

Abstract
Osterix (Osx, Sp7) is a zinc-finger transcription factor belonging to the specificity protein (Sp) family expressed in cells of the osteoblast lineage in the developing skeleton where it regulates expression of a number of osteoblastic genes. We previously reported inhibition of osterix mRNA and protein by parathyroid hormone (PTH) stimulation of cAMP in osteoblasts. We here show that Osx expression in osteoblasts is regulated by Sp proteins as demonstrated by mithramycin A inhibition of Osx mRNA and OSX protein levels. Mutation of putative transcription factor binding sites within the Osx promoter demonstrated a tandem repeat sequence that selectively binds OSX but not other Sp factors expressed in osteoblasts (Sp1, Sp3, or Tieg (Klf10)). Mutation of either or both the repeat sequences inhibited 90% of the promoter activity and also abrogated some of the PTH-mediated inhibition of the promoter. Previous studies have shown growth factor regulation of Osx expression by MAPK proteins, particularly p38 phosphorylation of OSX that increases its transcriptional activity. PTH stimulation of osteoblasts inhibits MAPK components (ERK, JNK, and p38) but inhibition of Osx mRNA and protein expression by PTH was selectively mimicked by p38 inhibition and expression of constitutively active MKK6, which stimulates p38, blocked PTH inhibition of OSX. Together, our studies suggest that OSX autoregulation is a major mechanism in osteoblasts and that PTH stimulation inhibits osterix by inhibition of p38 MAPK regulation of OSX.
Keywords: osterix, Sp factor, osteoblasts, parathyroid hormone, autoregulation
Introduction
Osterix (Osx, Sp7) is a zinc-finger containing transcription factor of the specificity protein (Sp) family that is essential for osteoblast differentiation and embryonic skeletal development. Osx-deficient mice accumulate preosteoblasts, arrested in their differentiation and unable to express key osteoblastic genes, such as osteocalcin, bone sialoprotein, and osteopontin (Nakashima et al. 2002). In the absence of OSX, the prenatal cartilaginous skeleton fails to mineralize, and these mice die at birth (Nakashima et al. 2002). There is also increasing evidence to suggest that OSX plays a role in the homeostasis of adult bones. When Osx is knocked down in postnatal mice, the animals accumulate bone microfractures, have decreased osteoblastic activity, and impaired osteocyte maturation and function (Baek et al. 2009, Zhou et al. 2010). In humans, several genome-wide association studies have correlated certain polymorphisms of the OSX gene with decreased bone mineral density in children and adults (Timpson et al. 2009, Lapunzina et al. 2010).
OSX is a transcriptional activator that drives the expression of target genes through its association with Sp binding motifs. Forced expression of OSX in vitro induces the expression of several osteoblastic genes, including collagen type 1A1 and osteocalcin (Fu et al. 2007, Kurata et al. 2007). The Osx gene is organized into two exons and one intron and is driven by a TATA-less promoter (Gao et al. 2004). Mouse and rat OSX proteins are expressed as both long and short isoforms that appear as two major bands of 56 and 46 kDa on immunoblots of bone protein extracts (Hong et al. 2009). It is not clear whether there are any differences in the activities of these isoforms. There is also evidence that OSX can undergo posttranslational modification. Ser-73 and Ser-77 on OSX can be phosphorylated by p38 MAPK, resulting in greater activation of two target promoters, fibromodulin and bone sialoprotein (Ortuno et al. 2010).
A number of factors regulate OSX levels and as such may contribute to the regulation of osteoblast differentiation. Bone morphogenic protein-2 (BMP2), one of the several BMPs important in osteoblast differentiation, can directly induce OSX expression through a number of pathways. BMP2 can induce expression of the runt-domain factor Runx2, a transcription factor expressed before OSX in the osteoblast lineage and shown to stimulate OSX expression (Lee et al. 1999). In addition, BMP2 can also activate the homeodomain-containing transcription factor Dlx5, which can then bind and activate the Osx promoter (Lee et al. 2003, Ulsamer et al. 2008). IGFs acting through IGF1 tyrosine kinase receptors can also stimulate OSX levels in human mesenchymal cells (Celil & Campbell 2005).
Other factors have been shown to inhibit OSX expression. The inflammatory cytokine tumor necrosis factor α (TNF-α) inhibits osteoblast differentiation and negatively regulates the expression of OSX (Gilbert et al. 2000, Lu et al. 2006). The effects of TNF-α on the Osx promoter have recently been shown to be mediated through EGF receptors by regulation of the transcription factor paired related homeobox protein (Prx1 (Prrx1); Lu et al. 2011). Parathyroid hormone (PTH), which is both catabolic and anabolic to bone, has receptors on osteoblasts and can regulate both osteoblast activity and osteoblast differentiation. We have reported that prolonged stimulation with PTH inhibits Osx mRNA and protein expression in osteoblastic UMR106-01 cells and murine calvaria via a cAMP-dependent pathway that causes downstream transcriptional repression (Hong et al. 2009). PTH responses were localized to the proximal −304/+91 region of the Osx promoter. Within the PTH-responsive region, NFκB, Ets1, and NF-Y sites and two contiguous putative Sp binding sites were identified. We have shown that deletion of the NFκB site had no effect on PTH regulation of the Osx promoter (Hong et al. 2009). The aim of this study was to identify transcriptional regulators of Osx within the PTH-responsive region and to further determine the mechanisms by which PTH inhibits OSX expression.
