

Abstract
There is a clear need for novel, effective therapeutic approaches to hemorrhagic fever due to filoviruses. Ebola virus hemorrhagic fever is associated with robust interferon (IFN)–α production, with plasma concentrations of IFN-α that greatly (60- to 100-fold) exceed those seen in other viral infections, but little IFN-β production. While all of the type I IFNs signal through the same receptor complex, both quantitative and qualitative differences in biological activity are observed after stimulation of the receptor complex with different type I IFNs. Taken together, this suggested potential for IFN-β therapy in filovirus infection. Here we show that early postexposure treatment with IFN-β significantly increased survival time of rhesus macaques infected with a lethal dose of Ebola virus, although it failed to alter mortality. Early treatment with IFN-β also significantly increased survival time after Marburg virus infection. IFN-β may have promise as an adjunctive postexposure therapy in filovirus infection.
Keywords: Filovirus, IFN-β, Ebola virus, Marburg virus, type I interferon
The filoviruses have a well-deserved reputation for lethality [1]. Mortality due to the Zaire species of Ebola virus (EBOV-Z) has been approximately 80% in human outbreaks and >95% in rhesus macaque models [2]. Similarly, the Angola strain of Marburg virus (MARV) has a human case mortality rate of approximately 85% [3], something mirrored in experimental macaque infection [4]. There are no approved therapies for filovirus hemorrhagic fever (HF), nor any approved approaches to postexposure prophylaxis—although postexposure vaccination and small interfering RNA administration have shown promise in macaque models [5, 6].
Although the pathogenesis of filovirus HF remains underdefined, light has been shed on that of EBOV in recent years. In fatalities, a refractory shock syndrome is integral to terminal disease [1]. Viral replication clearly plays an important role, yet the relative contributions of virus and host to pathogenesis remain unclear. As with bacterial septic shock, the fulminant end of the disease spectrum triggered by EBOV is thought to involve maladaptive innate immune responses. Mediators and pathways dysregulated in both EBOV HF and septic shock include the coagulation cascade, lymphocyte apoptosis, and proinflammatory cytokine production.
The coagulopathy associated with EBOV HF is associated with dysregulation of both the clotting system (including tissue factor overexpression) and the fibrinolytic system (including reductions in protein C) [7]. Recognition of the latter led to validation of the ability of a nematode-derived inhibitor of tissue factor-initiated coagulation to ameliorate disease in EBOV and MARV infection in macaque models [4, 8].
Widespread lymphocyte apoptosis, associated with lymphopenia and lymphoid depletion of lymph nodes and spleen, is a prominent feature of EBOV HF [9–11]. The underlying mechanisms remain unclear. It is not due to direct infection; EBOV does not infect lymphocytes. In fatalities, such apoptosis occurs prior to the development of an adequate adaptive immune response [10, 11]. In survivors, adaptive immunity, arising in the absence of such pathological lymphocyte apoptosis, controls viral replication [10, 11]. Other human HF viruses also have a marked propensity for inducing lymphocyte death visible pathologically [9, 12–14]. Largely antedating recognition of apoptosis, the literature demonstrating this refers to “widespread lymphocyte necrosis”; there is little doubt now that this represents apoptosis. Thus, lymphoid apoptosis may represent a final common pathogenic pathway in diseases that are otherwise clinically diverse, and may provide a therapeutic target in filovirus HF.
As for cytokine responses, dysregulation of proinflammatory cytokine production by macrophages, primary sites of filovirus replication, is thought to be as central to the immunopathogenesis of EBOV HF as it is to bacterial sepsis [15]. In the case of the former, the cytokines that stand out are the type I interferons (IFNs). The human type I IFN superfamily includes 13 functional IFN-α genes (14 in rhesus macaque), along with single IFN-β, -ω, and -κ genes. Like most viruses, the filoviruses have mechanisms for inhibiting type I IFN production and responsiveness [16, 17]. Despite this, in vitro infection of macrophages leads to marked secretion of IFN-α, and EBOV HF is associated with robust IFN-α production in humans [18, 19] and macaques [20]. Indeed, in the latter study, plasma concentrations of IFN-α remained in considerable excess of 5 ng/mL for days, up until the time of death— levels that greatly (60- to 100-fold) exceed those seen in other viral infections, including systemic infections such as measles, symptomatic acute human immunodeficiency virus, and acute mononucleosis, as well as prostrating “local” infections such as influenza [21–25]. This is also greatly in excess of the plasma levels (often undetectable) achieved during IFN-α therapy for viral hepatitis [26]. However, little IFN-β (or IFN-γ) is produced in response to EBOV infection, either in vitro or in vivo. Further, similar to data reported in other systems [27], preliminary data, using universal primers and sequencing of polymerase chain reaction products, suggest that EBOV infection leads to production of a highly restricted subset of IFN-α subtypes (Howard Young, unpublished data). While type I IFNs all signal through the same receptor, both quantitative and qualitative differences in biological activities are observed—something thought to be due to differential binding affinity of IFN-α subtypes (weaker) and IFN-β (stronger) [28]. Notably, a previous therapeutic trial of high-dose IFN-α2 (20 × 106 IU/kg/day) in cynomolgus macaques infected with EBOV-Z failed to demonstrate clinical benefit [29]. Moreover, in vitro screening of available type I IFN subtypes revealed that IFN-β was especially effective in reducing EBOV-Z replication in cell culture (unpublished data).
