L. acidophilus NCFM to BMDMs and analyzed the localization of the bacterium after 2 h. As can be observed in Fig. 4A, a large proportion of intracellular L. acidophilus bacteria did not colocalize with either EEA1 or LAMP-1 compartments. Indeed, we determined that after 2 h of incubation, approximately only 7% and 30% of intracellular L. acidophilus NCFM were contained within endosomal and lysosomal compartments, respectively (Fig. 4C). The remainder of bacteria were not associated with either marker (*, P < 0.05; **, P < 0.01). In contrast, we detected the formation of megasomes and a significant increase in L. acidophilus colocalized in the endosomal and lysosomal compartments (19% and 50%, respectively, after 2 h incubation) when H. pylori was added, compared to results for BMDMs that were stimulated with L. acidophilus alone (Fig. 4B and C). Figure 4D presents a magnified image of L. acidophilus localized in lysosomes (white arrow) in either the presence or absence of H. pylori. Our results indicate a greater release of L. acidophilus from the phagocytic compartments in the absence of H. pylori. We suggest that the addition of H. pylori leads to an accumulation of L. acidophilus in the endosomal and lysosomal compartments, thereby preventing the induction of IFN-β by L. acidophilus.
FIG 4 .

H. pylori redirects the endocytotic pathway of L. acidophilus. Phagocytic uptake of bacteria into early endosomes and lysosomes is shown. (A) BMDMs were exposed to red-labeled L. acidophilus NCFM for 2 h. Cells were either labeled with an EEA1 antibody for early endosomes or transfected with Lamp1-YFP for lysosomes. (B) As per panel A, with the addition of labeled or unlabeled H. pylori 251 (magenta). Cells were prelabeled with Lysotracker DND-26 to label lysosomes, incubated for 2 h, fixed, permeabilized, and stained with the EEA-1 antibody or anti-H. pylori serum for unlabeled H. pylori (HP). (C) Percentage of the total L. acidophilus NCFM located in the endosomal compartment or lysosomal compartment after 1 h and 2 h of incubation (with or without H. pylori 251). SEM are shown (*, P < 0.05; **, P < 0.01). (D) Magnified image of L. acidophilus NCFM (red) localized in lysosomes labeled with Lyso-DND26 (green) (2-h time point). Yellow indicates colocalization (white arrow) of signal or L. acidophilus NCFM located within the lysosomes. Data are representative of three independent experiments.
DISCUSSION
In the present work, we aimed to investigate whether L. acidophilus is able to modulate the inflammatory response induced by H. pylori in bone marrow-derived macrophages (BMDMs) by examining the cellular and molecular mechanisms involved in uptake and processing of these organisms. Whole-genome microarray analysis revealed that while L. acidophilus induces T-cell recruiting chemokines (Cxcl10 and Cxcl11), proinflammatory (IL-1β and IL-6), and Th1-skewing cytokines (IFN-β and IL-12), H. pylori predominantly induced cytokines indicative of innate immune responses (IL-1β and IL-1α) and neutrophil-recruiting chemokines (Cxcl1, Cxcl2, and Cxcl3). Most importantly, microarray analysis revealed that when H. pylori is added to BMDMs prestimulated with L. acidophilus, Th1-skewing cytokines induced by L. acidophilus are strongly inhibited. Real-time PCR confirmed that H. pylori blocks the gene expression of IFN-β, IL-12, Cxcl10, and TNF-α, while it has an additive effect on the cytokine IL-1β. Thus, our results show that H. pylori prevents the production of factors induced by L. acidophilus in macrophages that contribute to initiating the adaptive immune response.
Interestingly, our microarray analysis also revealed that addition of H. pylori to macrophages prestimulated with L. acidophilus had a major impact on regulators of cytoskeletal arrangements and actin filament depolarization, since the genes encoding Pdrx2 and Pdxp were strongly induced upon incubation with both bacteria. In contrast, the genes Rgs1/2, Fgd2, and Dock8, regulators of the Rho, Rac, and Cdc42 GTPases, which are molecular switches controlling the organization and dynamics of the actin cytoskeleton, were strongly suppressed. It is well established that many bacterial pathogens regulate host trafficking pathways by the selective inclusion or retention of GTPases in host cells during infection and use these important cellular regulators as part of their overall virulence strategy (36, 37). Thus, our data suggest that H. pylori modifies GTPases in macrophages to promote bacterial persistence in the host.