Materials and methods
Materials
Rat PTH(1–34) was from Bachem Bioscience, Inc. (King of Prussia, PA, USA). Mithramycin A (MA) and forskolin were from Tocris Bioscience (Ellisville, MO, USA). Primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). MAPK inhibitors U0126, SP600125, and SB203580 were from Selleck Chemicals (Burlington, ON, Canada). The luciferase Osx promoter plasmid −1269/+91 was kindly provided by Dr Mark Nanes (Emory University, Atlanta, GA, USA), and the cDNA encoding constitutively active MKK6(Glu) (Raingeaud et al. 1996) was developed by Dr Roger Davis (University of Massachusetts, Worcester, MA, USA) and obtained from Addgene (Cambridge, MA, USA). The cDNA encoding constitutively active JNKK2-JNK1 (Zheng et al. 1999) was kindly provided by Dr Anning Lin (The University of Chicago, Chicago, IL, USA). Antibodies used were as follows: osterix (ab22552) and actin (ab1801) from Abcam (Boston, MA, USA); Sp1 (07-645) from EMD Millipore (Billerica, MA, USA); Sp3 (sc-644), TIEG (sc-67062), MKP-1 (sc-2027), and IgG (sc-2027) from Santa Cruz Technology Santa Cruz, CA, USA; and Erk1/2 (9102), pERK1/2 (9101), JNK (9252), pJNK (9251), p38 (9212), and phospho-p38 (9211) from Cell Signaling Technology (Danvers, MA, USA).
Cell preparations and treatments
UMR106-01 osteosarcoma cells (from Dr Nicola Partridge UMDNJ-Robert Wood Johnson Medical School, Piscataway, NJ, USA) were cultured as described previously (Hong et al. 2009). Primary mouse osteoblasts were prepared from calvaria of neonatal CD1 mice by collagenase digestion as described previously (Wong & Cohn 1975). In some experiments, whole calvaria were dissected and cultured as described previously (Hong et al. 2009).
Cells were plated at a density of 5×105 cells/well in 6-well plates for RNA or protein extractions, at 2×105 cells/well in 12-well plates for luciferase assays or 106 cells in 100 mm dishes for chromatin preparations. Following treatments, cells were lysed and proteins were extracted as described previously (Lai et al. 2005) or RNA was extracted using TRIzol (Life Technologies) according to the manufacturer's instructions.
Real-time PCR
RNA samples were reverse transcribed and real-time PCR was performed as described previously (Hong et al. 2009). Primers used were glyceraldehyde-3-phosphate dehydrogenase (GAPDH): rat forward, 5′-CAT GGC CTT CCG TGT TCC TA-3′ and reverse, 5′-GCG GCA CGT CAG ATC CA-3′; mouse GAPDH forward, 5′-CAG CCT CCC GTA GAC A-3′ and reverse: 5′-CGC CCA ATA CGG CCA AA-3′; rat Osx forward, 5′-CAG CCT GCA GCA AGT TT GG-3′ and reverse, 5′-TTT TCC CAG GGC TGT TGA GT-3′; mouse Osx forward, 5′-GGT CCC CAG TCG AGG AT-3′ and reverse, 5′-CTA GAG CCG CCA AAT TTG CT-3′. The quantified individual RNA expression was normalized to GAPDH.
Immunoblotting
Designated amounts of protein were separated on 11% SDS–PAGE gels, transferred to nitrocellulose membranes, and probed with antibodies to specific proteins followed by HRP-labeled anti-rabbit IgG antibody (GE Healthcare, Mississauga, ON, Canada). Proteins were visualized using ECL reagent (GE Healthcare) and exposed to X-ray film. Protein bands were quantified by densitometry imaging using ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA). All target protein expression levels were normalized to the amount of α-actin detected in the same sample.
Site-directed mutagenesis of reporter constructs and luciferase assays
Osx promoter reporter mutants were made using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Palo Alto, CA, USA) according to the manufacturer's guidelines. Mutations that were introduced are highlighted in bold: m34/38, CAC CCC CAC CCC CAA AAA CCA CCC C; m34/44, CAC CCC CAC CCC CAC CCC AAA AAC C; m40/44, CAC CCC CAC CCC CAA AAA CAA AAC C; m114/118: TCA TTG GAT TCT CGC TAT; and m155/159, AGG AAA TAA CTA. All constructs were verified by DNA sequencing. The base pair numbering is in reference to the predicted transcription start site of the Osx promoter (Lu et al. 2006). Luciferase assays were performed as described previously (Hong et al. 2009) using a luciferin reagent (Promega). All luciferase results were corrected for transfection efficiency using β-gal activity assessed as described previously (Hong et al. 2009).
