

Abstract
Purpose
Despite intensive treatment regimens, overall survival for high risk neuroblastoma (HRNB) is still poor. This is in part due to an inability to cure the disease once a patient has reached clinical relapse. Identifying plasma biomarkers of active disease may provide a way of relapse monitoring in HRNB.
Experimental Design
In this study, we developed an integrated proteomic approach to identify plasma biomarkers for HRNB.
Results
We identified seven candidate biomarkers (SAA, APOA1, IL-6, EGF, MDC, sCD40L and Eotaxin) for HRNB. These biomarkers were then used to create a multivariate classifier of HRNB, which showed a specificity of 90% (95%CI, 73%, 98%), and a sensitivity of 81% (95%CI, 54%, 96%) for classifying HRNB in a training set. When evaluated on independent test samples, the classifier exhibited 86% accuracy (95% CI, 42%, 100%) of identifying diagnostic samples, and 86% accuracy (95% CI, 70%, 100%) of detecting post-diagnosis longitudinal samples that having active disease.
Conclusion and clinical relevance
Further validation of these biomarkers may improve patients‘ outcomes by developing a simple blood test for the detection of relapse prior to the development of clinically evident disease. Understanding the role of these biomarkers in immune surveillance of neuroblastoma may also provide a new direction of therapeutic strategies.
Keywords: Neuroblastoma, Relapse, Biomarkers, Cytokines, Chemokines
1 Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor of childhood, accounting for 8-10% of all cases. The complex biological and clinical features of neuroblastoma have allowed for the development of a risk-based model for staging and treatment of these patients [1,2]. This risk stratification system developed by the Children‘s Oncology Group classifies tumors as low, intermediate or high risk. Approximately 45 – 50% of patients will have high risk neuroblastoma (HRNB). Despite intensive treatment regimens including chemotherapy, surgery, radiation and biologic agents, the overall survival for HRNB is 30 – 40% [3,4]. This is in part due to an inability to cure the disease once a patient has reached clinical relapse [5].
Currently, monitoring for relapse in HRNB involves radiographic studies including MIBG, bone scan, and computed tomography (CT), as well as bone marrow aspirates and biopsies [6]. Ideally, a systemic marker of relapse would identify patients early, prior to clinical relapse; however, no such peripheral blood tumor marker for neuroblastoma exists. The tumor markers most commonly used now are the urinary catecholamines vanillylmandelic acid (VMA) and homovanillic acid (HVA). However, VMA and HVA have been shown to be elevated in only 50% of patients at the time of relapse [7]. Recently, circulating biomarkers for detecting relapse or prognosis in NB have been described, including circulating tumor cells [8], methylated-DCR2 [9], RASSF1A hypermethylation [10], and MYCN DNA [11]. However, the clinical value of these methods for detecting or monitoring for relapse in NB is still unclear. Since relapse patients have an active disease, we hypothesized that plasma biomarkers that are associated with active HRNB are also useful as biomarkers for monitoring for relapse. As a first step for a better detection of the relapse patients, the main goal of our study is to identify such biomarkers for active HRNB.
In NB, multiple cytokines and chemokines have been published in relationship to the immune-related effects caused by active tumor [12,13,14,15,16]. These proteins are also important for disease progression and metastasis in NB. One example of this is an increase in IL-6 levels in the peripheral blood in the presence of HRNB [12,17]. On the other hand, an acute phase protein, SAA, has also been described as associated with HRNB [18]. We, therefore, further hypothesized that combining the information of inflammatory proteins with cytokines and chemokines will allow us to detect HRNB from the low risk neuroblastoma (LRNB) and healthy subjects. To test this hypothesis, we developed a novel integrated approach, which consists of two complementary proteomic platforms for screening and identifying biomarkers and a bioinformatic classification approach for integrating the proteomic results. One platform was surface enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS), which has been used for global proteomic profiling and has the capability to identify abundant plasma proteins and inflammatory proteins [19,20]. The other is the Luminex Suspension Bead Array platform. This platform provides an ability to simultaneously identify multiple cytokines and chemokines that are present at low concentrations in the plasma proteome, which are often difficult to be detected by other proteomic technologies [21,22].
Using this unique integrated approach of detecting high- and low-level of plasma proteins, we identified seven peripheral blood biomarkers in HRNB by comparing them with the low risk neuroblastoma (LRNB) and pediatric healthy controls (HC). In this study, we further demonstrated that these candidate biomarkers can reliably detect active HRNB and have promise for relapse monitoring in HRNB patients.
2 Materials and Methods
2.1 Sample Collection
All the work described within this study was performed with the approval of the Baylor College of Medicine Institutional Review Board. Texas Children‘s Cancer Center has banked peripheral blood samples from patients with neuroblastoma and ganglioneuroblastoma. We retrospectively identified 35 plasma samples from HRNB patients, 20 samples from LRNB patients and 20 HC samples collected between 1995 and 2005 at the time of diagnosis (NB patients) or at the time of elective surgery. Risk categorization was defined by the Children‘s Oncology Group risk stratification system [4]. We also identified an additional 28 samples from 7 HRNB patients drawn at various times in their clinical course under a protocol for collection of blood samples in a longitudinal fashion at times of tumor evaluation. All peripheral blood samples were obtained as whole blood in heparin and separated into plasma. These were then aliquotted and stored at −80°C.