Expression of constitutively active Rac1, a member of the Rho family of small GTPases, potentiates the activity of the H. pylori VacA toxin in two different epithelial cell lines (SCC12F keratinocytes and MDCK cells) (32). Furthermore, pinocytosis of VacA is regulated by the Cdc42 GTPase in HeLa and gastric AGS cells (31). Hence, we postulated that the same mechanism may be utilized in macrophages and that the H. pylori VacA toxin is involved in the immune-inhibitory effect observed. Indeed, the addition of H. pylori vacA mutant and VacA-containing supernatant from wild-type H. pylori 251 to L. acidophilus-stimulated BMDMs showed that VacA is essential for the inhibition of IFN-β responses.
Upon internalization in macrophages, H. pylori induces the fusion of the individual phagosomes, resembling giant multivesicular bodies (13), an effect that we observed in our microscopy (Fig. 4B). Since VacA leads to cell vacuolation in the cell membranes of the late endosomal compartments in epithelial cells (33), we used the endosomal acidification blockers chloroquine and bafilomycin A1 to determine whether phagosomal maturation and processing of L. acidophilus is required. Both inhibitors completely abrogated the expression of IFN-β in BMDMs stimulated with L. acidophilus, indicating that both phagocytic uptake and phagolysosomal maturation is a prerequisite for IFN-β signaling in response to this bacterium. Our microscopy data showed that H. pylori changes the endosomal transport of L. acidophilus from the endosomal and lysosomal compartments in BMDMs, suggesting that a specific endocytotic pathway is a key step for the induction of IFN-β.
The immunosuppressive ability of H. pylori is further corroborated by our microarray data. We identified that Socs3, a negative regulator of cytokine signaling, was among the most strongly expressed genes in macrophages stimulated with H. pylori (see Table S2 in the supplemental material). Socs3 is upregulated by several viruses, e.g., herpes simplex virus 1 (HSV-1) (38), hepatitis virus C (HCV) (39), influenza A virus (40), human immunodeficiency virus type 1 (HIV-1) (41), severe acute respiratory syndrome (SARS) virus (42), and respiratory syncytial virus (RSV) (43, 44), resulting in the inhibition of the type I interferon signaling pathway. Hence, the expression of Socs3 in macrophages stimulated with H. pylori might be an additional mechanism utilized by this pathogen to suppress cytokine signaling and thereby the activation of adaptive immunity.
We therefore suggest that the H. pylori VacA toxin causes a redirection of the endocytic pathway in macrophages that prevents the activation of adaptive immunity, exemplified by the Th1-skewing commensal L. acidophilus in our present study. Collectively, this suppression will limit the effectiveness of L. acidophilus NCFM as a probiotic during treatment of H. pylori-induced inflammation, symptoms, or eradication from the host. Although additional investigations are required to determine how L. acidophilus may encounter macrophages in vivo, it is plausible that this occurs during inflammatory states. Identification of the molecular mechanisms underlying VacA action will lead to a better understanding of the pathogenesis of H. pylori-associated diseases, as they are possible targets for the treatment of H. pylori infection and for vaccination.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and preparation.
The Gram-positive lactic acid bacterium Lactobacillus acidophilus NCFM (Danisco, Copenhagen, Denmark) was grown anaerobically overnight at 37°C in de Man Rogosa Sharp (MRS) broth (Merck, Darmstadt, Germany). The Gram-negative bacterium Helicobacter pylori was grown on solid agar and liquid broth as previously described (45). The following strains and mutants were used: the clinical isolate Helicobacter pylori 251 (46) and the Helicobacter pylori 251 CagPAI deletion mutant (45). The Helicobacter pylori 251 VacA (HP0887) mutant was constructed in this study by natural transformation using the plasmid pCTB8::Km (47), a kind gift from Tim Cover (Vanderbilt University, Nashville, TN). Confirmation of allelic exchange of VacA with vacA::Km in the 251 vacA mutant was performed by PCR. Heat killing of H. pylori was performed by incubating H. pylori at 60°C for 20 min and confirming the bacteria were no longer viable by testing their growth on solid horse blood agar as previously described (45).
Generation of murine macrophages from bone marrow progenitor cells.