Chromatin immunoprecipitation assays
Cells were treated with either vehicle or 1 nM rPTH(1–34) for 2–24 h. Following treatment, cellular proteins were cross-linked and chromatin was prepared and immunoprecipitated as described previously (Matthews et al. 2005). To measure total input of DNA, 5 μl of each sample were incubated in the same buffer. DNA was uncross-linked by incubating overnight at 66 °C and purified on spin columns (Life Technologies). Osx promoter fragments were quantified by real-time PCR using Power SYBR Green, 300 nM forward and reverse ChIP primers (Osx promoter primer: forward, CTC ATT GGA TCC GGA GTC TTC T; reverse, TGT CTG TAG GGA TCC ACC CTC TA), and 2 μl DNA sample, in a final volume of 15 μl. Input DNA and immunoprecipitated samples were assayed in triplicate. Total amounts of binding by a specific antibody were determined by expressing the amount of DNA obtained for each immunoprecipitated sample as a percentage of total DNA using the following formula: 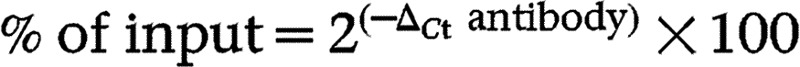 , where ΔCt antibody=Ct total DNA−Ct Ab.
, where ΔCt antibody=Ct total DNA−Ct Ab.
Statistical analysis
Data from three independent experiments are expressed with bars indicating s.e.m. Statistical analyses were performed using the GraphPad Prism software (San Diego, CA, USA). One-way ANOVA, where P<0.05 was considered statistically significant, followed by Bonferroni post hoc analysis was used to determine the effect of reagents on Osx expression. Statistical significance was considered with P<0.05.
Results
Sp binding sites are important for Osx promoter activity
To examine the importance of the potential transcription factor binding sites on Osx promoter activity, luciferase constructs containing the entire Osx promoter (−1269/+91) were mutated sequentially at each of the identified sites (Fig. 1A). Mutation of the NF-Y site (m114/118) increased promoter activity by 60%, while mutation of the Ets1 site (m155/159) had no effect. Conversely, mutation of either or both of the two Sp sites resulted in >90% decrease in basal promoter activity (Fig. 1B). This indicated that the two Sp sites in the proximal region of the Osx promoter were vital for promoter activity. All the three Sp site mutants displayed significantly higher activity than the baseline activity of empty PGL3 vector-transfected cells (PGL3basic), suggesting that promoter activity was not completely abolished. To determine whether mutation of the Sp sites altered PTH responsiveness of the Osx promoter, cells were treated with 1 nM PTH for 16 h. PTH inhibited the activity of the wild-type Osx promoter luciferase construct by ∼70%. Disruption of either or both the Sp sites resulted in significant impairment of PTH inhibition of Osx promoter activity, although activity was still reduced by as much as 50% by PTH (Fig. 1C). To determine whether the importance of the Sp sites on promoter activity could also be seen in primary osteoblasts, neonatal mouse osteoblasts were transfected with either the wild-type −1269/+91 Osx promoter or the promoter with one Sp site mutated (m40/44) and activity assessed with or without treatment with PTH. As shown in Fig. 1D, mutation of the Sp site reduced promoter activity by 70% and completely blocked PTH inhibition in these cells.
Figure 1.

Mutational analysis of the Osx promoter. (A) Schematic representation of the relative positions of putative transcription factor binding sites in the −1269/+91 Osx promoter. (B) UMR106-01 cells transfected with the −1269/+91 Osx promoter reporter; promoter constructs with mutations in the regions indicated in brackets or empty vector (PGL3basic). Cells were assayed for luciferase and β-galactosidase activities 24 h after transfection. Corrected luciferase activities expressed relative to the wild-type promoter are shown. (C) PTH regulation of the indicated promoter luciferase constructs or empty vector was assessed using 10 nM rPTH(1–34) for 16 h before measuring luciferase activity. The values shown indicate luciferase activity in the presence of PTH relative to that in the basal state. (D) Luciferase activity of −1269/+91 Osx promoter reporter and the mutation of the first Sp site (−40/−44) reporter were tested for basal and PTH-regulated activity in primary mouse osteoblasts. Cells were treated with or without 10 nM rPTH(1–34) for 16 h before measuring luciferase activity. The values shown indicate luciferase activity in the presence of PTH relative to that in the basal state, cells transfected with empty vector were assessed without PTH stimulation. Statistical significance in B and C: *P<0.001 relative to −1269/+91; in D, statistical differences between bars are indicated by differences in letters P<0.05.
