

Abstract
Background
Gitelman syndrome (GS) is a rare inherited disorder caused by mutations in SLC12A3, encoding the thiazide-sensitive transporter NCCT (sodium chloride co-transporter) in the distal tubule. It is characterized by renal potassium (K) and magnesium (Mg) wasting, relative hypotension and hypocalciuria. However, there is phenotypic variability and long-term studies are scarce.
Methods
We retrospectively assessed clinical and genetic characteristics, and electrolyte requirements, in a cohort of 36 patients with genetically proven GS.
Results
The 21 males and 15 females were of median age 39.5 years, range 17–66 years. Six were diagnosed in childhood. Among the 72 mutant alleles, 41 different sequence alterations were identified, of which 13 were previously unreported. Surprisingly, 44% (n = 16) of the cohort has developed hypertension (13 males, 3 females, P = 0.019; median age 53 versus 57 years, P = 0.95). One was already hypertensive by age 23 years. Currently normotensive patients were significantly younger: median 37 versus 55 years (P = 0.005). Hypertensive patients were more likely to harbour mutations in the C-terminal half of the NCCT protein (P = 0.016). Females required more K (median 128 versus 72 mmol/day; P = 0.01) but not Mg. Those with exon 26 and/or at least one destructive mutation had higher K requirements than those with neither: 108 versus 72 mmol (P = 0.016) and a tendency towards higher Mg needs: 30 versus 7.4 mmol (P = 0.07).
Conclusions
Our findings suggest that the development of secondary hypertension may be an expected feature of the ageing GS population despite the obligate salt wasting that characterizes the disorder. We hypothesize that this may be related to chronic secondary hyperaldosteronism. The apparently more severe phenotype in women may be related to the effects of female sex hormones on expression or function of NCCT.
Keywords: distal convoluted tubule, Gitelman syndrome, hypokalaemia, hypomagnesaemia, hypertension
BACKGROUND
Gitelman syndrome (GS) is a rare inherited tubulopathy associated with mutations in SLC12A3, which encodes the thiazide-sensitive sodium chloride co-transporter (NCCT) in the distal tubule [1, 2]. GS is characterized clinically by renal potassium and magnesium wasting, metabolic alkalosis, relative hypotension and hypocalciuria. However, there is phenotypic variability, with even members of the same family being affected with differing severity [3]. It has been suggested that males manifest a more severe phenotype, ascribed to the absence of female sex hormones that may attenuate the loss of NCCT function [4]. There is also considerable phenotypic overlap with Type 3 Bartter syndrome; this is caused by mutations in CLCNKB, which encodes the basolateral chloride channel in the thick ascending limb of Henle [5].
More than 170 different mutations in SLC12A3 have been described, and with improving techniques the detection rate in some series exceeds 90% [6]. Attempts have been made to establish genotype–phenotype correlations, with earlier authors noting that patients with two mutant alleles exhibit a more severe phenotype than those with only one or no mutations [7]. Attention has focused on examining which mutations may lead to hypomorphism rather than complete loss of function [4], and the potential role of other variables that may modify the phenotype.
This retrospective study examined the clinical and molecular genetic characteristics of patients with mutation-proven GS attending a specialist multidisciplinary clinic, and explores reasons for phenotypic variability.
SUBJECTS AND METHODS
Patients and genotyping
With ethical approval of the Cambridge Local Research Ethics Committee (08/H0306/62), all those known to our specialist adult nephrology service with phenotypic features in keeping with GS and two identified mutations in SLC12A3 were recruited for study. Mutation screening of SLC12A3 was performed by the East Anglian Regional Genetics Service by direct sequencing of polymerase chain reaction (PCR)-amplified coding exons and exon–intron boundaries, together with gene-dosage assessment by multiplex ligation-dependent probe amplification if only one allele was initially identified or apparent homozygosity was observed. Patient ethnicity was self-reported.
Clinical parameters
Initial biochemical assessment included serum urea and electrolytes, magnesium, bone profile, bicarbonate, ambulant random renin and aldosterone and 24-h urinary calcium measurements, with the patient off supplements and/or related drugs for at least 3 days in 34 of 36 patients.
Blood pressure (BP) measured in clinic using an automated sphygmomanometer, with the patient seated and following a short rest, or historical records, were used to evaluate hypertension. Each patient's 24-h magnesium and potassium requirement was recorded at their most recent clinic visit.
Statistical analysis
Data were expressed as median (interquartile range), or mean ± standard error where normally distributed. Mann–Whitney U test or Fisher's exact test was used as appropriate, with significance set at P < 0.05.