C57BL/6 mice were bred and housed under specific-pathogen-free (SPF) conditions within the animal facility of Monash Medical Centre. Mice were free of all Helicobacter species. All animal experiments were approved by and performed in accordance with the Monash Medical Centre Animal Ethics Committee. Female mice were used in these studies because they have been reported to be more susceptible to Helicobacter-induced pathology than male mice (48).
For the generation of murine bone marrow-derived macrophages (BMDMs), bone marrow from female 6- to 10-week-old C57BL/6 mice was flushed out from the femur and tibia and washed in phosphate-buffered saline (PBS). The cells were resuspended in RPMI 1640 medium (Gibco, Life Technologies, Singapore) containing 10% (vol/vol) heat-inactivated fetal calf serum (FCS) (Thermo Electron, VIC, Australia) supplemented with 1% penicillin-streptomycin (Gibco), 1% GlutaMAX (Gibco), and 20% of growth supernatant of L929 cells. The cells were seeded at a concentration of 7 × 106 cells in 10-cm2 nontreated cell culture plates (Nunc, Roskilde, Denmark) in 10 ml of conditioned medium and incubated at 37°C in 5% CO2. On day 4, 10 ml of complete medium containing 20% L929 cell-conditioned medium was added. On day 6, cells were harvested in conditioned medium, and 1 × 106 cells were seeded in 24-well tissue culture plates (Nunc, Roskilde, Denmark). The next day, the medium was removed, and 400 µl of fresh RPMI 1640 medium (without antibiotics and FCS) was added.
Stimulation of murine bone marrow-derived macrophages with bacteria or bacterial-free supernatants.
Overnight cultures of L. acidophilus and H. pylori were harvested by centrifugation at 1,250 × g for 10 min at 4°C and washed twice in sterile PBS. Bacteria were resuspended in RPMI 1640 medium (without FCS and antibiotics) and added (100 µl/well) to the BMDMs at an MOI of 1:1 (or medium, for a final volume of 500 µl/well). Bacterial-free supernatants (SN) from H. pylori 251 and the H. pylori 251 vacA mutant were prepared as previously reported (49). In brief, H. pylori grown for 24 h on agar plates was collected in 1 ml of sterile distilled water, vortexed for 30 s, and incubated at room temperature for 30 min. Bacteria were removed by centrifugation, and the supernatant was filter sterilized using a 0.2−µm filter. The VacA toxin contained within the water extracts was activated using hydrochloric acid, and the pH of the supernatant was subsequently neutralized prior to addition to BMDMs as previously described (50). Supernatants were added to BMDMs at various dilutions to a final volume of 500 µl/well. The cell cultures were incubated at 37°C in 5% CO2. In the inhibitor experiments, BMDMs were preincubated for 30 min with the endosomal acidification blockers chloroquine (final concentration, 20 µM) and bafilomycin A1 (final concentration, 25 µM) prior to stimulation with L. acidophilus. Both inhibitors were purchased from InvivoGen (San Diego, CA).
Microarray analysis.
Immature murine BMDMs from three female 6-week-old C57BL/6 mice were incubated with L. acidophilus NCFM, H. pylori 251, or H. pylori 251 added to BMDMs prestimulated with L. acidophilus NCFM for 1 h. The BMDMs were harvested 5 h after addition of H. pylori, and RNA was extracted using a Qiagen RNeasy minicolumn with on-column DNase digestion. For each sample, 200 ng of RNA was Cy3 labeled with a one-color low-RNA-input quick amplification kit (Agilent). The resulting cRNA was fragmented and hybridized to Agilent whole-mouse-genome expression microarrays (G4122F), and the slides were washed according to the manufacturer’s instructions. Slides were scanned immediately after washing using an Agilent DNA microarray scanner (G2505B). Feature extraction was performed with Agilent Feature Extraction 9.5 software using default parameters excluding nonuniform flagged outliers. Percentile normalization and differential gene expression analysis were performed using the program GeneSpring GX 11 (Agilent Technologies).
RNA extraction and quantitative real-time PCR analysis.