These data suggested the twinned hypotheses that dysregulated production of IFN-α, in the absence of IFN-β, leads to inefficient antiviral activity against EBOV, and that this imbalanced type I IFN production itself contributes to lymphocyte apoptosis and disease pathogenesis. Here we show that, whereas delayed blockade of type I IFN signaling resulted in acceleration of disease in EBOV-infected rhesus macaques, early postexposure treatment with IFN-β significantly increased survival time, although it failed to alter mortality. Further, early treatment with IFN-β significantly increased the survival time of rhesus macaques infected with lethal doses of MARV. IFN-β may have promise as an adjunctive postexposure therapy in filovirus infection.
MATERIALS AND METHODS
Infection Models
Healthy, filovirus-seronegative adult male and female rhesus macaques (2–9 kg) were inoculated intramuscularly in the caudal thigh with 1 mL of virus stock containing 1000 plaque-forming units of EBOV-Z [30] or MARV (Musoke isolate) [31], which were propagated and titered on Vero cells [32]. Macaques were monitored twice daily for clinical signs of illness [33]. To minimize discomfort, animals were euthanized when moribund, as previously described [33]. Animals were anesthetized with ketamine or telazol prior to weighing, clinical examination, administration of therapy, and phlebotomy [29].
Animal studies took place in a Biosafety Level 4 laboratory at the US Army Medical Research Institute of Infectious Diseases (USAMRIID) under protocols approved by the USAMRIID Laboratory Animal Care and Use Committee, in compliance with the Animal Welfare Act and other US federal statutes and regulations relating to animal experiments, adherent to principles of the Guide for the Care and Use of Laboratory Animals [34]. The USAMRIID facility was fully accredited by Association for Assessment and Accreditation of Laboratory Animal Care International.
Therapeutic Protocols
To inhibit type I IFN signaling, 5 mg/kg of monoclonal antibody to IFNAR1 (64G12; Coulter Pharmaceutical) was administered subcutaneously 2 and 5 days after infection with EBOV-Z. For IFN-β treatment, human recombinant IFN-β (Rebif; EMD Serono) was administered subcutaneously as follows: (A) EBOV-Z experiment 1: 10.5 µg/kg at 18 hours, 1 day, 3 days, 5 days, 7 days, and 9 days after infection; (B) EBOV-Z experiment 2: 35 µg/kg, 12 hours after infection and then daily until 14 days after infection; (C) MARV experiment: 35 µg/kg, 1 hour after infection and then daily until 14 days after infection. Control animals received injections of an equivalent volume of sterile saline at identical time points. Day 0 weight was used to calculate dosages.
Given the stereotypical course of experimental filovirus infection in rhesus macaques and the desire to minimize animal usage for ethical reasons, experimental controls (2 in the IFNAR blockade study, 1 in each of the IFN-β studies) were supplemented by historical controls matched for viral seed stock, dose, and infection route (vide infra). The 5 historical controls for MARV infection had been vaccinated with a control vesicular stomatitis virus vector encoding an irrelevant, nonfilovirus glycoprotein [35, 36].
RNA Processing and DNA Microarray Analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-treated peripheral blood [33]. RNA was extracted using TRIzol (Invitrogen), and amplified using the MessageAmp II Amino Allyl kit (Ambion). Fluorescently labeled RNA was hybridized to human exonic evidence-based oligonucleotide arrays (http://www.microarray.org./sfgf/heebo.do) in a 2-color comparative format. Experimental samples were labeled with Cy-5; a reference pool (Universal Human Reference, Stratagene) was labeled using Cy-3, providing an internal standard enabling comparison across multiple samples [37]. Previous studies have demonstrated that such microarrays can reliably assess gene expression profiles in nonhuman primates [38, 39].
Arrays were scanned using an Axon Scanner 4000A; image analysis was performed using GenePix Pro version 6.1 (Axon Instruments). Data were filtered to exclude elements that did not have a regression correlation of Cy5 to Cy3 signal over the pixels across the array element of ≥0.6 and an intensity background of ≥2.5 in at least 80% of the arrays.
To analyze the effects of EBOV-Z infection, and treatment for same, on gene expression, data from each sample for IFN-β–treated and control animals were normalized to baseline (preinfection) samples for that animal [40]. To compare interexperimental group changes in gene expression, these data (from 3 control animals: 3 arrays representing day 3 and 1 array representing day 6; from 4 IFN-β–treated animals: 3 arrays representing day 3 and 4 arrays representing day 6) were hierarchically clustered using the Cluster program and displayed using TREEVIEW (http://rana.lbl.gov/EisenSoftware.htm). Functional annotation of gene expression by GO term enrichment and calculation of statistical significance of enrichment were done using the GOrilla tool [41, 42].