The normal pediatric controls were collected upon permission from the parent or guardian. Whole blood was obtained from otherwise well children undergoing elective surgery such as hernia repair or circumcision. They had no identified infectious process at the time of surgery. The blood was processed in the same manner as described above.
2.2 Proteomic Profiling
As a part of the integrated approach, we used SELDI-TOF MS to identify abundant plasma proteins and inflammatory proteins that were associated with active HRNB. In brief, the plasma samples were immunedepleted by the Proteoprep Blue Albumin and IgG Depletion kit (Sigma, St. Louis, MO) and then fractionated into six fractions. The proteins in each fraction were captured by CM10 arrays and analyzed by the ProteinChip System, Series 4000 Enterprise Edition (Bio-Rad, Hercules, CA). The detail of the method is described in the Supporting Information.
2.3 Protein Purification and Protein ID
Four plasma samples with high expression of the peak with the m/z value of 28,089 were pooled and used as the positive sample while 4 plasma samples with low or no expression of the peak were used as a negative control. The candidate protein at 28KDa was purified from gel electrophoresis and analyzed by nano-LC/MS/MS peptide sequencing analysis. (Nextgen Science, Ann Arbor, MI; See Supporting Information for details).
2.4 Enzyme-Linked Immunosorbent Assay
Serum Amyloid A (SAA) levels were determined using a human SAA enzyme-linked immunosorbent assay (ELISA) (Invitrogen, Carlsbad CA). APOA1 levels were determined using a human APOA1 ELISA (Cayman Chemical, Michigan). All samples were tested in duplicate, and the mean was used for the final sample value (See Supporting Information for details).
2.5 Luminex Suspension Bead Array
The second proteomic platform of our integrated approach is Luminex Suspension Bead Array (Luminex Corp., Austin, TX), which permits simultaneous analysis of numerous analytes in a single sample. Milliplex Human 39-Plex Cytokine (Millipore, Billerica, MA) antibody beads and reagents were used in this study for the following the 39 human cytokines and chemokines: GM-CSF, G-CSF, IFNg, IL-1a, IL-1ra, IL-1b, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-12p40, IL-13, IL-15, IL-17, MCP-1, MCP-3, MDC, TNFa, TNFb, TGFa, Eotaxin, IFNa2, IP-10, MIP-1a, MIP-1b, EGF, FGF-2, Flt-3 Ligand, Fractalkine, GRO, VEGF, sCD40L, and sIL-2Ra. The assay was performed according to the manufacturer‘s protocol. Plates were analyzed using the Bio-Plex 200 system and the Bio-Plex manager software 4.1 (Bio-Rad). Fifty beads per analyte were collected and a timeout of 60 seconds was set while analyzing the plate. Cytokine concentrations were calculated based on standard curve data using 5-parameter logistic-fitting method in Bio-Plex manager software 4.1. We repeated the experiment after three weeks using plasma aliquots prepared at the same time with the same number of freeze/thaw cycles as the first plate. The average values of the replicates were used in the final analysis.
2.6 Multivariate Classifier
The last step of our integrated approach is to construct a HRNB classifier based on the proteomic results. Protein concentrations between two groups were compared using the nonparametric Mann-Whitney U-statistics, which were the area under the empirical ROC curves (AUC). Candidate biomarkers were chosen for classification if their concentrations were significantly different between the two groups with a p-value at or smaller than 0.05.
To create a multivariate classifier, the subjects were split into training data and the validation data. The data from the significant protein biomarkers from the two platforms were combined as a summation score. If the AUC value was less than 0.5 for a protein, the concentrations were transformed with a minus sign to ensure that higher values of protein concentrations were associated with higher probability of disease. The classification score is defined as: SAA + IL-6 – APO –MDC – sCD40L– EGF – Eotaxin. Before calculating the scores, the protein concentrations were normalized to their means and standard deviation. Such normalization would not change the AUC value of the individual protein. The empirical ROC curve was then calculated for the classification score, and a cutoff for classification was chosen to achieve the optimal sensitivity and the specificity rates in the training set. The classifier was then tested on the validation dataset, which contained both diagnostic and longitudinal samples.
Other statistical methods used in this study were described in the Supporting Information.
3 Results
3.1 Patient Characteristics
Demographic and staging characteristics for the patient populations used in each portion of this report are listed in Table 1.
Table 1.