Murine BMDMs were harvested and homogenized by using a QIAshredder instrument (Qiagen, Australia), and RNA was extracted using the RNeasy Plus minikit (Qiagen) according to the manufacturer’s instructions. The RNA concentration was determined with a NanoDrop instrument (Thermo, Wilmington), and 500 ng of total RNA was reverse transcribed with the TaqMan reverse transcription reagent kit (Applied Biosystems, Foster City, CA) using random hexamer primers according to the manufacturer’s instructions. The obtained cDNA was stored in aliquots at −80°C. The following 6-carboxyfluorescein (FAM)-labeled TaqMan gene expression assays were purchased (Applied Biosystems): Cxcl10 (assay identifier [ID] Mm99999072_m1), IFN-β (assay ID Mm00439546_s1), IL-12b (p40) (assay ID Mm00434174_m1), TNF-α (assay ID Mm00443258_m1), IL-1β (assay, ID Mm00434228_m1), and 18S (assay, ID 4319413E). The amplifications were prepared in a total volume of 10 µl containing 1× TaqMan universal PCR master mix (Applied Biosystems) and diluted cDNA. All PCR reactions were carried out in triplicate in MicroAmp optical 384-well reaction plates (Applied Biosystems). The cycling parameters were initiated by 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C, and 60°C for 1 min using the 7900HT fast real-time PCR system (Applied Biosystems). The amplifications were normalized to the expression of 18S. Relative transcript levels were calculated, applying the 2−ΔΔCT method.
Cytokine quantification by ELISA.
Protein production of IFN-β was quantified by enzyme-linked immunosorbent assay (ELISA) in the cell culture supernatants using rat anti-mouse IFN-β antibody (7F-D3; Abcam, USA) followed by rabbit anti-mouse IFN-β antibody (PBL Biomedicals, USA) for detection. Recombinant IFN-β was used as a standard (made in-house). IL-12p40 (OptEIA kit; BD, USA) was analyzed in the cell culture supernatants as per the manufacturer’s instructions.
Microscopy.
Immature murine BMDMs were seeded at a density 2 × 105 cells on 12-mm coverslips (Menzelglass, Germany) in 24-well tissue culture plates (Nunc) in a total volume of 500 µl. LAMP-1 yellow fluorescent protein (YFP) (concentration, 100 ng/1 × 105 cells; Rohan Teasdale, Institute for Molecular Bioscience, University of Queensland) was transfected into half of the wells containing BMDMs using the jetPEI macrophage in vitro DNA transfection kit (Polyplus Transfection SA, France), according to the manufacturer’s instructions. The next day, medium was removed, and 200 µl of fresh RPMI 1640 medium (without antibiotics and FCS) was added. L. acidophilus NCFM was labeled red with the dye Dil (Vybrant cell-labeling solutions; Molecular Probes, Life Technologies, USA), and H. pylori 251 was labeled green with the dye DiO (Vybrant cell-labeling solutions) according to the manufacturer’s protocol. The bacteria (or medium) were added (100 µl/well) to the BMDMs at an MOI of 1:1 (total volume, 400 µl). To determine the uptake of the bacteria in the early endosomes, Dil-labeled L. acidophilus NCFM and DiO-labeled H. pylori 251 were added to the BMDMs. The medium was removed, the cells were rinsed with PBS, fixed in 20% Formalin for 20 min, and stored at 4°C in PBS. For antibody labeling, cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min, blocked in 5% bovine serum albumin (BSA)-PBS for 30 min, and labeled with the early endosome marker EEA1 antibody (Abcam, United States). For lysosomal studies with coinfection, Dil-labeled L. acidophilus NCFM and unlabeled H. pylori 251 were added to the BMDMs prelabeled with LysoTracker green DND-26 (Molecular Probes, Life Technologies, USA) according to the manufacturer’s instructions. Antibody labeling with purified anti-H. pylori serum (in-house) was performed as described previously (45). Cells were visualized by both confocal laser microscopy (Leica SP5) and wide-field deconvolution microscopy (Deltavision core system; Applied Precision, USA). Images are presented as a two-dimensional (2D) maximum-intensity projection or as single-plane images.
Statistical analysis.
The GraphPad Prism software program, version 4.03 (GraphPad Software, San Diego, CA), was used, and one-way analysis of variance (ANOVA) and Bonferroni tests were applied (P < 0.01). The microarray data were analyzed applying one-way ANOVA with Benjamini-Hochberg multiple testing correction (P < 0.01). Colocalization studies were performed using the built-in feature in the Bitplane Imaris 7.5 software program with a threshold of background plus 2 standard deviations (SDs). Colocalization data were accumulated using the GraphPad Prism program as mentioned previously, and an unpaired t test was used to determine significance (*, P < 0.05; **, P < 0.01).