MA inhibits Osx transcription
To further investigate the importance of Sp factors in osterix expression, cells were treated with MA, a reagent that blocks interaction of Sp factors with their recognition sequences (Miller et al. 1987, Blume et al. 1991). MA decreased the activity of the wild-type −1269/+91 luciferase construct in a concentration-dependent manner (Fig. 2A). Moreover, similar to PTH, MA treatment also resulted in decreased expression of Osx mRNA in both UMR cells and in whole calvaria (Fig. 2B). Western blots also showed that cells treated with MA, PTH, or the combination of both decreased both OSX isoforms to similar levels, while actin levels in the cells were unaffected (Fig. 2C).
Figure 2.

Mithramycin A (MA) inhibits Osx promoter activity and OSX expression. (A) UMR106-01 cells were transfected with −1269/+91 Osx promoter and incubated in the presence of increasing concentrations of MA or vehicle control for 8 h before assessment of luciferase activity corrected for β-galactosidase activity in the same cells. (B) UMR106-01 cells or neonatal mouse calvaria were incubated in the presence of 10 nM rPTH(1–34) (PTH), 2 μM MA (MA), or vehicle control (Con) for 16 h before extraction of RNA and assessment of Osx mRNA levels by real-time PCR relative to the levels of GAPDH in each sample. Bars indicate Osx mRNA levels relative to Con for each cell/tissue type. Statistical significance in A and B: *P<0.001 relative to control values. (C) The effect of incubation of UMR106-01 cells with 1 μM MA (+MA), 10 nM rPTH(1–34) (+PTH), 1 μM MA and 10 nM rPTH(1–34) (+MA+P) or vehicle control (Con) for 16 h on OSX and α-actin protein expression were assessed by SDS–PAGE and immunoblot. Two isoforms of OSX were expressed and both were suppressed by MA or PTH by 50%. Duplicate samples were assessed in each experiment and each experiment was repeated twice with representative blots shown.
Osterix binds to its own promoter
As the Sp sites were responsible for much of the activity of the Osx promoter, it is likely that transcription factors acting at these sites are crucial for basal OSX expression. We assessed Sp1, Sp3, and TIEG1 expression and binding to the Osx promoter. In UMR cells, immunoblotting showed expression of Sp1, Sp3, and TIEG1, and this was unaffected by treatment with PTH for 8 h (Fig. 3C). Association of Sp1, Sp3, TIEG1 (KLF10), or OSX with the Osx promoter at the Sp sites was assessed by ChIP assays. As shown in Fig. 3A, OSX was the only one of these transcription factors that was significantly associated with the Osx promoter in the area proximal to the two Sp sites, indicating that OSX may drive its own expression in osteoblasts. Furthermore, OSX binding to its own promoter was significantly reduced following 2–24 h of PTH treatment. The decrease in Osx ChIP (Fig. 3B) preceded the reduction in OSX protein that is seen with PTH treatment (Fig. 3D), suggesting some additional mechanism of regulation by PTH.
Figure 3.

Osterix but not other Sp factors bind to the Osx promoter. (A) Chromatin prepared from UMR106-01 cells was assessed for transcription factor binding to the Osx promoter proximal to the two Sp binding sites (−44/−34). Following immunoprecipitation with antibodies specific to the indicated transcription factors, the amount of Osx promoter binding to each factor was assessed by real-time PCR. Bars indicate the amount of Osx DNA in each sample relative to the total input amount in the assay. Nonspecific binding was assessed using IgG in the immunoprecipitation assay. *Statistical significance: P<0.0001 relative to IgG. (B) UMR106-01 cells were incubated with 10 nM rPTH(1–34) for the indicated times before cell extraction and chromatin preparation. PTH stimulation significantly decreased OSX binding to its own promoter within 2 h of stimulation and this continued to decrease over 24 h. Statistical significance: *P<0.05 relative to control values. (C) Immunoblots of 25 μg samples of UMR106-01 cell extracts showing expression of each of the transcription factors assessed in the ChIP assays. Incubation of the cells with 10 nM rPTH(1–34) for 8 h had no effect on the levels of any of these proteins. (D) Immunoblots of OSX expression in UMR106-01 cells treated with 10 nM rPTH(1–34) over the same time periods as used for ChIP assays show that OSX protein was decreased with treatments of 4 h or longer.