Characteristics of the patients and individuals used in the proteomic profiling and cytokine bead array Investigations.
| Proteomic Profiling | Cytokine Bead Array | |||||
|---|---|---|---|---|---|---|
| High Risk |
Low Risk |
Healthy Control |
High Risk |
Low Risk |
Healthy Control |
|
| Number of samples |
35 | 20 | 20 | 18 | 15 | 15 |
| Median age (months) |
29 | 14 | 54 | 30 | 13 | 48 |
| Average Age |
40 | 21 | 68 | 43 | 22 | 48 |
| Range | 12–159 | 0.5–165 | 12–204 | 12–153 | 0.5–165 | 12–96 |
| Gender | ||||||
| Female | 15 (43%) | 7 (35%) | 6 (30%) | 8 (44%) | 5 (33%) | 4 (27%) |
| Male | 20 (57%) | 13 (65%) | 14 (70%) | 10 (56%) | 10 (67%) | 11 (73%) |
| INSS Stage | ||||||
| 1 | 0 (0%) | 9 (45%) | 0 (0%) | 5 (33%) | ||
| 2 | 1 (3%) | 7 (35%) | 0 (0%) | 7 (47%) | ||
| 3 | 5 (14%) | 0 (0%) | 2 (11%) | 0 (0%) | ||
| 4 | 29 (83%) | 0 (0%) | 16 (89%) | 0 (0%) | ||
| 4S | 0 (0%) | 4 (20%) | 0 (%) | 3 (20%) | ||
3.2 Proteomic Profiling
In order to detect patients with active HRNB, we developed an integrated proteomic approach to identify circulating biomarkers that are associated with HRNB. The integrated approach is composed of three separate but complementary steps: proteomic profiling, bead array analysis, and a classification model. For the proteomic profiling, we first immunodepleted albumin and IgG from the plasma samples and performed SELDI-TOF MS on the depleted plasma from three different groups of samples, i.e. HRNB, LRNB, and HC. Using this strategy, we identified totally 1,153 ion peaks in the proteomic profiling. These ion peaks were used to perform a comparative analysis of the three groups. First, unsupervised hierarchical clustering of the HRNB and LRNB or HC groups using the complete set of peaks showed that a majority of the samples from these three groups were clustered distinctively from each other, suggesting that the plasma proteins of the patient samples contain information of classifying HRNB (Fig. S1 in the Supporting Information).
To identify specific biomarkers for HRNB, we first selected the ion peaks that can classify HRNB from LRNB. We found that four ion peaks together had a specificity of 0.914 and sensitivity of 0.6 of identifying HRNB (Supporting Information). Two of the four peaks appeared to be peak variants from one protein and the other two appeared to be peak variants from another protein based on their m/z values (Table 2A). One of the peak clusters had m/z values between 11,544 and 11,711. These values correspond with the previously described protein, serum amyloid protein A (SAA) [18,23]. The second set of peaks had m/z values between 4,964 and 4,966. These low molecular weight peaks have not been previously reported. Next, we identified ion peaks that can classify HRNB from HC using a similar classification approach. We found that six ion peaks together gave a sensitivity of 0.94 and a specificity of 0.9 for classifying HRNB from HC samples (Supporting Information). Within these six ion peaks, a cluster of three peaks appeared to be variants of a single protein with m/z values of 27,962, 28,093, and 28,900 (Table 2B). This cluster of peaks in the plasma of the HC was higher than the one in plasma of the HRNB patients. This suggests a loss of expression of the protein in the presence of neuroblastoma. The other peaks used in this classifier formed another cluster; however, they were not distinct enough to isolate and identify as an individual protein after spectra review.
Table 2.
Informative peaks identified in the proteomic profiling when comparing A. HRNB and LRNB samples and B. HRNB and HC samples.
| A. | ||||
|---|---|---|---|---|
| Average Relative Peak Intensity |
Fold Change | |||
| p-value | m/z | High | Low | High/Low |
| 0.000046 | 11,544 | 3.0 | 1.0 | 3.0 |
| 0.000054 | 11,711 | 3.4 | 1.0 | 3.4 |
| 0.0001541 | 4,964 | 14.1 | 3.2 | 4.4 |
| 0.0004939 | 4,966 | 19.2 | 4.0 | 4.8 |
| B. | ||||
|---|---|---|---|---|
| Average Relative Peak Intensity |
Fold Change | |||
| p-value | m/z | High | Healthy | High/Healthy |
| < 0.0000001 | 27,962 | 3.3 | 10.8 | 0.31 |
| < 0.0000001 | 28,093 | 5.1 | 17.9 | 0.29 |
| < 0.0000001 | 28,900 | 1.9 | 4.9 | 0.39 |
| < 0.0000001 | 17,328 | 2.9 | 8.8 | 0.33 |
| < 0.0000001 | 17,212 | 1.6 | 3.9 | 0.41 |
| < 0.0000001 | 17,263 | 1.5 | 4.2 | 0.36 |
3.3 Protein Identification and Confirmation
Based on these observations, we selected peak clusters of m/z 28,093 and m/z 4,965 for protein identification. Pooled plasma samples from samples expressing high peaks of the m/z 27,962, m/z 28,093, and m/z 28,900 were first analyzed on protein electrophoresis gels where band and spots at the approximate molecular weight were purified and the identity of the protein in the gel slice was identified by LC/MS/MS. The mass spectrometry analysis scored 19 unique MS/MS spectra with Mascot ion score ranged from 29 to 101. Sixteen unique peptide sequences showed homology to apolipoprotein A1 precursor (APOA1) (Fig. S2 in the Supporting Information). The protein coverage was 60%. Attempts to identify the protein at the m/z 4,965 cluster have not been successful to date, suggesting that these low molecular weight peaks may not constitute a protein or peptide.