Microarray data accession number.
The data generated in this work have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE42622.
SUPPLEMENTAL MATERIAL
Top 20 induced genes by L. acidophilus NCFM in murine BMDMs.
Top 20 induced genes by H. pylori 251 in murine BMDMs.
Heat map of BMDMs stimulated with L. acidophilus and H. pylori. Murine BMDMs from three individual mice were stimulated for 5 h with either L. acidophilus NCFM or H. pylori 251 or prestimulated with L. acidophilus NCFM for 1 h prior to the addition of Helicobacter pylori 251. Heat map of differentially expressed genes (P < 0.01; fold change greater than ±2) clustered hierarchically on expression using Euclidean distance and centroid linkage. Replicates are shown as columns, with blue and red representing 4-fold change down or up, respectively, compared to results for the untreated condition. Download
Addition of H. pylori to L. acidophilus-stimulated BMDMs downregulates the expression of Cxcl10 and Tnf-α but upregulates IL-1β. BMDMs were stimulated with either L. acidophilus NCFM or H. pylori 251 alone or with L. acidophilus NCFM for 1 h prior to addition of H. pylori 251. RNA was extracted after 4 h, cDNA was synthesized, and the expression of Cxcl10, Tnf-α and IL-1b was measured by RT-PCR (L. acidophilus NCFM plus H. pylori 251 versus L. acidophilus NCFM alone; *, P < 0.01). Data are means and SEM for triplicate cultures and are representative of three independent experiments. Download
Addition of VacA-containing supernatant abrogates induction of IFN-β. (A) BMDMs were stimulated with L. acidophilus NCFM, H. pylori 251, or supernatant (SN) from H. pylori 251 (1:10, 1:20, and 1:30 diluted) alone or with L. acidophilus NCFM for 1 h prior to addition of H. pylori 251 or supernatant from H. pylori 251 (1:10, 1:20, and 1:30 diluted). IFN-β was measured after 10 h of stimulation in the supernatants by ELISA (L. acidophilus NCFM plus H. pylori 251/H. pylori supernatant [SN] versus L. acidophilus NCFM alone; *, P < 0.01). (B) BMDMs were stimulated with L. acidophilus NCFM, the H. pylori 251 vacA mutant, or supernatant (SN) from the H. pylori 251 vacA mutant (1:10, 1:20, and 1:30 diluted) alone or with L. acidophilus NCFM for 1 h prior to addition of the H. pylori 251 vacA mutant or supernatant from the H. pylori 251 vacA mutant (1:10, 1:20, and 1:30 diluted). IFN-β was measured after 10 h of stimulation in the supernatants by ELISA (L. acidophilus NCFM plus the H. pylori 251 vacA mutant/H. pylori 251 vacA mutant supernatant [SN] versus L. acidophilus NCFM alone; *, P < 0.01). Represented are the means ± SEM for triplicate macrophage cultures; macrophages were pooled from 4 mice. Download
ACKNOWLEDGMENTS
Gudrun Weiss was supported by a postdoctoral fellowship and a travel grant to visit Australia from the University of Copenhagen, the Augustinus Foundation, the A.P. Møllers and Wife Chastine Mc-Kinney Møllers Foundation for Medical Science Promotion, the Christian and Ottilia Brorsons Travel Foundation for Young Scientists, the Sophus Jacobsen and Wife Astrid Jacobsens Foundation, the Oticon Foundation, and the Torben and Alice Frimodts Foundation. This work was also supported by funding from the Australian Research Council (to M.K.-L.), Victorian Life Sciences Computation Initiative, Monash e-Research, the National Health & Medical Research Council (NH&MRC) of Australia (to R.L.F. and P.H.), and the Victorian Government’s Operational Infrastructure Support Program. R.L.F. and P.H. are senior and professorial research fellows, respectively, of the NH&MRC.
We thank Jodee Gould for invaluable technical assistance.
Footnotes
Citation Weiss G, Forster S, Irving A, Tate M, Ferrero RL, Hertzog P, Frøkiær H, Kaparakis-Liaskos M. 2013. Helicobacter pylori VacA suppresses Lactobacillus acidophilus-induced interferon beta signaling in macrophages via alterations in the endocytic pathway. mBio 4(3):e00609-12. doi:10.1128/mBio.00609-12.