RESULTS
Demographics and clinical management
We identified 36 patients from 35 unrelated families; 21 males and 15 females (Table 1). Their median age at the time of data collection was 39.5 years (range 17–66), with no significant gender difference (male 42 years, female 37 years, P = 0.26). Thirty-four patients were Caucasian, one Pakistani and one Filipino. Twenty of 21 patients who were specifically asked expressed a long-standing preference for salty foods over sweet. Anecdotal evidence regarding extreme salt-craving behaviours included one who licked salt from potato chips in childhood, another whose nickname was ‘Peanuts’ and several accounts of drinking the vinegar from pickle jars.
Table 1.
Clinical and pharmacological characteristics of study subjects
| Patient | Sex | Age (years) | Age at symptom onset (years) | BP >130/80 mmHg? | Renin (mU/L; normal <78) | Urinary calcium (mmol/24 h; normal 2.5–7.5) | Urine ACR >2.5? | K dose/24 h (mmol) | Mg dose/24 h (mmol) | Current other electrolyte-protective/antihypertensive medications (mg daily) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 17 | 10 | N | 88 | Unknown | N | 192 | 30 | |
| 2 | F | 25 | 4 | N | 607 | Unknown | N | 128 | 50 | Losartan 50 |
| 3 | F | 28 | 23 | N | 216 | 3.3 | N | 192 | 80 | Spironolactone 50, Lisinopril 5b |
| 4 | F | 28 | 21 | N | 697 | Unknown | N | 64 | 30 | Spironolactone 75 |
| 5 | F | 31 | Childhood | N | 425 | Unknown | N | 80 | 33.6 | Amiloride 20 |
| 6 | F | 33 | 27 | N | 100 | Unknown | N | 96 | 140 | |
| 7 | F | 33 | Unknown | Y | 94 | 1.5 | N | 192 | 30 | Lisinopril 10, Atenolol 50 |
| 8 | F | 37 | 6 | N | 251 | Unknown | N | 192 | 30 | |
| 9 | F | 38 | 24 | N | 125 | 1.2 | N | 32 | 0 | Amiloride 5 |
| 10 | F | 44 | 29 | N | 263 | 1.5 | Y | 128 | 15 | Amiloride 5 |
| 11 | F | 47 | 37 | N | 177 | 0.8 | N | 96 | 15 | |
| 12 | F | 50 | 45 | N | 237 | 1.7 | N | 108 | 0 | Amiloride 10 |
| 13 | F | 50 | Teenager | N | 99 | 2.7 | N | 144 | 45 | Lisinopril 2.5 |
| 14 | F | 57 | 33 | Y | Unknown | 3.3 | Y | 144 | 98 | Spironolactone 25,Atenolol 50 b |
| 15 | F | 66 | 56 | Y | 29c | 0.4 | N | 16 | 6.7 | b |
| 16 | M | 18 | 14 | N | 282 | Unknown | N | 0 | 8 | |
| 17 a | M | 19 | 13 | N | 220 | 0.52 | N | 32 | 20 | Amiloride 10 |
| 18 a | M | 24 | 3 | N | 236 | Unknown | N | 32 | 26 | |
| 19 | M | 27 | 20 | Y | 376 | Unknown | N | 96 | 30 | Amiloride 30 |
| 20 | M | 31 | Childhood | Y | 556 | 1.5 | N | 48 | 40 | Spironolactone 200 |
| 21 | M | 35 | 21 | Y | 1432 | Unknown | N | 72 | 15 | Spironolactone 300, Perindopril 2 |
| 22 | M | 36 | 32 | N | 315 | Unknown | N | 128 | 56 | Amiloride 20 |
| 23 | M | 38 | 32 | Y | 108 | 1.9 | N | 32 | 0 | Irbesartan 75 |
| 24 | M | 39 | 33 | N | 318 | Unknown | Y | 96 | 0 | |
| 25 | M | 40 | 13 | Y | 4422 | 4.4 | N | 156 | 6.7 | Amiloride 10, Irbesartan 150 |
| 26 | M | 42 | Teenager | N | 90 | Unknown | Y | 96 | 30 | Lisinopril 7.5, Epleronone 25 |
| 27 | M | 46 | 43 | N | 249 | 1.2 | N | 0 | 0 | |
| 28 | M | 49 | 41 | Y | 71 | 8.8 | Y | 120 | 26 | Ramipril 5b |
| 29 | M | 53 | Childhood | Y | 238 | Unknown | N | 72 | 15 | Amiloride 5 |
| 30 | M | 58 | Childhood | Y | 71c | 1.2 | N | 64 | 40 | Aliskiren 150, Irbesartan 150 |
| 31 | M | 59 | 51 | N | 200 | <1.0 | N | 72 | 0 | |
| 32 | M | 63 | 58 | Y | Unknown | 1.3 | Y | 96 | 0 | Lisinopril 5 |
| 33 | M | 65 | 57 | Y | 113 | 1.7 | N | 32 | 15 | b |
| 34 | M | 65 | 23 | Y | 103 | Unknown | N | 80 | 60 | Lisinopril 30, Amlodipine 5b |
| 35 | M | 67 | 62 | Y | 98 | 4.2 | N | 72 | 40 | Amiloride 20 |
| 36 | M | 67 | 61 | Y | 82 | 2.9 | N | 96 | 0 | Amiloride 10 |
aSiblings.
bAlso on proton pump inhibitor.
cUpper limit at time of assay 60 mU/L.