To confirm the results of the proteomic profiling, we used ELISAs to measure the levels of SAA and APOA1 in raw plasma samples from the same set of patients. SAA levels were significantly higher in the HRNB patients (mean = 41.7 ug/ml) as compared to either the LRNB patients (mean = 7.2 ug/ml, p < 1×10−7) or the HC (mean = 11.6 ug/ml, p = 1×10−5, Fig. 1A). For APOA1, HRNB plasma levels (mean = 93.9 ug/ml) were significantly lower than those of the normal controls (mean = 138 ug/ml; p < 1×10−5) as well as those of the LRNB patients (mean = 136.5 ug/ml; p < 0.001, Fig. 1B). In both instances, no significant differences were observed between the LRNB and HC samples.
Figure 1.
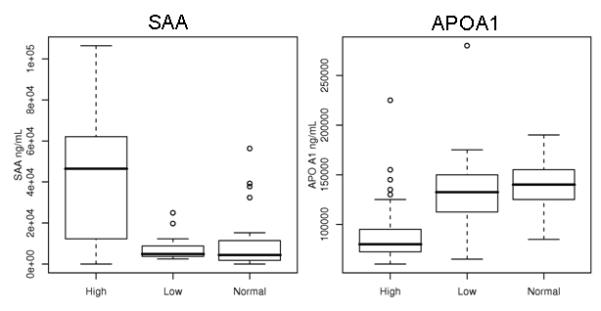
Box plots showing the plasma concentrations (mg/ml) of A. SAA and B. APOA1 in High (HRNB), Low (LRNB) and Normal (HC) groups. The box represents values between the 25th and 75th percentile with the horizontal line indicating the median value. ° indicates outlier values.
3.4 Identification of Cytokines and Chemokines Biomarkers
The second step of the integrated approach is to use a Luminex Bead Array system to identify cytokines and chemokines that were associated with HRNB. Due to sample limitation, a subset of the plasma samples was selected for this analysis (See Table 1 for the patient characteristics). To simplify the classification problem, we further combined the LRNB and HC groups to create a single control group (CC, Combined Control) to compare with HRNB in order to identify biomarkers that can distinguish HRNB from both sets of controls. Furthermore, identification of LRNB has a very limited clinical value. Using AUC analysis, five proteins (IL-6, EGF, MDC, sCD40L and eotaxin) were identified as being able to significantly classify between the HRNB and the CC groups (p < 0.05, Fig. 2). We also confirmed that SAA and APOA1 were significant of classifying between the HRNB and CC groups under the AUC analysis (p < 0.05, Fig. 2). Among all the seven proteins tested, APOA1 exhibited the highest AUC value (AUC = 0.84). These biomarkers exhibited various degrees of correlation among each other with a low average correlation of 0.19 (r = −0.32 to 0.78, Table S1 in the Supporting Information).
Figure 2.

Receiver Operating Characteristic (ROC) analysis of the seven proteins used in the HRNB classifier for the training set samples. A higher Area Under the ROC Curve (AUC) value indicates increased accuracy for a given marker to predict the presence of HRNB. The p-value of the each of the classification is also indicated. For the biomarkers (Eotaxin, MDC, SCD40L and EGF) that were lower in the high-risk neuroblastoma group, the concentration values multiplied by −1 were used to generate the ROC plots.
3.5 Development of the HRNB Classifier
The final step of our integrated approach is to develop a multivariate classifier for HRNB by combining the information of the seven plasma proteins identified from the two proteomic platforms. We first standardized the concentration values across all seven proteins and then developed a classifier by summing their values as described in Materials and Methods. The performance of the classifier for classifying the HRNB and CC samples was statistically significant (p = 3.6×10−8) with an AUC value of 0.94 (exact 95%CI, 0.88, 1.0). Using an optimized cutoff, the classifier has a specificity of 90% (exact 95%CI, 73%, 98%), and a sensitivity of 81% (exact 95%CI, 54%, 96%) for classifying HRNB in the training set. 87% of subjects were correctly classified (exact 95%CI, 74%, 95%).
We next evaluated the classifier with an independent test set, which contained both diagnostic and longitudinal samples from patients known to have HRNB. Some of these longitudinal samples were collected prior to the clinical detection of relapse. Table 3 shows the patient characteristics and the prediction results for each of the 7 patients. The expression patterns of the biomarkers in these patients are illustrated in Supporting Figure S3. The HRNB classifier correctly predicted 6 of 7 diagnostic samples as having active disease (85.7% accuracy, 95% exact CI, 42%, 100%). 18 of the 21 post-diagnosis longitudinal samples were also correctly predicted based on their clinical features (85.7% accuracy, 95% exact CI, 70%, 100%). Together, these results suggest that the HRNB classifier is potentially useful for monitoring HRNB disease progression and relapse.
Table 3.