Previous studies showing OSX phosphorylation by p38 MAPK suggested a potential mechanism by which PTH could be regulating Osx. As shown in Fig. 4A, PTH treatment rapidly inhibited the levels of activated, phosphorylated, ERK1/2, JNK, and p38, while increasing the expression of the MAPK phosphatase MKP-1. We therefore examined the effects of pharmacological inhibitors of MAPKs on the levels of Osx mRNA. As shown in Fig. 4B, incubation with the MEK1/2 inhibitor U0126 or the JNK inhibitor SP600125 had no effect on Osx RNA, indicating that the loss of pERK or pJNK by PTH was not likely to mediate its inhibition of Osx. However, incubation of the cells with SB203580, an inhibitor of p38 MAPK, significantly inhibited Osx mRNA (Fig. 4B) and inhibited luciferase activity of the wild-type Osx promoter −1269/+91 construct (Fig. 4C). Our previous work showed that the effect of PTH on OSX is mediated through the cAMP pathway (Hong et al. 2009), and we further explored here whether stimulation of adenylyl cyclase through forskolin could also mimic the effects of PTH. As shown in Fig. 5, forskolin produced similar effect on the Osx promoter, OSX expression at the mRNA and protein levels, and also inhibited pp38 levels to a similar degree as PTH treatment of the same cells. Thus, PTH inhibition of p38 MAPK through a cAMP-mediated mechanism could contribute to the inhibition of Osx in these cells. To further test this possibility, we transfected UMR cells with constitutively active MKK6 to stimulate p38 MAPK and tested whether this blocked the inhibition of Osx by PTH. As shown in Fig. 6A, caMKK6 but not caJNK did block some of the PTH inhibition of Osx mRNA. Similar to the effects of the MAPK regulators on PTH inhibition of OSX protein and as seen in Fig. 6B, caMKK6 blocked most of the effect of PTH while caJNK had no effect. When we examined the effects on PTH regulation of the wild-type Osx promoter, again caMKK6 but not caJNK blocked some of the PTH inhibition of the promoter, particularly at the early time point (Fig. 6C). Finally, we examined whether increasing pp38 could increase expression of the full-length Osx promoter, and indeed, this was the case in the UMR cells where cells transfected with caMKK6 showed 40% increase in basal luciferase activity compared with cells transfected with a control plasmid (Fig. 6D). The promoter construct in which the two Sp binding sites were mutated (−1269/+91m34/44) was also stimulated by co-expression of caMKK6, demonstrating that sites in the promoter outside the −34/44 domain also likely contribute to stimulation by p38MAPK. Together, these results suggest that PTH regulation of OSX is at least partially mediated by its effects on p38 MAPK.
Figure 4.

Effect of PTH stimulation on the levels of MAPKs and the effect of MAPK inhibitors on Osx mRNA and Osx promoter luciferase activity. (A) UMR106-01 cells were incubated for the indicated times with 10 nM rPTH(1–34) before extraction of cellular proteins. 10 μg samples of cellular extracts were analyzed by SDS–PAGE and immunoblots using specific antibodies to each of the indicated MAPKs and their activated (phosphorylated) forms as shown on the side of each blot. Levels of MKP-1, a phosphatase that can dephosphorylate MAPKs, are also shown in the bottom panel. (B) UMR106-01 cells were incubated with 10 μM concentrations of U0126, a specific inhibitor of ERK1/2; SP600125, a specific inhibitor of JNK (SP); SB203580, a specific inhibitor of p38 MAPK (SB), or vehicle control for 16 h before extraction of cellular RNA and assessment of Osx mRNA by real-time PCR. (C) UMR106-01 cells were transfected with −1269/+91 promoter luciferase construct and the cells were then incubated with either 10 μM SB203580 for the indicated times or 10 nM rPTH(1–34) for 24 h before assessment of luciferase activity. Statistical significance in B and C indicated by *P<0.05 relative to control values.
Figure 5.

Effects of PTH on OSX and p38MAPK are also seen with forskolin stimulation of UMR cells. (A) UMR106-01 cells were transfected with the −1269/+91 Osx promoter reporter construct and treated with 10 nM rPTH(1–34), 10 μM forskolin, or vehicle control for 16 h before measuring luciferase activity. The values shown indicate luciferase activity relative to that in the basal state. (B) UMR106-01 cells were incubated in the presence of 10 nM rPTH(1–34) (PTH), 10 μM forskolin (FSK), or vehicle control (Con) for 16 h before extraction of RNA and assessment of Osx mRNA levels by real-time PCR relative to the levels of GAPDH in each sample. Bars indicate Osx mRNA levels relative to Con for each cell/tissue type. Statistical significance in A and B *P<0.001 relative to control values. (C) UMR106-01 cells were incubated in the presence of 10 nM rPTH(1–34) (PTH), 10 μM forskolin (FSK), or vehicle control (Con) for 16 h before extraction of protein and assessment of OSX protein levels by immunoblot relative to the levels of actin in each sample. (D) UMR106-01 cells were incubated for the 90 min with 10 nM rPTH(1–34) (PTH), 10 μM forskolin (FSK), or vehicle control (Con) before extraction of cellular proteins. Samples of cellular extracts were analyzed by SDS–PAGE and immunoblots using specific antibodies to the indicated p38MAPK and its activated form (pp38) as shown on the side of each blot.