Hematological, Immunological, Virological, and Pathological Analysis
Complete blood counts were quantified using a Coulter ACT10 (Coulter Electronics). Blood chemistries were quantified using a Piccolo Point-of-Care Blood Analyzer (Abaxis). Serum cytokine concentrations were quantified using a human cytokine multiplex-25 bead array assay kit (BioSource International) [43]. Plasma virus burden was quantified on Vero cells as previously described; for MARV titering, Vero cell monolayers were stained on day 8 and read on day 9 [32, 44].
At necropsy, tissues were immersion-fixed in 10% neutral-buffered formalin, and processed for histopathology and immunohistochemistry as previously described [33]. For quantification of viral burden, tissues were stored at −70°C until assayed [33].
Statistical Analysis
Survival data were analyzed using the Wilcoxon rank-sum test with continuity correction. Kaplan–Meier survival analysis was used to construct graphs of percent survival over time. For ethical reasons, relevant historical controls were used to reduce the number of animals. Twenty-three rhesus macaques from studies performed over the last 9 years were used to supplement the 4 EBOV-Z–positive untreated controls in the current study to increase statistical power for comparing mean time to death. Similarly, 5 historical controls were used to supplement the single MARV-infected, untreated control in the current study.
The Kruskal–Wallis test was used for analysis of nonsurvival data whenever possible. Unless otherwise noted, differences among treated and control groups were not statistically significant or the dimensions of the data available did not permit nonparametric statistical analysis. For statistical analysis of microarray data, see above.
RESULTS
IFN-β Therapy Provides a Survival Benefit in Experimental EBOV-Z Infection
We took complementary approaches to investigate the hypothesis that imbalanced type I IFN production (robust production of IFN-α in the absence of evident IFN-β production) leads to both inefficient antiviral activity and dysregulated lymphocyte apoptosis during EBOV HF—delayed inhibition of type I IFN signaling, and therapy with IFN-β. To inhibit type I IFN signaling, macaques were treated with a monoclonal antibody to human IFNAR1, administered 2 and 5 days after infection. For IFN-β therapy, infected macaques were treated with human recombinant IFN-β (10.5 µg/kg) 18 hours, 1 day, 3 days, 5 days, 7 days, and 9 days after infection. As shown in Figure 1A, whereas inhibition of type I IFN signaling resulted in a significant decrease in survival (mean time to death = 6.25 days; P = .0016, Wilcoxon rank-sum test), treatment with IFN-β significantly prolonged mean time to death (13.8 days; P = .0097, Wilcoxon rank-sum test) relative to intraexperimental and historical controls (mean time to death = 8.33 days for intraexperimental controls, and 8.35 days for historical controls). Remarkably, 1 IFN-β-treated macaque survived for 29 days following EBOV-Z challenge.
Figure 1.
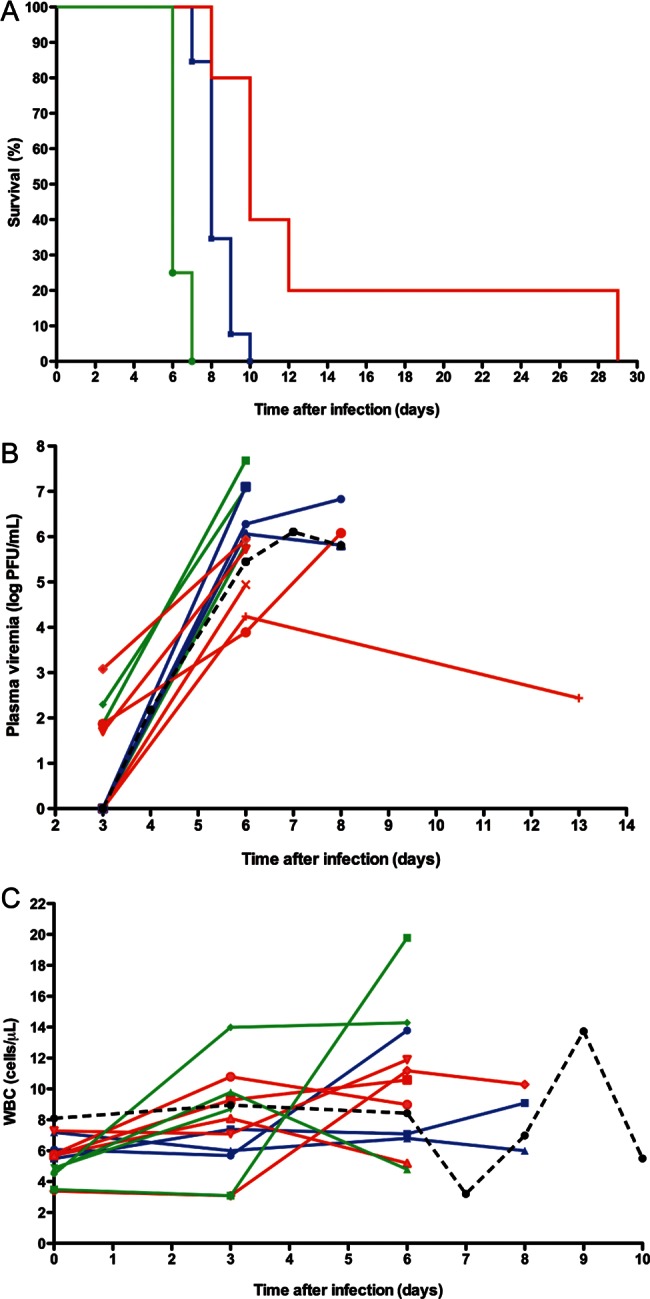
Therapy with interferon beta (IFN-β) provides a survival benefit in experimental Ebola virus infection. A, Mortality. Rhesus macaques were infected with 1000 plaque-forming units of the Zaire species of Ebola virus (EBOV-Z) and treated with (i) 5 mg/kg monoclonal antibody to the IFNAR1 chain of the human type I IFN receptor administered 2 and 5 days after infection (n = 4; green line); (ii) (in a separate experiment) recombinant human IFN-β 10.5 µg/kg, 18 hours and 1, 3 d, 5, 7, and 9 days after infection (n = 5; red line); or (iii) an equivalent volume of sterile saline at identical time points as a control (n = 2, IFNAR blockade; n = 1, IFN-β administration— supplemented with 23 historical controls: total n = 26; blue line). The mean time to death was 8.3 days in controls (intraexperimental controls succumbed on day 9 [IFNAR blockade] and day 7 [IFN-β treatment]). The mean time to death was significantly decreased by blockade of the type I IFN receptor (6.25 days; P = .0016, Wilcoxon