Longitudinal samples used for the independent validation of the HRNB classifier.
| Pt No. |
Pt Info | Time Point | Disease Status |
Sites of Disease |
Prediction | Correct? |
|---|---|---|---|---|---|---|
| 1 | M, 12 mo, MNA | |||||
| Diagnosis | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Present | ST, B | Disease | Yes | ||
| Progression | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Not Present | Disease | No | |||
| Relapse | Present | ST, B, BM | Disease | Yes | ||
|
| ||||||
| 2 | F, 12 mo, MNA | |||||
| Diagnosis | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Not Present | No Disease | Yes | |||
| Off Therapy | Not Present | No Disease | Yes | |||
|
| ||||||
| 3 | F, 52 mo, MA | |||||
| Diagnosis | Present | ST, B | Disease | Yes | ||
| On Therapy | Not Present | No Disease | Yes | |||
| On Therapy | Not Present | No Disease | Yes | |||
|
| ||||||
| 4 | M, 24 mo, MA | |||||
| Diagnosis | Present | ST1 | Disease | Yes | ||
| On Therapy | Not Present | No Disease | Yes | |||
| On Therapy | Not Present | No Disease | Yes | |||
| Off Therapy | Not Present | No Disease | Yes | |||
|
| ||||||
| 5 | F, 22 mo, MA | |||||
| Diagnosis | Present | ST1 | Disease | Yes | ||
| Relapse | Present | ST | Disease | Yes | ||
| Progression | Present | ST, B, BM | Disease | Yes | ||
|
| ||||||
| 6 | M, 14 mo, MA | |||||
| Diagnosis | Present | ST, B2 | No Disease | No | ||
| On Therapy | Not Present | No Disease | Yes | |||
| On Therapy | Not Present | No Disease | Yes | |||
| Off Therapy | Not Present | Disease | No | |||
|
| ||||||
| 7 | M, 55 mo, MA | |||||
| Diagnosis | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Present | ST, B, BM | Disease | Yes | ||
| On Therapy | Present | ST, BM | Disease | Yes | ||
| On Therapy | Present | BM | Disease | Yes | ||
| On Therapy | Present | ST, BM | No Disease | No | ||
Pt info: M=male, F=female; mo=age in months at diagnosis; MA=MYCN amplified; MNA=MYCN not amplified. Sites of Disease: ST=soft tissue; B=Bone; BM=Bone Marrow.
Stage 3
No Primary Tumor.
4 Discussion
Identification of circulating biomarkers for HRNB is one of the most important but challenging areas for improving the clinical management of this deadly disease. Using the SELDI-TOF MS, He et al showed that a unique protein signature existed in the peripheral blood samples from a small set of HRNB patients [24]. Using a larger number of patient samples, Combaret et al identified SAA as a protein present in the face of metastatic disease [18]. We improved their methods by using the extra steps of sample immunodepletion and fractionation, as well as using samples from healthy children along with samples from those with LRNB. Our results validated their finding that SAA is indeed a biomarker of high risk disease, as well as a biomarker which is present at the time of relapse. Additionally, we have identified another candidate plasma biomarker, APOA1, which was not found in their study.
SAA is an acute phase reactant, a marker of inflammation and infection [25]. Its presence in the face of high risk disease and again at relapse is likely due to the body‘s response to neuroblastoma. SAA has been described by others in NB, as well as in other cancers including osteosarcoma, prostate, ovarian, and melanoma [23,26,27,28,29]. Similarly, APOA1 is an abundant protein in plasma that serves as the major protein component of high-density lipoprotein (HDL) and a key element of the reverse cholesterol transport pathway, a process that protects against the development of atherosclerosis [30]. APOA1 is has been described previously with low levels being a marker for disease in ovarian, pancreatic, and breast cancer [31,32,33]. However, the identification of APOA1 as a candidate biomarker for HRNB is novel.
The Luminex Suspension Bead Array platform is a new and useful tool for screening a variety of biomarkers using a small amount of sample. The platform has been used in to assess potential serum biomarkers in breast cancer and to assess potential biomarkers in the saliva of patients with oral cancer [22,21]. Of the five cytokines and chemokines identified as differentially abundant in HRNB using the Luminex platform, many of them are known to play a role in NB. IL-6 has been previously described as being increased in the peripheral blood of patients at the time of diagnosis [12]. IL-6 has been implicated in changing the boney microenvironment in HRNB, however the elevated plasma levels are more likely due to an acute inflammatory response [17,34]. CD40L is a component of multiple immune responses, including dendritic cell maturation and antitumor activity [16]. CD40L has been shown in a mouse model to confer a protective effect by decreasing tumor size when directly injected in to a mouse with active NB [35]. Lower CD40L levels in the patients with active HRNB would be consistent with this finding. MDC is produced by dendritic cells and may be involved in host anti-tumor response [36]. This has not been evaluated in NB. Eotaxin-1 (CCL11) is a chemokine that recruits eosinophils into tissues. It has also not been previously implicated in neuroblastoma. Its role in tumor immune surveillance was described by Simson et al [37]. This group found an increase in methylcholanthrene induced fibrosarcomas in CCL11 deficient mice. As with CD40L, a low level of circulating eotaxin-1 may be associated with HRNB‘s active presence. EGF has also been associated with the growth of NB [13]. We postulate that the low circulating plasma levels may be due to excessive binding at the tumor site. Our findings warrant further work to discover the roles of these chemokines in tumor immune surveillance and growth.