Potassium and magnesium supplements were prescribed towards targets of K >3.0 mmol/L and Mg >0.6 mmol/L, subjective improvement in symptoms, or both, and/or to the limit of patient tolerance. Where systolic BP was >100 mmHg, one or more of amiloride, spironolactone, eplerenone, angiotensin-converting enzyme (ACE) inhibitor/angiotensin receptor blocker (ARB) or β-blocker were often prescribed (Table 1) as potassium-conserving agents.
Genetic diagnosis
In all 36 patients, Sanger sequencing and/or multiplex ligation-dependent probe amplification had identified two pathogenic alleles (Table 2), of which 56 (78%) were missense (one introducing a premature stop codon) while 9 (13%) disrupted splice sites and 5 (7%) were very small out-of-frame insertions or deletions that introduced frame-shifts. The commonest was Gly741Arg, present as 14 alleles in 12 individuals. Two heterozygous intragenic deletions removed exon 26 and the first seven exons, respectively. In all, 41 different mutations were found, of which 13 were novel. The majority of patients (33 of 36, 92%) were compound heterozygotes. A history of consanguinity was evident in only one of the three homozygotes (Patient 5).
Table 2.
Genetic details of study subjects
| Patient | Exon | Genomic sequence (1 = start of coding sequence) | Predicted protein change |
|---|---|---|---|
| 1 | 13 | 1664C>T | Ser555Leu |
| 26 | Exon deleted | Thr985Xc | |
| 2 | 10 | 1196_1202dupGTGATGC | Ser402X |
| 22 | 2576T>C | Leu859Pro | |
| 3 | 3 | 363G>C | Glu121Asp |
| 5 | 626G>A | Arg209Gln | |
| 4 | 14 | 1763C>T | Ala588Val |
| 25 | 2893C>T | Gln965Xc | |
| 5a | 10 | Homozygous 1195C>T | Arg399Cys |
| 6 | 3 | 497C>T | Ala166Val |
| 22 | 2576T>C | Leu859Pro | |
| 7 | 18 | 2221G>A | Gly741Arg |
| 26 | 2981G>A | Cys994Tyr | |
| 8 | 14 | 1825+1delG | Intron 14 5′ splice site lost |
| 26 | 2981G>A | Cys994Tyr | |
| 9 | 7 | 961C>T | Arg321Trp |
| 18 | 2221G>A | Gly741Arg | |
| 10 | 7 | 911C>T | Thr304Met |
| 24 | 2883+1G>T | Intron 24 5′ splice site lost | |
| 11 | 16 | 1963C>T | Arg655Cys |
| 26 | 2990_2993dupCGCT | Leu998fsc | |
| 12 | 13 | 1664C>T | Ser555Leu |
| 18 | 2221G>A | Gly741Arg | |
| 13 | 18 | 2221G>A | Gly741Arg |
| 24 | 2883+1G>T | Intron 24 5′ splice site lost | |
| 14 | 26 | 2996A>G | Tyr999Cysc |
| 26 | 3089A>G | Gln1030Arg | |
| 15 | 9 | 1145C>T | Thr382Met |
| 18 | 2221G>A | Gly741Arg | |
| 16 | 5 | 710G>A | Gly237Aspc |
| 22 | 2576T>C | Leu859Pro | |
| 17b | 10 | 1196G>T | Arg399Leuc |
| 14 | 1825+1G>T | Intron 14 5′ splice site lost | |
| 18b | 10 | 1196G>T | Arg399Leuc |
| 14 | 1825+1G>T | Intron 14 5′ splice site lost | |
| 19 | 4 | 539-543delCGGTG | Thr180fs |
| 24 | 2883+1G>T | Intron 24 5′ splice site lost | |
| 20 | 11 | 1390G>A | Ala464Thr |
| 22 | 2576T>C | Leu859Pro | |
| 21 | 18 | 2221G>A | Gly741Arg |
| 26 | 3053G>C | Arg1018Proc | |
| 22 | 4 | 506-1G>A | Intron 3 3′ splice site lost |
| 17 | 2089_2095delACCAAGT | Thr697fs | |
| 23 | 14 | 1763C>T | Ala588Val |
| 22 | 2576T>C | Leu859Pro | |
| 24 | 10 | 1258G>A | Ala420Thrc |
| 25 | 2947G>A | Val983Ilec | |
| 25 | 8 | 1061_1062dupTC | Leu355fsc |
| 26 | 2965G>A | Gly989Arg | |
| 26 | 10 | 1315G>A | Gly439Ser |
| 18 | 2221G>A | Gly741Arg | |
| 27 | 18 | Homozygous 2221G>A | Gly741Arg |
| 28 | 18 | 2221G>A | Gly741Arg |
| 26 | 2981G>A | Cys994Tyr | |
| 29 | 16 | 2037+1G>A | Intron 16 5′ splice site lost |
| 26 | 3053G>A | Arg1018Gln | |
| 30 | 1-7 | Exons deleted | Probable loss |
| 18 | 2221G>A | Gly741Arg | |
| 31 | 5 | 626G>A | Arg209Gln |
| 14 | 1763C>A | Ala588Gluc | |
| 32 | 22 | 2581C>T | Arg861Cys |
| 23 | 2723T>C | Ile908Thrc | |
| 33 | 11 | 1351T>A | Ser451Thrc |
| 18 | 2221G>A | Gly741Arg | |
| 34 | 10 | 1315G>A | Gly439Ser |