rank-sum test), and significantly increased by IFN-β treatment (13.8 days; P = .0097, Wilcoxon rank-sum test). B, Viremia. Plasma EBOV-Z kinetics in individual macaques treated with monoclonal antibody to IFNAR1 (n = 4; green lines), IFN-β (n = 5; red lines), and saline (n = 3; intraexperimental controls; blue lines). Mean plasma viremia for 4 available matched, untreated EBOV-Z historical control animals is provided for reference (black dashed line). C, Peripheral blood leukocytes. Peripheral blood white blood cell kinetics in experimental macaques and available (n = 7) historical controls. Symbols as in B. Abbreviations: PFU, plaque-forming units; WBC, white blood cell.
As might be expected, kinetic analysis of plasma viremia (Figure 1B) revealed a trend toward lower viral burdens with IFN-β treatment, and a reciprocal trend with inhibition type I IFN signaling. It will be noted that wide interanimal variability in viral burden, together with the small sample size, undermined statistical power in these studies. Terminal quantification of tissue viral burden revealed a similar trend toward a lower viral load in IFN-β–treated macaques, but no trend toward increased viral load in macaques subjected to type I IFN receptor inhibition (Supplementary Figure 1A). Quantification of peripheral leukocyte (Figure 1C) and lymphocyte (Supplementary Figure 1B) concentrations and/or percentages provided no evidence that either therapy altered lymphocyte apoptosis during EBOV infection, something mirrored by analysis of apoptosis in lymph nodes using terminal deoxynucleotidyl transferase-dUTP nick end labeling assays (data not shown). It is acknowledged that the fact that lymph nodes were harvested at autopsy inhibits firm negative conclusions being drawn from the results of the latter assay.
Analysis of serum cytokine concentrations revealed a trend toward earlier and greater proinflammatory cytokine production in recipients of type I IFN receptor blockade, with a reciprocal trend in IFN-β–treated macaques (Supplementary Figure 1C). The long-term survivor in the IFN-β treatment group had unusually low serum cytokine concentrations, with levels undetectable in our assay. Similar trends were observed in biochemical measures of liver and kidney damage (Supplementary Figure 1D).
Control animals exhibited characteristic clinical features of EBOV-Z infection, including fever and cutaneous rashes, by the sixth day after challenge; animals treated with type I IFN receptor blockade exhibited such by the fifth day after challenge (Supplementary Table 1). Clinical symptoms appeared to be milder or delayed in IFN-β–treated animals, with a notable delay in the development of the severe rashes that are characteristic of EBOV-Z infection. Of note, no rash was observed in the long-term survivor. Three of 4 IFN-β–treated animals exhibited substantial lymphadenopathy, compared to controls. Two IFN-β–treated animals also showed evidence of brain congestion at necropsy.
Immunohistochemical analysis revealed no apparent differences in EBOV-Z antigen distribution or load in macaques succumbing on or before 10 days after infection—in either therapeutic group or controls. However, IFN-β–treated macaques that succumbed after day 10 revealed apparent clearing of EBOV from lymphoid organs including spleen (Figure 2A) and lymph nodes (data not shown), something confirmed by analysis of organ viral burden (Supplementary Figure 1A). Of note, the long-term survivor (previously reported, in part [45]) exhibited EBOV-Z staining in atypical, nontarget tissues including brain, lung, and pancreas (Figure 2B). In addition, infectious virus was recovered from cerebrospinal fluid (Supplementary Figure 1A). This macaque also developed neurological symptoms of disease beginning 17 days after infection, including hind leg stiffness and tremors. This prolonged survivor also exhibited pneumonia, with widespread infiltrates (Figure 2B). Immunostaining revealed numerous EBOV-Z–positive cells that appeared to be alveolar macrophages (Figure 2B, inset).