The HRNB classifier created using all seven of the biomarkers has promise as a tool for detecting relapse via peripheral blood. Of the 28 samples from 7 patients, 24 predictions were correct, including all of the samples at relapse and many samples during the treatment regimens but prior to the relapse. To explore the false predictions further, 2 of the 4 false predictions occurred in patient 6. One was at diagnosis (false negative) and one was at 3 months after completion of therapy (false positive). Interestingly, this patient had an unusual presentation consisting of HRNB with no primary mass and a single boney metastatic lesion with a surrounding soft tissue component. This suggests that this case may be an atypical form of HRNB that resulted in lack of detection using our model.
Another false prediction occurred in patient 7 who had refractory disease and was being treated with his second salvage regimen. This patient had multiple correct predictions prior to this misclassification. The cause for this is unclear, but may reflect an evolution of the tumor‘s interaction with its host as it overcame the effects of each chemotherapy regimen. The fourth false prediction was patient 1 who was predicted to have disease when the radiologic and bone marrow exams showed no disease. Upon further review we found that this patient relapsed clinically weeks after the sample was drawn. This intriguing information suggests that while no disease could be found using standard current detection techniques, it is possible that our model detected a subtle level of disease-related changes of our biomarkers in this patient that was consistent with active disease. If we control for these observations, our prediction shows promising potential for relapse monitoring in HRNB.
As mentioned above, since both SAA and APOA1 have been described in other cancers, we conclude that these two biomarkers are not HRNB-specific. However, because of their relationship with the acute phase response, they can be used as sensitive reporters of how the body reacted to active and relapsed cancers. To test if the other biomarkers used in the classifier are more tumor specific, we have expanded our biomarker analysis on a series of plasma samples collected from osteosarcoma patients at diagnosis and control plasma samples from children with noncancerous diseases. Among the five cytokines, two of them (EGF and sCD40L) were significantly increased in the osteosarcoma samples relative to the control samples (Table S2). These results are opposite to the lower expression of these two cytokines in HRNB when compared to the control. The expressions of two biomarkers (Eotaxin and IL-6) in the osteosarcoma samples were in the same directions as in the HRNB samples, but the differences between the osteosarcoma and control samples were not statistically significant (p > 0.05, Table S2). Together, our results suggest that some of the biomarkers used in the classifier have certain tumor-specificity. The cytokine biomarkers seem to be more HRNB-specific than the two plasma proteins.
It is also important to recognize that the main goal of the current study is to identify a set of plasma biomarkers that are associated with the active HRNB disease, so that one can further test if they can be used in monitoring disease progression and relapse. These biomarkers are used only after initial diagnosis is made, i.e. patients have already diagnosed with HRNB. They are not intended to be used as diagnostic markers to screen general pediatric population for the presence of HRNB. Therefore, the issue of tumor specificity is not as relevant or severe as those biomarkers that are used for initial diagnosis or early detection. In addition, the development of second malignancies in HRNB is a very rare event. Long-term follow-up data for the landmark trial that defined what has become standard of care for HRNB was published in 2010. There were 539 patients enrolled; of the 405 first events reported, 378 were relapse or progression of disease, as compared to 4 secondary malignancies [38]. The utility of the classifier in distinguish relapses versus second malignancies in HRNB patients will need to be properly designed and tested in future studies with patients containing both types of tumors. Lastly, tumor progression in neuroblastoma is tightly linked to body‘s immune system [12,17]. Identification of biomarkers that are related to the immune response is biologically significant in this tumor. The classifier can be used to augment the conventional clinical methods for relapse detection in HRNB. Patients who show positive results in our assay may warrant further clinical confirmation, such as bone scan. Thus, despite the non-tumor specific nature of some of the biomarkers identified in this study, we believe that our finding is a significant step towards effective relapse detection in HRNB.
In summary, our study demonstrated that the integrated proteomic approach was able to identify plasma biomarkers that are associated with HRNB. We also provide strong evidence that the HRNB classifier was able to detect longitudinal samples from relapse NB patients. Despite the encouraging results shown in this study, we realize that the utility of the HRNB classifier need to be evaluated independently in many more patients. With a larger sample size, a sub-analysis of utility in various high risk presentations e.g. MYCN amplification or the presence of metastatic disease, would also be possible. Our results also warrant more studies to test if the classifier can detect the active NB disease before clinical relapse. Using a stepwise removal procedure of the biomarkers, we found that the performance of the classifier remained the same in the training set until only four biomarkers (SAA, APOA1, MDC and IL6) were left in the model. This suggests that we may be able to further reduce the number of proteins in the classifier without sacrificing the performance. In addition, since the biomarkers identified from both the SELDI-TOF MS and Luminex platforms are used in the reduced model, it suggests that integrating the biomarker information from the two platforms is indeed important for the HRNB classification. After the model is validated, the development of a multiplex ELISA-based blood test would allow this assay to be used in multiple facilities and improve the clinical management of this deadly childhood cancer.
Supplementary Material
Statement of clinical relevance.