Figure 6.

Expression of constitutively active MKK6 but not JNK abrogates PTH inhibition of Osx mRNA, protein, and Osx promoter activity. (A) UMR106-01 cells were transfected with cDNAs encoding caMKK6, an upstream kinase that activated p38 MAPK or JNKK2-JNK1, a constitutively active JNK1. The following day, cells were incubated with or without 10 nM rPTH(1–34) for 8 h before RNA extraction and assessment of Osx mRNA by real-time PCR. *Values significantly different from basal levels, P<0.05; ¶values significantly different from control +PTH, P<0.05. (C and D) UMR106-01 cells were transfected with caMKK6, JNKK2-JNK1 (caJNK), or a control cDNA (control) in addition to −1269/+91 promoter luciferase construct. Cells were then incubated with 10 nM rPTH(1–34) for 8 or 16 h and compared with cells incubated without PTH (Basal). Luciferase activities relative to control basal are plotted. *Values significantly different from control basal levels, P<0.01. (B) UMR106-01 cells were transfected with caMKK6, JNKK2-JNK1 (caJNK), or a control cDNA (control) and then incubated with 10 nM rPTH(1–34) for 8 h before extraction of cellular proteins. 25 μg samples of protein extracts were run on SDS–PAGE followed by immunoblot for OSX or actin. The numbers on top of the blots indicate the levels of OSX protein relative to control basal levels.
Discussion
In this study, we examined areas in the murine Osx promoter that might be important for its expression and regulation by PTH in osteoblasts. We provide evidence for the importance of a domain containing tandem CCACCC elements within the murine proximal promoter that are crucial for Osx expression in osteoblasts. This sequence motif does not conform to the traditional Sp consensus sequence (GGGCGGG), and using ChIP assays, we found no evidence of other Sp factors binding to this area of the Osx promoter. However, Osx binding to this area was quite strong and OSX has been previously reported to bind to these unorthodox Sp sites to drive the transcription of collagen 5a1 (Wu et al. 2010). Thus, the CCACCC motif is emerging as a preferred OSX binding site in some osteoblastic gene promoters. A comparison of the rat, mouse, and human Osx promoters revealed that the upstream CCACCC site is highly conserved across all the three species, indicating a requirement for at least one of these elements within the Osx promoter. However, in the mouse promoter used in our studies, mutation of either of the CCACCC sites eliminated similar amounts of basal promoter activity, suggesting that both sites are required for full expression of the murine gene. It will be important to test whether this positive OSX feedback mechanism is also observed in human osteoblasts and whether it is regulated in a similar manner through the single CCACCC site in the human OSX promoter.
Our previous work, as well as data shown here, has demonstrated that activation of the PTH receptor for 3 h or longer results in inhibition of Osx mRNA and later protein levels in osteoblasts are also decreased. Using ChIP assays, we here show that PTH decreased OSX binding to its own promoter within 2 h. This consequently leads to significant loss of Osx mRNA first seen after 3 h and later loss of OSX protein 4–6 h after initiation of PTH stimulation (Hong et al. 2009). This temporal pattern suggested that there must be a signal downstream of the PTH receptor that initiates loss of stimulation on the Osx promoter or recruitment of an inhibitor. Rapid inhibition of multiple MAPKs, particularly phospho-p38 MAPK that has been shown to activate OSX, emerged as one such pathway that could begin inhibiting Osx. Indeed, PTH inhibition of Osx was blocked when cells were transfected with the constitutively active MKK6 (MKK6Glu) that is a selective activator of p38 MAPK (Raingeaud et al. 1996). Four isoforms of p38 MAPKα-δ have been identified in mammalian cells. Inhibition of p38α and p38β in mouse calvaria by daily s.c. injection of the p38 inhibitor SB203580 over the calvaria for 1 week or selective deletion of either p38α or p38β or the upstream kinases MKK3 and MKK6 resulted in reduced osteoblast differentiation and bone mineralization that included reduced expression of Osx (Greenblatt et al. 2010). Stimulation by PTH resulted in increased expression of MKP-1, one of the four MAPK phosphatases that can dephosphorylate and thus inactivate p38α and p38β (Cuadrado & Nebreda 2010). Thus, our data are consistent with PTH stimulation leading to dephosphorylation of p38 MAPK resulting in diminished Osx transcriptional activity. Over several hours, this would lead to loss of OSX protein binding to its own promoter leading to further loss of its own autoregulation. Our data demonstrate an inhibitory effect of PTH on p38 as well as ERK and JNK MAPKs in the UMR106-01 cells, which is likely mediated by cAMP, in agreement with studies shown here with direct activation of adenylyl cyclases by forskolin and in previous studies by our laboratory and others using this cell line (Doggett et al. 2002, Lai & Mitchell 2009) and others (Zhen et al. 2001, Lai et al. 2005). Others have reported positive effects of PTH on p38 MAPK in differentiating MC3T3 osteoblastic cells by mechanisms that have not been identified (Rey et al. 2007). p38 MAPK signaling was also increased in primary mouse calvarial osteoblasts that were treated with PTH intermittently over a 2-week period (Bianchi & Ferrari 2009). In both these experimental paradigms, Osx expression was increased by the anabolic effects of PTH; however, this is likely the result of increased osteogenesis that occurs with long-term intermittent PTH treatment in vivo. It is not clear whether p38 or other MAPKs play a direct role in regulating Osx expression in mature osteoblasts; however, UMR106-01 cells express osteocalcin, a protein expressed by mature osteoblasts. Thus, it is possible that PTH regulates p38 MAPK differently in osteoblasts at different stages of differentiation and depending on the mode of PTH delivery in either a continuous catabolic or an intermittent anabolic fashion. Our data showing that inhibitors of p38 MAPK downregulate Osx expression are consistent with the reported essential role of p38 MAPK on skeletogenesis in mice (Greenblatt et al. 2010).