Figure 2.

Atypical, multiorgan foci of the Zaire species of Ebola virus (EBOV-Z) replication in the long-term survivor treated with interferon beta (IFN-β). A, Clearance of EBOV-Z antigen from the spleen. Comparative immunohistochemical analysis of EBOV-Z antigen in the spleen of the prolonged survivor (lower right panel), a control animal (upper left), and 3 macaques that received IFN-β and died on day 10 (upper right), day 12 (lower left), and day 29 (lower right). Intracellular EBOV-Z antigen is visible as brown staining. B, Multiorgan involvement. Histological examination of the lung (hematoxylin-and-eosin staining, upper left) reveals diffuse leukocytic infiltration. Immunohistochemical analysis of the lung (upper right) reveals EBOV-Z antigen in cells similar in appearance to alveolar macrophages (inset). EBOV-Z antigen is also evident in unusual anatomical sites such as the pancreas (lower left) and brain (lower right). Abbreviations: EBOV, Ebola virus; H&E, hematoxylin-and-eosin; rIFN, recombinant interferon; Tx, treatment.
Functional Genomic Analysis of IFN-β Treatment
In an attempt to gain further insight into the effects of manipulating the type I IFN axis during EBOV-Z infection, we performed comparative gene expression profiling of PBMCs in a subset of EBOV-Z–infected macaques, treated with IFN-β or not treated, analyzing the enrichment of gene ontology term representation in sets of genes whose expression profiles clustered by treatment group. Of interest, despite the lack of demonstrable treatment-specific differences in peripheral blood lymphocyte counts or lymphocyte apoptosis in lymphoid tissue, there was a major transcriptional effect of IFN-β therapy on genes important in adaptive immune regulation (Supplementary Table 2). While the nature of these studies precluded further functional analysis of EBOV-Z–specific T-cell and B-cell responses, these differential gene expression patterns suggest the hypothesis that an important therapeutic effect of IFN-β therapy in this infection occurs via bolstering of the vigor and/or breadth of the adaptive immune response—in this infection whose usual fatal course is marked by immune dysregulation and a failure to mount effective adaptive immune responses [10].
More Intensive IFN-β Therapy Provides No Additive Survival Benefit
We subsequently tested the utility of more intensive (daily, higher dose) IFN-β therapy for EBOV-Z infection. As shown in Supplementary Figure 2A, and in confirmation of the data described above, IFN-β treatment led to significant prolongation of the mean time to death (10.8 days) compared with controls (8.45 days; the intraexperimental control succumbed on day 11; P = .0019 by Wilcoxon rank-sum test). However, such treatment intensification did not provide any additive benefit (compare with Figure 1). As with lower dose IFN-β therapy, a trend toward lower plasma burden was observed in treated macaques (Supplementary Figure 2B). Of interest, a trend toward higher peripheral leukocyte and lymphocyte counts was observed in such animals (Supplementary Figure 2C and Supplementary Figure 2D), Notably, however, there was a marked trend toward increased concentrations of serum interleukin 6 with intensive IFN-β treatment (Supplementary Figure 2E), along with a parallel trend in biochemical measures of liver and kidney compromise (Supplementary Figure 2F), something that may provide insight into the lack of additive benefit of more intensive therapy (Supplementary Figure 1C).
IFN-β Therapy Has Clinical Benefit in MARV Infection
Given the prolongation of survival time in experimental EBOV-Z infection, we tested the efficacy of IFN-β in experimental MARV infection. Four macaques were infected with a lethal dose of MARV-Musoke. Three were treated with IFN-β (35 µg/kg) administered 1hour after infection, and thence daily for 14 days; 1 received saline as a control. Importantly, one of the macaques treated with IFN-β was fully protected from the MARV infection (Figure 3A), surviving past the study endpoint (day 28) and ultimately being euthanized 112 days after infection. At this time, immunohistological analysis revealed no evidence of MARV persistence (data not shown). The autopsy was otherwise notable for mild meningoencephalitis (data not shown). Omitting this survivor, the mean time to death for the IFN-β–treated group was 14.5 days, compared to 10.8 days in the control group when supplemented with 5 historical controls. When the survivor animal was censored from the analysis, the mean time to death between the experimental and control groups was statistically significantly different (Figure 3A, P = .0186, Wilcoxon rank-sum test). IFN-β therapy was associated with a marked trend toward suppression of viremia (Figure 3B) as well as with a trend toward lower peripheral leukocyte counts (Figure 3C), but an increased peripheral lymphocyte percentage (Figure 3D). As with EBOV infection, the IFN-β–treated long-term survivor of MARV exhibited marked suppression of systemic cytokine production (data not shown). Clinical data are presented in Supplementary Table 1. As opposed to intensive treatment of EBOV, intensive IFN-β treatment of MARV was not associated with a trend toward increases in serum biochemical markers of liver or kidney compromise (data not shown).
Figure 3.