| 24 | 2883+1G>T | Intron 24 5′ splice site lost | |
| 35 | 18 | Homozygous 2221G>A | Gly741Arg |
| 36 | 5 | 626G>A | Arg209Gln |
| 26 | 2981G>A | Cys994Tyr |
aParental consanguinity.
bSiblings.
cNot previously identified. The missense mutations were absent from SNP databases and/or were in residues previously reported to be differently substituted.
Symptom onset
Twenty-nine patients ascribed a definite age to symptom onset, at median 29 years (range 3–62, Table 1). Of the remaining seven, most subjectively felt that they had been unwell for many years, but this could not be detailed more objectively. Of the six who presented in childhood (some with symptom onset in infancy), four were male (P = 0.67). Three of these individuals were initially diagnosed with Bartter syndrome before a genetic diagnosis of GS was made, and as a result were prescribed non-steroidal anti-inflammatory drugs during childhood.
Blood pressure
Surprisingly, 16 of the 36 (44%) have developed hypertension, as defined by BP >130/80 mmHg on at least three occasions, or a diagnosis of hypertension prior to referral, now treated (Table 1). Median systolic and diastolic BP readings were both significantly higher (Figure 1): 138 (133–143) versus 116 (109–122) and 86 (77–88) versus 73 (64–77) mmHg (P < 0.0001 and P = 0.0007, respectively). All but two of these 16 have had clinic BPs >130/80 despite being on potassium-conserving agents that have antihypertensive action, suggesting that their untreated BP would have been higher. The hypertensive subgroup was more likely to be male: 13 versus 3 females (P = 0.018) but of similar current age (median 53 versus 57 years, P = 0.95) and was significantly older than normotensive subjects: median 55 versus 36.5 years (P = 0.005). However, one female was already hypertensive by the age of 23 years. Patient 32's initial presentation was with hypertension at age 58 years; secondary hyperaldosteronism was discovered and the diagnosis of GS followed, other causes having been excluded.
FIGURE 1:

Systolic and diastolic blood pressures in study subgroups. BP, blood pressure; HT, hypertensive; NT, normotensive; S, systolic; D, diastolic.
Potassium and magnesium requirements
Among the whole cohort, median daily potassium supplementation was 96 (60–128) mmol. Females required significantly more than males; 128 versus 72 mmol in 24 h (P = 0.01) (Figure 2A). The top quartile of patients ingested more than 128 mmol potassium every day, and the majority of these were females (80%, P = 0.007). Two patients did not take any potassium.
FIGURE 2:

Biochemical differences among study subgroups. (A) Potassium dosage was higher in females, while (B) magnesium requirements were similar. (C) Renin levels did not differ significantly between normo- and hypertensive subjects. (D) Those with at least one exon 26 mutation required significantly more potassium. (E and F) ‘Severe’ (destructive and/or exon 26) mutations were associated with higher potassium requirements and trended towards higher magnesium needs.
As expected, hypomagnesaemia was common, with 28 of 36 patients requiring supplementation. The median daily magnesium dose was 26 (6.7–40) mmol and there was a non-significant trend towards women consuming larger doses than men; 30 versus 15 mmol (P = 0.10) (Figure 2B). Magnesium-requiring individuals tended to be older (48 versus 36.5 years, P = 0.07). Their potassium requirements were not significantly different (84 versus 96 mmol, P = 0.20). The top quartile of patients took more than 40 mmol in 24 h with no difference between the sexes. The six patients taking proton-pump inhibitors trended towards higher magnesium requirements; median 43 versus 20 mmol in 24 h (P = 0.16).