REFERENCES
- 1. Blaser MJ, Atherton JC. 2004. Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113:321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dunn BE, Cohen H, Blaser MJ. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Telford JL, Covacci A, Rappuoli R, Chiara P. 1997. Immunobiology of Helicobacter pylori infection. Curr. Opin. Immunol. 9:498–503 [DOI] [PubMed] [Google Scholar]
- 4. Algood HM, Cover TL. 2006. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin. Microbiol. Rev. 19:597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allen LA, Schlesinger LS, Kang B. 2000. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J. Exp. Med. 191:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Allen LA. 2007. Phagocytosis and persistence of Helicobacter pylori. Cell. Microbiol. 9:817–828 [DOI] [PubMed] [Google Scholar]
- 7. Zheng PY, Jones NL. 2003. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell. Microbiol. 5:25–40 [DOI] [PubMed] [Google Scholar]
- 8. Kaparakis M, Walduck AK, Price JD, Pedersen JS, van Rooijen N, Pearse MJ, Wijburg OL, Strugnell RA. 2008. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect. Immun. 76:2235–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Backert S, Clyne M. 2011. Pathogenesis of Helicobacter pylori infection. Helicobacter 16:19–25 [DOI] [PubMed] [Google Scholar]
- 10. Ramarao N, Gray-Owen SD, Backert S, Meyer TF. 2000. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol. Microbiol. 37:1389–1404 [DOI] [PubMed] [Google Scholar]
- 11. Ramarao N, Meyer TF. 2001. Helicobacter pylori resists phagocytosis by macrophages: quantitative assessment by confocal microscopy and fluorescence-activated cell sorting. Infect. Immun. 69:2604–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cendron L, Zanotti G. 2011. Structural and functional aspects of unique type IV secretory components in the Helicobacter pylori cag-pathogenicity island. FEBS J. 278:1223–1231 [DOI] [PubMed] [Google Scholar]
- 13. Rittig MG, Shaw B, Letley DP, Thomas RJ, Argent RH, Atherton JC. 2003. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vacA and cag status. Cell. Microbiol. 5:887–899 [DOI] [PubMed] [Google Scholar]
- 14. Lesbros-Pantoflickova D, Corthésy-Theulaz I, Blum AL. 2007. Helicobacter pylori and probiotics. J. Nutr. 137:812S–818S [DOI] [PubMed] [Google Scholar]
- 15. Lionetti E, Miniello VL, Castellaneta SP, Magistá AM, De Canio A, Maurogiovanni G, Ierardi E, Cavallo L, Francavilla R. 2006. Lactobacillus reuteri therapy to reduce side-effects during anti-Helicobacter pylori treatment in children: a randomized placebo controlled trial. Aliment. Pharmacol. Ther. 24:1461–1468 [DOI] [PubMed] [Google Scholar]
- 16. Miki K, Urita Y, Ishikawa F, Iino T, Shibahara-Sone H, Akahoshi R, Mizusawa S, Nose A, Nozaki D, Hirano K, Nonaka C, Yokokura T. 2007. Effect of Bifidobacterium bifidum fermented milk on Helicobacter pylori and serum pepsinogen levels in humans. J. Dairy Sci. 90:2630–2640 [DOI] [PubMed] [Google Scholar]
- 17. Medeiros J, Gonçalves FO, Boyanova L, Pereira M, de Carvalho JN, Pereira AM, Cabrita AM. 2011. Evaluation of Helicobacter pylori eradication by triple therapy plus Lactobacillus acidophilus compared to triple therapy alone. Eur. J. Clin. Microbiol. Infect. Dis. 30:555–559 [DOI] [PubMed] [Google Scholar]
- 18. Weiss G, Christensen HR, Zeuthen LH, Vogensen FK, Jakobsen M, Frokiaer H. 2011. Lactobacilli and bifidobacteria induce differential interferon-beta profiles in dendritic cells. Cytokine 56:520–530 [DOI] [PubMed] [Google Scholar]
- 19. Weiss G, Maaetoft-Udsen K, Stifter SA, Hertzog P, Goriely S, Thomsen AR, Paludan SR, Frokiaer H. 2012. MyD88 drives the IFN-beta response to Lactobacillus acidophilus in dendritic cells through a mechanism involving IRF1, IRF3, and IRF7. J. Immunol. 5:2860–2866 [DOI] [PubMed] [Google Scholar]