In summary, we have demonstrated for the first time an autoregulatory pathway in which OSX stimulates its own promoter through a tandem repeat CCACCC element in its proximal promoter. Inhibition of Osx by PTH decreases OSX binding to these elements in its promoter and this appears to be initiated through stimulation of cAMP and inhibition of p38 MAPK.
Acknowledgements
The authors thank Dr Jason Matthews for his help with ChIP assays.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by a grant from the Canadian Institutes for Health Research (MOP11506) to J M.
References
- Baek WY, Lee MA, Jung JW, Kim SY, Akiyama H, de Crombrugghe B, Kim JE. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. Journal of Bone and Mineral Research. 2009;24:1055–1065. doi: 10.1359/jbmr.081248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi EN, Ferrari SL. β-Arrestin2 regulates parathyroid hormone effects on a p38 MAPK and NFkappaB gene expression network in osteoblasts. Bone. 2009;45:716–725. doi: 10.1016/j.bone.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. Journal of Clinical Investigation. 1991;88:1613–1621. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celil AB, Campbell PG. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. Journal of Biological Chemistry. 2005;280:31353–31359. doi: 10.1074/jbc.M503845200. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochemical Journal. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Doggett TA, Swarthout JT, Jefcoat SC, Jr, Wilhelm D, Dieckmann A, Angel P, Partridge NC. Parathyroid hormone inhibits c-Jun N-terminal kinase activity in rat osteoblastic cells by a protein kinase A-dependent pathway. Endocrinology. 2002;143:1880–1888. doi: 10.1210/en.143.5.1880. [DOI] [PubMed] [Google Scholar]
- Fu H, Doll B, McNelis T, Hollinger JO. Osteoblast differentiation in vitro and in vivo promoted by Osterix. Journal of Biomedical Materials Research. Part A. 2007;83:770–778. doi: 10.1002/jbm.a.31356. [DOI] [PubMed] [Google Scholar]
- Gao Y, Jheon A, Nourkeyhani H, Kobayashi H, Ganss B. Molecular cloning, structure, expression, and chromosomal localization of the human Osterix (SP7) gene. Gene. 2004;341:101–110. doi: 10.1016/j.gene.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Inhibition of osteoblast differentiation by tumor necrosis factor-α. Endocrinology. 2000;141:3956–3964. doi: 10.1210/en.141.11.3956. [DOI] [PubMed] [Google Scholar]
- Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, Lotinun S, Sano Y, Baron R, Park JM, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. Journal of Clinical Investigation. 2010;120:2457–2473. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Lu X, Nanes MS, Mitchell J. Regulation of osterix (Osx, Sp7) and the Osx promoter by parathyroid hormone in osteoblasts. Journal of Molecular Endocrinology. 2009;43:197–207. doi: 10.1677/JME-09-0012. [DOI] [PubMed] [Google Scholar]
- Kurata H, Guillot PV, Chan J, Fisk NM. Osterix induces osteogenic gene expression but not differentiation in primary human fetal mesenchymal stem cells. Tissue Engineering. 2007;13:1513–1523. doi: 10.1089/ten.2006.0374. [DOI] [PubMed] [Google Scholar]
- Lai LP, Mitchell J. Parathyroid hormone inhibits phosphorylation of mitogen-activated protein kinase (MAPK) ERK1/2 through inhibition of c-Raf and activation of MKP-1 in osteoblastic cells. Cell Biochemistry and Function. 2009;27:269–275. doi: 10.1002/cbf.1568. [DOI] [PubMed] [Google Scholar]
- Lai LP, DaSilva KA, Mitchell J. Regulation of Indian hedgehog mRNA levels in chondrocytic cells by ERK1/2 and p38 mitogen-activated protein kinases. Journal of Cellular Physiology. 2005;203:177–185. doi: 10.1002/jcp.20208. [DOI] [PubMed] [Google Scholar]
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R, Martinez-Glez V, Elhossini R, Amr K, Vilaboa N, et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. American Journal of Human Genetics. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Javed A, Kim HJ, Shin HI, Gutierrez S, Choi JY, Rosen V, Stein JL, van Wijnen AJ, Stein GS, et al. Transient upregulation of CBFA1 in response to bone morphogenetic protein-2 and transforming growth factor β1 in C2C12 myogenic cells coincides with suppression of the myogenic phenotype but is not sufficient for osteoblast differentiation. Journal of Cellular Biochemistry. 1999;73:114–125. doi: 10.1002/(SICI)1097-4644(19990401)73:1%3c114::AID-JCB13%3e3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lee MH, Kwon TG, Park HS, Wozney JM, Ryoo HM. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochemical and Biophysical Research Communications. 2003;309:689–694. doi: 10.1016/j.bbrc.2003.08.058. [DOI] [PubMed] [Google Scholar]
- Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. Journal of Biological Chemistry. 2006;281:6297–6306. doi: 10.1074/jbc.M507804200. [DOI] [PubMed] [Google Scholar]
- Lu X, Beck GR, Jr, Gilbert LC, Camalier CE, Bateman NW, Hood BL, Conrads TP, Kern MJ, You S, Chen H, et al. Identification of the homeobox protein Prx1 (MHox, Prrx-1) as a regulator of osterix expression and mediator of tumor necrosis factor α action in osteoblast differentiation. Journal of Bone and Mineral Research. 2011;26:209–219. doi: 10.1002/jbmr.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews J, Wihlen B, Thomsen J, Gustafsson JA. Aryl hydrocarbon receptor-mediated transcription: ligand-dependent recruitment of estrogen receptor α to 2,3,7,8-tetrachlorodibenzo-p-dioxin-responsive promoters. Molecular and Cellular Biology. 2005;25:5317–5328. doi: 10.1128/MCB.25.13.5317-5328.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Polansky DA, Thomas SD, Ray R, Campbell VW, Sanchez J, Koller CA. Mithramycin selectively inhibits transcription of G-C containing DNA. American Journal of the Medical Sciences. 1987;294:388–394. doi: 10.1097/00000441-198711000-00015. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/S0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- Ortuno MJ, Ruiz-Gaspa S, Rodriguez-Carballo E, Susperregui AR, Bartrons R, Rosa JL, Ventura F. p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. Journal of Biological Chemistry. 2010;285:31985–31994. doi: 10.1074/jbc.M110.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Molecular and Cellular Biology. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey A, Manen D, Rizzoli R, Ferrari SL, Caverzasio J. Evidences for a role of p38 MAP kinase in the stimulation of alkaline phosphatase and matrix mineralization induced by parathyroid hormone in osteoblastic cells. Bone. 2007;41:59–67. doi: 10.1016/j.bone.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Timpson NJ, Tobias JH, Richards JB, Soranzo N, Duncan EL, Sims AM, Whittaker P, Kumanduri V, Zhai G, Glaser B, et al. Common variants in the region around Osterix are associated with bone mineral density and growth in childhood. Human Molecular Genetics. 2009;18:1510–1517. doi: 10.1093/hmg/ddp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulsamer A, Ortuno MJ, Ruiz S, Susperregui AR, Osses N, Rosa JL, Ventura F. BMP-2 induces Osterix expression through up-regulation of Dlx5 and its phosphorylation by p38. Journal of Biological Chemistry. 2008;283:3816–3826. doi: 10.1074/jbc.M704724200. [DOI] [PubMed] [Google Scholar]
- Wong GL, Cohn DV. Target cells in bone for parathormone and calcitonin are different: enrichment for each cell type by sequential digestion of mouse calvaria and selective adhesion to polymeric surfaces. PNAS. 1975;72:3167–3171. doi: 10.1073/pnas.72.8.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YF, Matsuo N, Sumiyoshi H, Yoshioka H. Sp7/Osterix is involved in the up-regulation of the mouse pro-α1(V) collagen gene (Col5a1) in osteoblastic cells. Matrix Biology. 2010;29:701–706. doi: 10.1016/j.matbio.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Zhen X, Wei L, Wu Q, Zhang Y, Chen Q. Mitogen-activated protein kinase p38 mediates regulation of chondrocyte differentiation by parathyroid hormone. Journal of Biological Chemistry. 2001;276:4879–4885. doi: 10.1074/jbc.M004990200. [DOI] [PubMed] [Google Scholar]
- Zheng C, Xiang J, Hunter T, Lin A. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. Journal of Biological Chemistry. 1999;274:28966–28971. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, Darnay BG, de Crombrugghe B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. PNAS. 2010;107:12919–12924. doi: 10.1073/pnas.0912855107. [DOI] [PMC free article] [PubMed] [Google Scholar]