There were no significant differences in electrolyte requirements between hypertensives and normotensives; potassium 76 (60–102) versus 96 (56–128) mmol (P = 0.56), and magnesium 20.5 (5–40) versus 28 (13–37) (P = 0.85). In addition, the six manifesting the disorder in childhood did not subsequently have higher electrolyte requirements than those who present later, despite their apparently more severe phenotype: median 24 h potassium 80 versus 96 mmol (P = 0.90) and magnesium 23 versus 28 mmol (P = 0.95). Interestingly, we observed one female GS patient (no. 15) who started to use high-dose fish oil supplements and subsequently noted that her electrolyte requirements reduced 4-fold.
Patients taking aldosterone receptor antagonists tended towards higher daily magnesium requirements than those who did not (35 versus 17.5 mmol, P = 0.06) although there was no significant difference between their potassium doses (84 versus 96, P = 0.73). Those on amiloride had similar electrolyte requirements to those not taking it: potassium 96 versus 96 mmol (P = 0.81) and magnesium 15 versus 30 mmol (P = 0.35).
Renin and aldosterone
Renin measurements not confounded by treatment were available for 34 of the 36 study participants. Levels were elevated in all but two of these 34: median 218 mU/L (99–306). There was no significant difference in median renin between hypertensive and normotensive subjects: 105 (86–342) and 237 (164–290) mU/L, respectively (P = 0.19; Figure 2C). Aldosterone measurements are not included as they could not reliably be interpreted in light of variably low potassium levels.
Proteinuria
Six of the 36 had proteinuria (defined as random albumin:creatinine ratio >2.5); all six had preserved excretory renal function and none was diabetic. They were not of significantly different current age (46.5 versus 37.5 years; P = 0.19) or age at symptom onset (33 versus 23.5 years; P = 0.15) to the rest of the cohort, nor were magnesium (20.5 versus 28 mmol; P = 0.61) or potassium requirements different; 108 versus 76 mmol; P = 0.12). Three were hypertensive (P = 1.00) and four were male (P = 1.00).
Urinary calcium measurements
Urinary calcium data were available for 21 patients (Table 1). Notably, while 14 of these (67%) were hypocalciuric (<3 mmol/24 h), 6 (29%) were normocalciuric and 1 (5%) was hypercalciuric (>7 mmol/24 h). Seven of 8 (88%) women in this subgroup were hypocalciuric compared with 7 of 13 (54%) men (P = 0.17). Available data showed no significant difference in urinary biochemistry between BP groups; 8 of 12 hypertensives and 7 of 9 normotensives were hypocalciuric (P = 0.66).
Genotype–phenotype correlation
Mutations found in hypertensive individuals were more likely to be located downstream of exon 15, i.e. in NCCT's long intracellular C-terminal tail: 24 of 42 compared with 8 of 30 upstream alleles, P = 0.016 (Figure 3). Those with at least one mutation in exon 26 (6 alleles in 9 subjects) needed significantly more potassium: 132 versus 76 mmol (P = 0.007) (Figure 2D), although there was no significant magnesium difference: 20.5 versus 28 mmol (P = 0.78). Exon 26 mutations were not significantly more frequent in females (5 of 15, P = 0.71) nor those presenting in childhood (3 of 6, P = 0.31).