Interferon beta (IFN-β) therapy provides a survival benefit in experimental Marburg virus (MARV) infection. A, Mortality. Rhesus macaques were infected with 1000 plaque-forming units of MARV-Musoke and treated with (i) recombinant human IFN-β at 35 µg/kg, 1 hour postinfection and every day until day 14 (n = 3; red line) or (ii) an equivalent volume of sterile saline at identical time points as a control (n = 1, supplemented with 5 historical controls: total n = 6; blue line). The intraexperimental control animal succumbed on day 11 postchallenge, while 1 of the 3 macaques that received IFN-β treatment was protected from infection and survived past the study endpoint. Omitting this surviving animal, the mean time to death for the IFN-β–treated experimental group was 14.5 days, whereas the mean time to death was 10.8 days in the control group (intraexperimental control died on day 11, P = .0186, Wilcoxon rank-sum test.) B, Viremia. Plasma MARV-Musoke kinetics in individual macaques treated with IFN-β (n = 3; red lines), and saline (n = 1; intraexperimental control; blue line). Mean plasma viremia for 6 available matched, untreated MARV-Musoke historical control animals is provided for reference (black dashed line). C, Peripheral blood leukocytes. Peripheral blood white blood cell kinetics in experimental macaques and available (n = 6) historical controls. D, Peripheral blood lymphocytes. Kinetic analysis of lymphocytes in macaques infected with MARV and treated daily with IFN-β (n = 3; red lines) or saline (n = 1; blue line). Abbreviations: PFU, plaque-forming units; WBC, white blood cell.
DISCUSSION
The results presented here represent the first in vivo evaluation of IFN-β as a treatment for filovirus infection. These data indicate that IFN-β may have therapeutic promise: early postexposure treatment significantly increased survival time in macaques infected with EBOV-Z and MARV. Its role, however, is likely to be as an adjunct to other agents—no EBOV-Z–infected macaque survived, and the small sample size precludes assessment of the statistical significance of the observed successful clearance of MARV infection with IFN-β treatment. Long-term survival of EBOV-Z infection treatment was also associated with spread of viral replication to organs not normally productively infected. Whether IFN-β has therapeutic potential after disease onset remains to be defined.
Functional genomic analysis revealed that, despite the high amounts of circulating IFN-α observed in EBOV-Z infection, IFN-β treatment altered the regulation of immune response genes, a result consonant with previous literature demonstrating quantitative and qualitative differences in biological activities among the type I IFNs. The relevant mechanism(s) of action of IFN-β remain underdetermined, however. Mechanistic analysis in these studies was inhibited by the low number of animals used, for ethical reasons. As IFN-β treatment was associated with a trend toward lower plasma and tissue viral burden, it is reasonable to postulate direct effects on viral replication as well as on the adaptive immune response. As for immunoregulatory effects on events thought to be important in filovirus pathogenesis, findings are even less clear. No evidence that IFN-β treatment of EBOV-Z infection altered lymphocyte apoptosis was obtained. Blunted systemic proinflammatory cytokine production was seen in both the long-term survivor of EBOV-Z and the macaque that cleared MARV, findings consistent with previous studies correlating lower levels of proinflammatory cytokines with increased survival or survival time [4, 43, 46, 47]. Whether this occurred secondary to lower viral load [33] and/or effects of IFN-β on proinflammatory cytokine production [48, 49] remains unclear.
Delayed inhibition of type I IFN signaling provided complementary insight. The shortened mean time to death observed with such treatment underscores the critical role of type I IFNs in host defense against filoviruses, something evident as well in mouse models [50]. Such treatment had no observable effect on lymphocyte apoptosis. Taken together with the IFN-β treatment data, these findings suggest that dysregulated type I IFN production does not play a role in this feature of pathogenesis. Concordant with IFN-β treatment results, type I IFN receptor blockade was associated with a trend toward increases in plasma (though not tissue) viral burden, as well as increases in serum concentrations of proinflammatory cytokines.
The pathogenesis of filovirus HF remains poorly understood. There are practical reasons for this. Natural disease is rare and occurs in places that hamper investigation. Furthermore, filoviruses need to be studied in high-containment settings. Finally, whereas human filovirus HF is closely mirrored in macaques, human disease does not appear to be mimicked in experimentally tractable small animal models. Despite mechanistic lacunae, the current studies suggest a potential therapeutic role for IFN-β, in combination with other therapeutic and/or preventive agents. That said, the fact that prolonged survival was associated with viral spread to tissues not usually noted to exhibit productive viral replication is cause for caution. Follow-up studies will be necessary to define both mechanism(s) of action of IFN-β as well as therapeutic utility in combination with other agents.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank P. Jahrling for facilitation of these studies.
Disclaimer. The opinions, interpretations, conclusions, and recommendations contained herein are those of the authors and are not necessarily endorsed by the US Army.