- 20. Weiss G, Rasmussen S, Zeuthen LH, Nielsen BN, Jarmer H, Jespersen L, Frøkiaer H. 2010. Lactobacillus acidophilus induces virus immune defence genes in murine dendritic cells by a toll-like receptor-2-dependent mechanism. Immunology 131:268–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boylan MT, Crockard AD, Duddy ME, Armstrong MA, McMillan SA, Hawkins SA. 2001. Interferon-beta1a administration results in a transient increase of serum amyloid A protein and C-reactive protein: comparison with other markers of inflammation. Immunol. Lett. 75:191–197 [DOI] [PubMed] [Google Scholar]
- 22. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA. 2012. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36:239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brake AJ, Wagenbach MJ, Julius D. 1994. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature 371:519–523 [DOI] [PubMed] [Google Scholar]
- 24. Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ. 2004. The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J. Leukoc. Biol. 75:1173–1182 [DOI] [PubMed] [Google Scholar]
- 25. Verhoef PA, Estacion M, Schilling W, Dubyak GR. 2003. P2X7 Receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1β release. J. Immunol. 170:5728–5738 [DOI] [PubMed] [Google Scholar]
- 26. Huang TY, DerMardirossian C, Bokoch GM. 2006. Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 18:26–31 [DOI] [PubMed] [Google Scholar]
- 27. Cotton M, Claing A. 2009. G protein-coupled receptors stimulation and the control of cell migration. Cell. Signal. 21:1045–1053 [DOI] [PubMed] [Google Scholar]
- 28. Ruusala A, Aspenström P. 2004. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. 572:159–166 [DOI] [PubMed] [Google Scholar]
- 29. Huber C, Mårtensson A, Bokoch GM, Nemazee D, Gavin AL. 2008. FGD2, a CDC42-specific exchange factor expressed by antigen-presenting cells, localizes to early endosomes and active membrane ruffles. J. Biol. Chem. 283:34002–34012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9:690–701 [DOI] [PubMed] [Google Scholar]
- 31. Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. 2005. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol. Biol. Cell 16:4852–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hotchin NA, Cover TL, Akhtar N. 2000. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J. Biol. Chem. 275:14009–14012 [DOI] [PubMed] [Google Scholar]
- 33. Cover TL, Blanke SR. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320–332 [DOI] [PubMed] [Google Scholar]
- 34. Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo JP, Tock EP, Toh BH. 1995. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine "fingers" and contains a calmodulin-binding IQ motif. J. Biol. Chem. 270:13503–13511 [DOI] [PubMed] [Google Scholar]
- 35. Chen JW, Cha Y, Yuksel KU, Gracy RW, August JT. 1988. Isolation and sequencing of a cDNA clone encoding lysosomal membrane glycoprotein mouse LAMP-1. Sequence similarity to proteins bearing onco-differentiation antigens. J. Biol. Chem. 263:8754–8758 [PubMed] [Google Scholar]
- 36. Aktories K. 2011. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 9:487–498 [DOI] [PubMed] [Google Scholar]
- 37. Stein MP, Muller MP, Wandinger-Ness A. 2012. Bacterial pathogens commandeer Rab GTPases to establish intracellular niches. Traffic 13:1565–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Miura S, Jimbow K, Fujii N. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282–6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bode JG, Ludwig S, Ehrhardt C, Erhardt A, Albrecht U, Schaper F, Heinrich PC, Häussinger D. 2003. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 17:488–490 [DOI] [PubMed] [Google Scholar]
- 40. Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, Ludwig S. 2008. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 4:e1000196 http://dx.doi.org/10.1371/journal.ppat.1000196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clements JE, Benveniste EN. 2010. Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J. Immunol. 185:2393–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okabayashi T, Kariwa H, Yokota S, Iki S, Indoh T, Yokosawa N, Takashima I, Tsutsumi H, Fujii N. 2006. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 78:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oshansky CM, Krunkosky TM, Barber J, Jones LP, Tripp RA. 2009. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a Toll-like receptor pathway. Viral Immunol. 22:147–161 [DOI] [PubMed] [Google Scholar]