FIGURE 3:
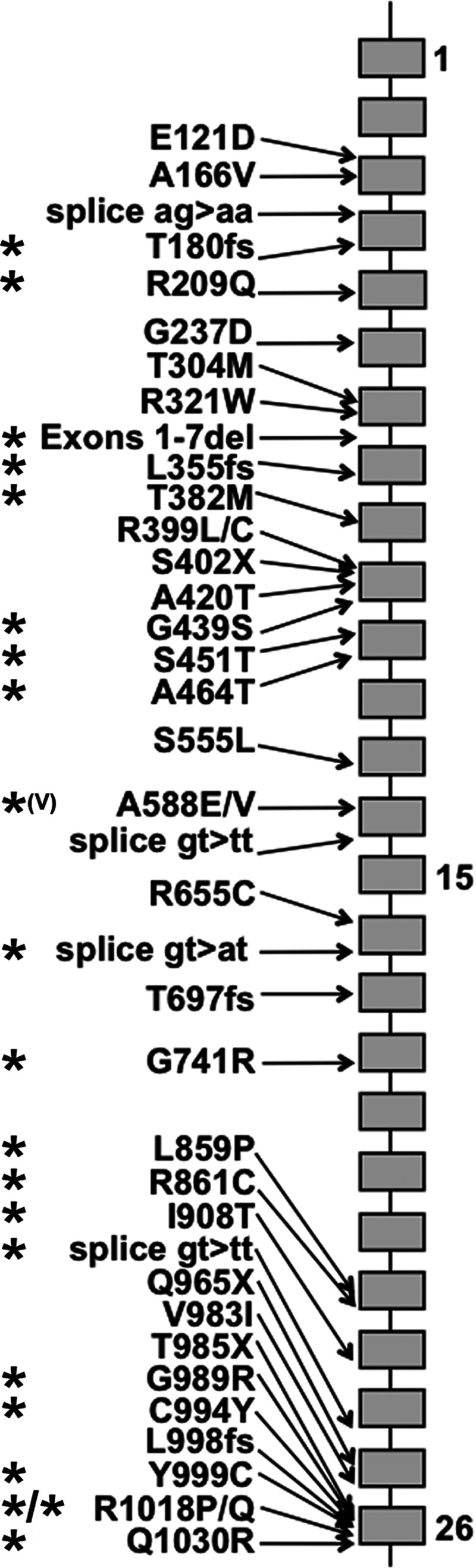
Mutations found in NCCT. Grey blocks indicate exons 1–26. The C-terminal tail is encoded by exons 16–26. Mutant alleles found in one or more hypertensive subjects are asterisked.
In addition, those with at least one destructive (splice site, truncation or frame-shift) mutation had higher potassium requirements than those with two missense mutations outside of exon 26: 96 versus 76 mmol in 24 h (P = 0.12) and magnesium 30 versus 11.5 mmol (P = 0.04). Five of the six patients presenting in childhood had at least one destructive mutation (P = 0.06). Those with a mutation in exon 26 and/or at least one destructive mutation had significantly higher potassium requirements than those with neither: 108 versus 72 mmol (P = 0.016) (Figure 2E) and a tendency towards higher magnesium needs: 30 versus 7.4 mmol (P = 0.07; Figure 2F). These findings may reflect more severe loss of function compared with likely hypomorphism of point mutants.
Pregnancy
Two patients in the cohort (nos. 5 and 6) underwent successful pregnancies, both requiring substantially more electrolyte replacement, including hospital admission for intravenous magnesium and/or potassium treatment.
DISCUSSION
The two striking observations in this cohort of GS patients were the unexpectedly high rate of secondary hypertension, and the apparently more severe electrolyte disturbance in females. With respect to BP, an important feature of our study is that despite obligate salt-wasting in GS, hypertension has developed in nearly half of our patients, and although it was more prevalent in the older males, it was not always a feature of advancing age. Neither age, renin level, degree of hypokalaemia, duration of symptoms nor untreated disease, proteinuria or medications solely accounted for the hypertension. Although large for a disease of this rarity (estimated prevalence 1:40 000 [8]), the relatively small cohort size precluded useful multivariate analysis. Interestingly, hypokalaemia has previously been suggested to exacerbate essential hypertension, even in the absence of hyperaldosteronism [9]. However, the proposed mechanisms to account for this finding included sodium retention and calcium depletion, which would not apply to GS patients.
Both GS and Bartter syndrome are characterized by chronic hyper-reninaemia. We hypothesize that this ongoing stimulation of the renin–aldosterone axis results in chronic vasoconstriction, leading to a situation where the hypovolaemia associated with hypotension is overcome, and hypertension supervenes. In support of this, a study conducted in the Framingham Offspring group suggests that in the healthy population, increased aldosterone levels, even within the normal physiologic range, may predispose subjects to developing hypertension [10]. This effect was independent of left ventricular dimensions and urinary sodium excretion, but renin values were not reported. Aldosterone measurements in this cohort would have been uninterpretable in the face of varying levels of hypokalaemia. However, unconfounded baseline renin measurements did not show a significant difference between hypertensive and normotensive groups (in fact the opposite was seen). These data were collected at the time of presentation, often some years before hypertension developed. It is possible that persistent hyper-reninism might presage the development of hypertension in GS patients, but we do not have sufficient data to assess this at present, and we hope that further collaborative studies will shed light on this.
We also find that hypertensive GS patients are more likely to have mutations in the C-terminal domain of SLC12A3. Given that exon 26 mutations may also confer a more severe phenotype as evidenced by higher potassium requirements, we hypothesize that the C-terminal domain is vital for NCCT function mediated through its role in protein trafficking. More severe electrolyte wasting (as observed in exon 26 patients) may give rise to more exaggerated compensatory mechanisms (stimulation of the renin–aldosterone axis) and the subsequent development of hypertension.