HRNB continues to be a devastating disease in the relapse setting. This study uses a novel integrated proteomic approach to develop a classifier that is able to detect HRNB with a high accuracy and show promise in detecting or monitoring for relapse. Since our findings were generated in immunoassays, this would permit a rapid development of a blood test for future clinical uses. This biomarker-based test may add to the current methods used to evaluate these patients e.g. bone marrow biopsies, MIBG and CT scans. Further study of these biomarkers may also allow for a better detection of relapse in some patients prior to the development of clinically evident disease and, ultimately, increasing their survival chances. In addition, understanding the role of these biomarkers in inflammatory response and immune surveillance in the patient may lead to the development of a new therapeutic strategy.
Acknowledgments
The authors would like to thank Susan Burlingame with the Neuroblastoma Program at Texas Children‘s Cancer and Hematology Center (TXCH) for her help in identifying and preparing patient samples; Alexander Yu with TXCH for his help on data analysis and figure preparation; Myra Custorio at the Proteomics Core of Dan L. Duncan Cancer Center of Baylor College of Medicine for her technical help in the Luminex assays and analyzing the plates, and Carl Allen for his help in the Luminex experiment. Grant support was provided by the National Cancer Institute grant NIH-NCI 1 K12 CA90433-04.
Abbreviations
- NB
neuroblastoma
- HRNB
high risk neuroblastoma
- LRNB
low risk neuroblastoma
- HC
healthy control
- CC
combined control
- SELDI-TOF MS
Surface Enhanced Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry
Footnotes
The authors have declared no conflict of interest. TAD was a former employee of Ciphergen Biosystems, Inc and Vermillion, Inc.
5 References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Lancet. 2007 Jun 23;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, Black CT, Gerbing R, Haase GM, Stram DO, Swift P, Lukens JN. J. Clin. Oncol. 1998;16:1256–1264. doi: 10.1200/JCO.1998.16.4.1256. [DOI] [PubMed] [Google Scholar]
- 3.Lau L. Pediatr. Hematol. Oncol. 2002;19:79–89. doi: 10.1080/08880010252825669. [DOI] [PubMed] [Google Scholar]
- 4.Maris JM. Curr. Opin. Pediatr. 2005;17:7–13. doi: 10.1097/01.mop.0000150631.60571.89. [DOI] [PubMed] [Google Scholar]
- 5.Lau L, Tai D, Weitzman S, Grant R, Baruchel S, Malkin D. J. Pediatr. Hematol. Oncol. 2004;26:227–232. doi: 10.1097/00043426-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Brodeur GM, Maris JM. In: Neuroblastoma in Principles and practice of pediatric oncology. Pizzo P, Poplack D, editors. Lippincott, Williams and Wilkins; Philadelphia: 2006. pp. 993–1070. [Google Scholar]
- 7.Simon R, Radmacher MD, Dobbin K, McShane LM. J. Natl. Cancer Inst. 2003 Jan 1;95:14–18. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 8.Kuroda T, Morikawa N, Matsuoka K, Fujino A, Honna T, Nakagawa A, Kumagai M, Masaki H, Saeki M. J. Pediatr. Surg. 2008;43:2182–2185. doi: 10.1016/j.jpedsurg.2008.08.046. [DOI] [PubMed] [Google Scholar]
- 9.Yagyu S, Gotoh T, Iehara T, Miyachi M, Katsumi Y, Tsubai-Shimizu S, Kikuchi K, Tamura S, Tsuchiya K, Imamura T, Misawa-Furihata A, Sugimoto T, Sawada T, Hosoi H. Clin. Cancer Res. 2008 Nov 1;14:7011–7019. doi: 10.1158/1078-0432.CCR-08-1249. [DOI] [PubMed] [Google Scholar]
- 10.Misawa A, Tanaka S, Yagyu S, Tsuchiya K, Iehara T, Sugimoto T, Hosoi H. Br. J. Cancer. 2009 Jan 27;100:399–404. doi: 10.1038/sj.bjc.6604887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Combaret V, Audoynaud C, Iacono I, Favrot MC, Schell M, Bergeron C, Puisieux A. Cancer Res. 2002 Jul 1;62:3646–3648. [PubMed] [Google Scholar]
- 12.Egler RA, Burlingame SM, Nuchtern JG, Russell HV. Clin. Cancer Res. 2008 Nov 1;14:7028–7034. doi: 10.1158/1078-0432.CCR-07-5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Cancer Res. 2005 Nov 1;65:9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- 14.Raffaghello L, Cocco C, Corrias MV, Airoldi I, Pistoia V. Semin. Cancer Biol. 2009;19:97–102. doi: 10.1016/j.semcancer.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Russell HV, Hicks J, Okcu MF, Nuchtern JG. J. Pediatr. Surg. 2004;39:1506–1511. doi: 10.1016/j.jpedsurg.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Walker SR, Redlinger RE, Jr., Barksdale EM., Jr. J. Pediatr. Surg. 2005;40:244–249. doi: 10.1016/j.jpedsurg.2004.09.050. [DOI] [PubMed] [Google Scholar]