Financial support. This work was funded by JSTO-CBD/DTRA (Projects 19595 4.10009_06_RD_B and 19585 4.10037_07_RD_B to L. E. H.), National Institute of Allergy and Infectious Diseases (grants AI053539 to C. L. K. and AI057159 to L. E. H.), and Cincinnati Children's Hospital Medical Center (Translational Research Initiative Grant to C. L. K). L. M. S. was supported by an appointment to the Student Research Participation Program at the US Army Medical Research and Material Command administered by the Oak Ridge Institute for Science and Education, a Stanford URP Major Grant, and National Science Foundation Graduate Research Fellowship DGE-1147470.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Sanchez A, Kahn AS, ZS R, Nabel GL, Ksiazek TG, Peters CJ. Filoviridae. In: Knipe DM, Howley PM, editors. Fields virology. Philadelphia: Lippincott, Williams & Wilkins; 2001. pp. 1279–304. [Google Scholar]
- 2.Geisbert TW, Hensley LE. Ebola virus: new insights into disease aetiopathology and possible therapeutic interventions. Expert Rev Mol Med. 2004;6:1–24. doi: 10.1017/S1462399404008300. [DOI] [PubMed] [Google Scholar]
- 3.Bausch DG, Nichol ST, Muyembe-Tamfum JJ, et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N Engl J Med. 2006;355:909–19. doi: 10.1056/NEJMoa051465. [DOI] [PubMed] [Google Scholar]
- 4.Geisbert TW, Daddario-DiCaprio KM, Geisbert JB, et al. Marburg virus Angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J Infect Dis. 2007;196(uppl 2):S372–81. doi: 10.1086/520608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geisbert TW, Daddario-DiCaprio KM, Williams KJ, et al. Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J Virol. 2008;82:5664–8. doi: 10.1128/JVI.00456-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geisbert TW, Lee AC, Robbins M, et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet. 2010;375:1896–905. doi: 10.1016/S0140-6736(10)60357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. 2003;188:1618–29. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 8.Geisbert TW, Hensley LE, Jahrling PB, et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362:1953–8. doi: 10.1016/S0140-6736(03)15012-X. [DOI] [PubMed] [Google Scholar]
- 9.Geisbert TW, Hensley LE, Gibb TR, Steele KE, Jaax NK, Jahrling PB. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab Invest. 2000;80:171–86. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 10.Baize S, Leroy EM, Georges-Courbot MC, et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus–infected patients. Nat Med. 1999;5:423–6. doi: 10.1038/7422. [DOI] [PubMed] [Google Scholar]
- 11.Baize S, Leroy EM, Mavoungou E, Fisher-Hoch SP. Apoptosis in fatal Ebola infection. Does the virus toll the bell for immune system? Apoptosis. 2000;5:5–7. doi: 10.1023/a:1009657006550. [DOI] [PubMed] [Google Scholar]
- 12.Baskerville A, Satti A, Murphy FA, Simpson DI. Congo-Crimean haemorrhagic fever in Dubai: histopathological studies. J Clin Pathol. 1981;34:871–4. doi: 10.1136/jcp.34.8.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monath TP, Brinker KR, Chandler FW, Kemp GE, Cropp CB. Pathophysiologic correlations in a rhesus monkey model of yellow fever with special observations on the acute necrosis of B cell areas of lymphoid tissues. Am J Trop Med Hyg. 1981;30:431–43. doi: 10.4269/ajtmh.1981.30.431. [DOI] [PubMed] [Google Scholar]
- 14.Walker DH, McCormick JB, Johnson KM, et al. Pathologic and virologic study of fatal Lassa fever in man. Am J Pathol. 1982;107:349–56. [PMC free article] [PubMed] [Google Scholar]
- 15.Bray M, Geisbert TW. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol. 2005;37:1560–6. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 16.Basler CF, Amarasinghe GK. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res. 2009;29:511–20. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halfmann P, Neumann G, Kawaoka Y. The Ebolavirus VP24 protein blocks phosphorylation of p38 mitogen-activated protein kinase. J Infect Dis. 2011;204(uppl 3):S953–6. doi: 10.1093/infdis/jir325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta M, Mahanty S, Ahmed R, Rollin PE. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with Ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology. 2001;284:20–5. doi: 10.1006/viro.2001.0836. [DOI] [PubMed] [Google Scholar]
- 19.Villinger F, Rollin PE, Brar SS, et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis. 1999;179(suppl 1):S188–91. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 20.Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol Lett. 2002;80:169–79. doi: 10.1016/s0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 21.Masuyama T, Matsuo M, Ichimaru T, Ishii K, Tsuchiya K, Hamasaki Y. Possible contribution of interferon-alpha to febrile seizures in influenza. Pediatr Neurol. 2002;27:289–92. doi: 10.1016/s0887-8994(02)00452-6. [DOI] [PubMed] [Google Scholar]
- 22.Ronnblom LE, Perers A, Vallin HS, et al. Detection of serum interferon-alpha by dissociation-enhanced lanthanide fluoroimmunoassay. Studies of patients with acute viral and bacterial infections. Apmis. 1997;105:531–6. [PubMed] [Google Scholar]