While we cannot explicitly rule out a second diagnosis in young GS sufferers, this unexpectedly high frequency suggests that hypertension may be a predicted feature of the GS population as the natural history of the disorder emerges. Long-term follow-up studies in GS are rare, with only one recent publication noting coexistent hypertension in 4 of 15 genetically-proven subjects [7]. Other reports suggest a link between the secondary hyperaldosteronism of Bartter syndrome and subsequent focal segmental glomerulosclerosis [11, 12].
Our finding of an apparently more severe electrolyte-related phenotype in females is also novel; previous authors have suggested either that there is no significant difference between the sexes [13] or that men are more severely affected [3]. Our observation could not be explained by an increased frequency of destructive genetic alterations in women. We suggest that the difference may relate in part to the effect of female sex hormones on renal NCCT expression. Normally, high levels of progesterone compete for binding with, and block, aldosterone, resulting in net sodium and water loss [14]. In mitigation of this, a different progesterone-mediated mechanism promotes salt and potassium retention; it has been reported that progesterone stimulates the renal isoform of the H+/K+-ATPase [15]. Oestrogens may also increase NCCT expression in an attempt to conserve sodium; it has been shown that oestradiol reverses the decline in NCCT density observed following ovariectomy in rats [16]. Additionally, both progesterone and estradiol have been shown to cause relative hypokalaemia in rat models [17]. Thus up-regulation of NCCT by female sex hormones may be a compensatory mechanism to preserve circulating volume in the face of functional aldosterone antagonism, and failure to achieve this exacerbates the Gitelman phenotype. The observation that both pregnant patients in our cohort required additional electrolyte replacement supports this, and 28 of 47 patients in a larger series of pregnant GS patients had higher potassium requirements in pregnancy [18]. Hyperfiltration may also account for the greater degree of renal potassium wasting observed. The putative protective effects observed in rats of female sex hormones against those of hyperaldosteronism [19] could explain in part why female GS patients appear to develop secondary hypertension less frequently than males.
Qualitative data also suggest that symptoms attributed to GS vary throughout the menstrual cycle [13]. We are not currently able to assess differences in electrolyte requirements between pre- and post-menopausal women, because only four of our females were over 50 years old. However, this remains an intriguing prospect as our Gitelman cohort ages.
Although the apparent beneficial effect of fish oil supplements in one subject is anecdotal and is difficult to account for mechanistically, it is interesting that the potential role of omega oils in reducing pre-term labour is well reported [20] and that in ovine studies long-chain polyunsaturates have been shown to reduce estradiol levels in pregnancy [21]. This raises the additional possibility of hormonal manipulation mediated by omega oils that may affect the GS phenotype. Future collaborative studies involving larger numbers will be required to investigate this further.
Symptom onset predated genetic diagnosis of GS by many years in our cohort, partly because genetic testing is a relatively recent addition to the clinician's armamentarium. Such a time lag is, however, typical in the diagnosis of rare diseases; many of our patients commented upon the relief they felt at finally reaching a definite diagnosis. Patients often retrospectively recognize symptoms stretching as far back as childhood, and the childhood preference for salty foods is a good discriminating question when eliciting the history.
Hypocalciuria has previously been used to distinguish Bartter from GS [22], but we did not universally find low urinary calcium levels (Table 1). Hypocalciuria in GS is thought to result from compensatory up-regulation of proximal tubular calcium re-absorption [23, 24]. Hence it is a consequence of impaired NCCT rather than a primary defect, and thus might be expected to vary with the degree of hypomorphism of mutant alleles.
Finally, the high frequency of hypertension that we have observed suggests that GS should be considered not only in those with unexplained hypokalaemia, but also in the differential diagnosis of hypertension associated with secondary hyperaldosteronism. Future studies will reveal if this turns out to be the case in a significant proportion of such individuals.
AUTHORS’ CONTRIBUTIONS
C.R. and M.B. collected the data; M.B., C.R. and F.E.K.F. analysed it; M.B. and F.E.K.F. wrote the paper.
CONFLICT OF INTEREST STATEMENT
None declared. The results presented in this paper have not been published previously in whole or part, except in an abstract format.
ACKNOWLEDGEMENTS
We thank our patients and their referring physicians; the East Anglian Regional Genetics Service; Thomas Hiemstra for statistical assistance; Rona Smith and Menna Clatworthy; Deborah Spencer for additional data; and our funding bodies: the National Institute for Health Research Cambridge Biomedical Research Centre and Wellcome Trust (award 088489 to FEKF).