- 17.Ara T, Song L, Shimada H, Keshelava N, Russell HV, Metelitsa LS, Groshen SG, Seeger RC, DeClerck YA. Cancer Res. 2009 Jan 1;69:329–337. doi: 10.1158/0008-5472.CAN-08-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Combaret V, Bergeron C, Brejon S, Iacono I, Perol D, Negrier S, Puisieux A. Cancer Lett. 2005 Oct 18;228:91–96. doi: 10.1016/j.canlet.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 19.Liotta LA, Ferrari M, Petricoin E. Nature. 2003 Oct 30;425:905. doi: 10.1038/425905a. [DOI] [PubMed] [Google Scholar]
- 20.Petricoin EF, Liotta LA. Curr. Opin. Biotechnol. 2004;15:24–30. doi: 10.1016/j.copbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 21.rellano-Garcia ME, Hu S, Wang J, Henson B, Zhou H, Chia D, Wong DT. Oral Dis. 2008;14:705–712. doi: 10.1111/j.1601-0825.2008.01488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nolen BM, Marks JR, Ta’san S, Rand A, Luong TM, Wang Y, Blackwell K, Lokshin AE. Breast Cancer Res. 2008;10:R45. doi: 10.1186/bcr2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Dang TA, Shen J, Perlaky L, Hicks J, Murray J, Meyer W, Chintagumpala M, Lau CC, Man TK. Proteomics. 2006;6:3426–3435. doi: 10.1002/pmic.200500472. [DOI] [PubMed] [Google Scholar]
- 24.He QY, Zhu R, Ren Y, Tam PK, Chiu JF. J. Cell Biochem. 2005 May 1;95:165–172. doi: 10.1002/jcb.20417. [DOI] [PubMed] [Google Scholar]
- 25.Malle E, Sodin-Semrl S, Kovacevic A. Cell Mol. Life Sci. 2009;66:9–26. doi: 10.1007/s00018-008-8321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Findeisen P, Zapatka M, Peccerella T, Matzk H, Neumaier M, Schadendorf D, Ugurel S. J. Clin. Oncol. 2009 May 1;27:2199–2208. doi: 10.1200/JCO.2008.18.0554. [DOI] [PubMed] [Google Scholar]
- 27.Le L, Chi K, Tyldesley S, Flibotte S, Diamond DL, Kuzyk MA, Sadar MD. Clin. Chem. 2005;51:695–707. doi: 10.1373/clinchem.2004.041087. [DOI] [PubMed] [Google Scholar]
- 28.Moshkovskii SA, Serebryakova MV, Kuteykin-Teplyakov KB, Tikhonova OV, Goufman EI, Zgoda VG, Taranets IN, Makarov OV, Archakov AI. Proteomics. 2005;5:3790–3797. doi: 10.1002/pmic.200401205. [DOI] [PubMed] [Google Scholar]
- 29.Sandoval JA, Turner KE, Hoelz DJ, Rescorla FJ, Hickey RJ, Malkas LH. J. Surg. Res. 2007;142:268–274. doi: 10.1016/j.jss.2007.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fielding CJ, Fielding PE. J. Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- 31.Ehmann M, Felix K, Hartmann D, Schnolzer M, Nees M, Vorderwulbecke S, Bogumil R, Buchler MW, Friess H. Pancreas. 2007;34:205–214. doi: 10.1097/01.mpa.0000250128.57026.b2. [DOI] [PubMed] [Google Scholar]
- 32.Goncalves A, Esterni B, Bertucci F, Sauvan R, Chabannon C, Cubizolles M, Bardou VJ, Houvenaegel G, Jacquemier J, Granjeaud S, Meng XY, Fung ET, Birnbaum D, Maraninchi D, Viens P, Borg JP. Oncogene. 2006 Feb 16;25:981–989. doi: 10.1038/sj.onc.1209131. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z, Bast RC, Jr., Yu Y, Li J, Sokoll LJ, Rai AJ, Rosenzweig JM, Cameron B, Wang YY, Meng XY, Berchuck A, van Haaften-Day C, Hacker NF, de Bruijn HW, van der Zee AG, Jacobs IJ, Fung ET, Chan DW. Cancer Res. 2004 Aug 15;64:5882–5890. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 34.Sohara Y, Shimada H, Minkin C, Erdreich-Epstein A, Nolta JA, DeClerck YA. Cancer Res. 2005 Feb 15;65:1129–1135. doi: 10.1158/0008-5472.CAN-04-2853. [DOI] [PubMed] [Google Scholar]
- 35.Grossmann ME, Brown MP, Brenner MK. Hum. Gene Ther. 1997 Nov 1;8:1935–1943. doi: 10.1089/hum.1997.8.16-1935. [DOI] [PubMed] [Google Scholar]
- 36.Ishida T, Ueda R. Cancer Sci. 2006;97:1139–1146. doi: 10.1111/j.1349-7006.2006.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simson L, Ellyard JI, Dent LA, Matthaei KI, Rothenberg ME, Foster PS, Smyth MJ, Parish CR. J. Immunol. 2007 Apr 1;178:4222–4229. doi: 10.4049/jimmunol.178.7.4222. [DOI] [PubMed] [Google Scholar]
- 38.Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG. J. Clin. Oncol. 2009 Mar 1;27:1007–1013. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.