- 23.Shiozawa S, Yoshikawa N, Iijima K, Negishi K. A sensitive radioimmunoassay for circulating alpha-interferon in the plasma of healthy children and patients with measles virus infection. Clin Exp Immunol. 1988;73:366–9. [PMC free article] [PubMed] [Google Scholar]
- 24.von Sydow M, Sonnerborg A, Gaines H, Strannegard O. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses. 1991;7:375–80. doi: 10.1089/aid.1991.7.375. [DOI] [PubMed] [Google Scholar]
- 25.Stylianou E, Aukrust P, Bendtzen K, Muller F, Froland SS. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)—possible immunosuppressive role of IFN-alpha in HIV infection. Clin Exp Immunol. 2000;119:479–85. doi: 10.1046/j.1365-2249.2000.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cranberry Township, PA: Three Rivers Pharmaceuticals:1–10; 2012. Full prescribing information for INFERGEN® (interferon alfacon-1). [Google Scholar]
- 27.Li L, Sherry B. IFN-alpha expression and antiviral effects are subtype and cell type specific in the cardiac response to viral infection. Virology. 2010;396:59–68. doi: 10.1016/j.virol.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–72. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Jahrling PB, Geisbert TW, Geisbert JB, et al. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J Infect Dis. 1999;179(suppl 1):S224–34. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 30.Jahrling PB, Geisbert J, Swearengen JR, et al. Passive immunization of Ebola virus–infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch Virol Suppl. 1996;11:135–40. doi: 10.1007/978-3-7091-7482-1_12. [DOI] [PubMed] [Google Scholar]
- 31.Smith DH, Johnson BK, Isaacson M, et al. Marburg-virus disease in Kenya. Lancet. 1982;1:816–20. doi: 10.1016/s0140-6736(82)91871-2. [DOI] [PubMed] [Google Scholar]
- 32.Geisbert TW, Geisbert JB, Leung A, et al. Single-injection vaccine protects nonhuman primates against infection with Marburg virus and three species of Ebola virus. J Virol. 2009;83:7296–304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geisbert TW, Hensley LE, Larsen T, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003;163:2347–70. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Council NR. Guide for the care and use of laboratory animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 35.Daddario-DiCaprio KM, Geisbert TW, Stroher U, et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet. 2006;367:1399–404. doi: 10.1016/S0140-6736(06)68546-2. [DOI] [PubMed] [Google Scholar]
- 36.Geisbert TW, Hensley LE, Geisbert JB, et al. Postexposure treatment of Marburg virus infection. Emerg Infect Dis. 2010;16:1119–22. doi: 10.3201/eid1607.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 38.Dillman JF, 3rd, Phillips CS. Comparison of non-human primate and human whole blood tissue gene expression profiles. Toxicol Sci. 2005;87:306–14. doi: 10.1093/toxsci/kfi243. [DOI] [PubMed] [Google Scholar]
- 39.Chismar JD, Mondala T, Fox HS, et al. Analysis of result variability from high-density oligonucleotide arrays comparing same-species and cross-species hybridizations. Biotechniques. 2002;33:516–8. doi: 10.2144/02333st01. 520, 522 passim. [DOI] [PubMed] [Google Scholar]
- 40.Rubins KH, Hensley LE, Wahl-Jensen V, et al. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 2007;8:R174. doi: 10.1186/gb-2007-8-8-r174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ochs MF, Peterson AJ, Kossenkov A, Bidaut G. Incorporation of gene ontology annotations to enhance microarray data analysis. Methods Mol Biol. 2007;377:243–54. doi: 10.1007/978-1-59745-390-5_15. [DOI] [PubMed] [Google Scholar]
- 43.Hensley LE, Stevens EL, Yan SB, et al. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J Infect Dis. 2007;196(suppl 2):S390–9. doi: 10.1086/520598. [DOI] [PubMed] [Google Scholar]
- 44.Jahrling PB. Filoviruses and Arenaviruses. In: Murray PR, Baron EJ, Pfaller M, Tenover FC, Yolken RH, editors. Manual of clinical microbiology. Washington, DC: ASM Press; 1999. pp. 1125–36. [Google Scholar]
- 45.Larsen T, Stevens EL, Davis KJ, et al. Pathologic findings associated with delayed death in nonhuman primates experimentally infected with Zaire Ebola virus. J Infect Dis. 2007;196(suppl 2):S323–8. doi: 10.1086/520589. [DOI] [PubMed] [Google Scholar]
- 46.Baize S, Leroy EM, Georges AJ, et al. Inflammatory responses in Ebola virus–infected patients. Clin Exp Immunol. 2002;128:163–8. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geisbert TW, Young HA, Jahrling PB, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163:2371–82. doi: 10.1016/S0002-9440(10)63592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jungo F, Dayer JM, Modoux C, Hyka N, Burger D. IFN-beta inhibits the ability of T lymphocytes to induce TNF-alpha and IL-1beta production in monocytes upon direct cell-cell contact. Cytokine. 2001;14:272–82. doi: 10.1006/cyto.2001.0884. [DOI] [PubMed] [Google Scholar]
- 49.Khabar KS, Young HA. Post-transcriptional control of the interferon system. Biochimie. 2007;89:761–9. doi: 10.1016/j.biochi.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raymond J, Bradfute S,, Bray M. Filovirus infection of STAT-1 knockout mice. J Infect Dis. 2011;204(suppl 3):S986–90. doi: 10.1093/infdis/jir335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.