REFERENCES
- 1.Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. [PubMed] [Google Scholar]
- 2.Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 3.Lin SH, Cheng NL, Hsu YJ, et al. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. 2004;43:304–312. doi: 10.1053/j.ajkd.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Riveira-Munoz E, Chang Q, Bindels RJ, et al. Gitelman's syndrome: towards genotype–phenotype correlations? Pediatr Nephrol. 2007;22:326–332. doi: 10.1007/s00467-006-0321-1. [DOI] [PubMed] [Google Scholar]
- 5.Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 6.Vargas-Poussou R, Dahan K, Kahila D. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22:693–703. doi: 10.1681/ASN.2010090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balavoine AS, Bataille P, Vanhille P, et al. Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome. Eur J Endocrinol. 2011;165:665–673. doi: 10.1530/EJE-11-0224. [DOI] [PubMed] [Google Scholar]
- 8. Orphanet http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN .
- 9.Krishna GG, Kapoor SC. Potassium depletion exacerbates essential hypertension. Ann Intern Med. 1991;115:77–83. doi: 10.7326/0003-4819-115-2-77. [DOI] [PubMed] [Google Scholar]
- 10.Vasan RS, Evans JC, Larson MG, et al. Serum aldosterone and the incidence of hypertension in non-hypertensive persons. N Engl J Med. 2004;351:33–41. doi: 10.1056/NEJMoa033263. [DOI] [PubMed] [Google Scholar]
- 11.Su IH, Frank R, Gauthier BG, et al. Bartter syndrome and focal segmental glomerulosclerosis: a possible link between two diseases. Pediatr Nephrol. 2000;14:970–972. doi: 10.1007/s004670050054. [DOI] [PubMed] [Google Scholar]
- 12.Akil I, Ozen S, Kandiloglu AR, et al. A patient with Bartter syndrome accompanying severe growth hormone deficiency and focal segmental glomerulosclerosis. Clin Exp Nephrol. 2010;14:278–282. doi: 10.1007/s10157-009-0262-7. [DOI] [PubMed] [Google Scholar]
- 13.Cruz DN, Shaer AJ, Bia MJ, et al. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710–717. doi: 10.1046/j.1523-1755.2001.059002710.x. [DOI] [PubMed] [Google Scholar]
- 14.Wambach G, Higgins JR. Antimineralocorticoid action of progesterone in the rat: correlation of the effect on electrolyte excretion and interaction with renal mineralocorticoid receptors. Endocrinology. 1978;102:1686–1693. doi: 10.1210/endo-102-6-1686. [DOI] [PubMed] [Google Scholar]
- 15.Elabida B, Edwards A, Salhi A, et al. Chronic potassium depletion increases adrenal progesterone production that is necessary for efficient renal retention of potassium. Kidney Int. 2011;80:256–62. doi: 10.1038/ki.2011.15. [DOI] [PubMed] [Google Scholar]
- 16.Verlander JW, Tran TM, Zhang L, et al. Estradiol enhances thiazide-sensitive NaCl cotransporter density in the apical plasma membrane of the distal convoluted tubule in ovariectomized rats. J Clin Invest. 1998;101:1661–1669. doi: 10.1172/JCI601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng W, Shi M, You SE, et al. Estrogens contribute to a sex difference in plasma potassium concentration: a mechanism for regulation of adrenal angiotensin receptors. Gend Med. 2006;3:43–53. doi: 10.1016/s1550-8579(06)80193-2. [DOI] [PubMed] [Google Scholar]
- 18.Mascetti L, Bettinelli A, Simonetti GD, et al. Pregnancy in inherited hypokalemic salt-losing renal tubular disorder. Obstet Gynecol. 2011;117:512–516. doi: 10.1097/AOG.0b013e3182075317. [DOI] [PubMed] [Google Scholar]
- 19.Wambach G, Higgins JR. Antihypertensive effect of progesterone in rats with mineralocorticoid-induced hypertension. Am J Physiol. 1979;236:366–370. doi: 10.1152/ajpendo.1979.236.4.E366. [DOI] [PubMed] [Google Scholar]
- 20.Secher NJ. Does fish oil prevent preterm birth? J Perinat Med. 2007;35:S25–S27. doi: 10.1515/JPM.2007.033. [DOI] [PubMed] [Google Scholar]
- 21.Baguma-Nibasheka M, Brenna JT, Nathanielsz PW. Delay of preterm delivery in sheep by omega-3 long-chain polyunsaturates. Biol Reprod. 1999;60:698–701. doi: 10.1095/biolreprod60.3.698. [DOI] [PubMed] [Google Scholar]
- 22.Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992;120:38–43. doi: 10.1016/s0022-3476(05)80594-3. [DOI] [PubMed] [Google Scholar]
- 23.Loffing J, Vallon V, Loffing-Cueni D, et al. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman's syndrome. J Am Soc Nephrol. 2004;15:2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63. [DOI] [PubMed] [Google Scholar]
- 24.Nijenhuis T, Vallon V, van der Kemp AW, et al. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]