- 44. Zhao DC, Yan T, Li L, You S, Zhang C. 2007. Respiratory syncytial virus inhibits interferon-alpha-inducible signaling in macrophage-like U937 cells. J. Infect. 54:393–398 [DOI] [PubMed] [Google Scholar]
- 45. Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, Le Bourhis L, Karrar A, Viala J, Mak J, Hutton ML, Davies JK, Crack PJ, Hertzog PJ, Philpott DJ, Girardin SE, Whitchurch CB, Ferrero RL. 2010. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell. Microbiol. 12:372–385 [DOI] [PubMed] [Google Scholar]
- 46. Philpott DJ, Belaid D, Troubadour P, Thiberge JM, Tankovic J, Labigne A, Ferrero RL. 2002. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell. Microbiol. 4:285–296 [DOI] [PubMed] [Google Scholar]
- 47. Cover TL, Tummuru MK, Cao P, Thompson SA, Blaser MJ. 1994. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 269:10566–10573 [PubMed] [Google Scholar]
- 48. Court M, Robinson PA, Dixon MF, Jeremy AH, Crabtree JE. 2003. The effect of gender on Helicobacter felis-mediated gastritis, epithelial cell proliferation, and apoptosis in the mouse model. J. Pathol. 201:303–311 [DOI] [PubMed] [Google Scholar]
- 49. Letley DP, Rhead JL, Twells RJ, Dove B, Atherton JC. 2003. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J. Biol. Chem. 278:26734–26741 [DOI] [PubMed] [Google Scholar]
- 50. Raju D, Jones NL. 2010. Methods to monitor autophagy in H. pylori vacuolating cytotoxin A (VacA)-treated cells. Autophagy 6:138–143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Top 20 induced genes by L. acidophilus NCFM in murine BMDMs.
Top 20 induced genes by H. pylori 251 in murine BMDMs.
Heat map of BMDMs stimulated with L. acidophilus and H. pylori. Murine BMDMs from three individual mice were stimulated for 5 h with either L. acidophilus NCFM or H. pylori 251 or prestimulated with L. acidophilus NCFM for 1 h prior to the addition of Helicobacter pylori 251. Heat map of differentially expressed genes (P < 0.01; fold change greater than ±2) clustered hierarchically on expression using Euclidean distance and centroid linkage. Replicates are shown as columns, with blue and red representing 4-fold change down or up, respectively, compared to results for the untreated condition. Download
Addition of H. pylori to L. acidophilus-stimulated BMDMs downregulates the expression of Cxcl10 and Tnf-α but upregulates IL-1β. BMDMs were stimulated with either L. acidophilus NCFM or H. pylori 251 alone or with L. acidophilus NCFM for 1 h prior to addition of H. pylori 251. RNA was extracted after 4 h, cDNA was synthesized, and the expression of Cxcl10, Tnf-α and IL-1b was measured by RT-PCR (L. acidophilus NCFM plus H. pylori 251 versus L. acidophilus NCFM alone; *, P < 0.01). Data are means and SEM for triplicate cultures and are representative of three independent experiments. Download
Addition of VacA-containing supernatant abrogates induction of IFN-β. (A) BMDMs were stimulated with L. acidophilus NCFM, H. pylori 251, or supernatant (SN) from H. pylori 251 (1:10, 1:20, and 1:30 diluted) alone or with L. acidophilus NCFM for 1 h prior to addition of H. pylori 251 or supernatant from H. pylori 251 (1:10, 1:20, and 1:30 diluted). IFN-β was measured after 10 h of stimulation in the supernatants by ELISA (L. acidophilus NCFM plus H. pylori 251/H. pylori supernatant [SN] versus L. acidophilus NCFM alone; *, P < 0.01). (B) BMDMs were stimulated with L. acidophilus NCFM, the H. pylori 251 vacA mutant, or supernatant (SN) from the H. pylori 251 vacA mutant (1:10, 1:20, and 1:30 diluted) alone or with L. acidophilus NCFM for 1 h prior to addition of the H. pylori 251 vacA mutant or supernatant from the H. pylori 251 vacA mutant (1:10, 1:20, and 1:30 diluted). IFN-β was measured after 10 h of stimulation in the supernatants by ELISA (L. acidophilus NCFM plus the H. pylori 251 vacA mutant/H. pylori 251 vacA mutant supernatant [SN] versus L. acidophilus NCFM alone; *, P < 0.01). Represented are the means ± SEM for triplicate macrophage cultures; macrophages were pooled from 4 mice. Download