

Abstract
L-dopa-induced dyskinesias are a serious long-term side effect of dopamine replacement therapy for Parkinson’s disease for which there are few treatment options. Our previous studies showed that nicotine decreased L-dopa-induced abnormal involuntary movements (AIMs). Subsequent work with knockout mice demonstrated that α6β2* nicotinic receptors (nAChRs) play a key role. The present experiments were done to determine if α4β2* nAChRs are also involved in L-dopa-induced dyskinesias. To approach this, we took advantage of the finding that α6β2* nAChRs are predominantly present on striatal dopaminergic nerve terminals, while a significant population of α4β2* nAChRs are located on other neurons. Thus, a severe dopaminergic lesion would cause a major loss in α6β2*, but not α4β2* nAChRs. Experiments were therefore done in which rats were unilaterally lesioned with 6-hydroxydopamine, at a dose that lead to severe nigrostriatal damage. The dopamine transporter, a dopamine nerve terminal marker, was decreased by >99%. This lesion also decreased striatal α6β2* nAChRs by 97%, while α4β2* nAChRs were reduced by only 12% compared to control. A series of β2* nAChR compounds, including TC-2696, TI-10165, TC-8831, TC-10600 and sazetidine reduced L-dopa-induced AIMs in these rats by 23–32%. TC-2696, TI-10165, TC-8831 were also tested for parkinsonism, with no effect on this behavior. Tolerance did not develop with up to 3 months of treatment. Since α4a5β2 nAChRs are also predominantly on striatal dopamine terminals, these data suggest that drugs targeting α4β2 nAChRs may reduce L-dopa-induced dyskinesias in late stage Parkinson’s disease.
Keywords: Dyskinesia, L-dopa, nicotine, nicotinic, Parkinson’s disease, sazetidine
1. Introduction
Dopamine precursor treatment with L-dopa is the gold-standard for Parkinson’s disease therapy (Meissner et al., 2011; Obeso et al., 2010; Rascol et al., 2011; Schapira and Jenner, 2011). However, L-dopa-induced dyskinesias are a complication that arises in most Parkinson’s disease patients with continued treatment (Ahlskog, 2003; Bezard et al., 2001; Cenci and Lindgren, 2007; Fox et al., 2008; Jenner, 2008; Obeso et al., 2000). Since these abnormal involuntary movements (AIMs) are a major limitation with long term L-dopa therapy, an intense research effort is underway to identify drugs that may reduce their occurrence. Compounds directed to various CNS neurotransmitter systems have proven beneficial against L-dopa-induced dyskinesias in different parkinsonian animal models, including glutamate, adenosine, noradrenaline, 5-hydroxytryptamine, cannabinoid, and opioid receptor drugs (Brotchie and Jenner, 2011; Carta et al., 2008a; Cenci and Konradi, 2010; Jenner, 2008; Morin et al., 2010). Nicotine, a drug that acts at nicotinic acetylcholine receptors (nAChRs), also reduces L-dopa-induced dyskinetic-like movements in parkinsonian rats, mice and nonhuman primates, without affecting the antiparkinsonian effect of L-dopa (Bordia et al., 2008; Bordia et al., 2010; Huang et al., 2011a; Quik et al., 2007). Declines in dyskinesia are observed with several modes of administration including oral gavage, subcutaneous minipumps or injection, suggesting that an oral formulation (pill or solution) or a slow release mode (nicotine patch) may be useful in patients. Moreover, a small clinical trial showed that nicotine (NP002) reduced several components of L-dopa-induced dyskinesias in Parkinson’s disease patients (http://www.neuraltus.com/pages/news.html).
Nicotine reduces L-dopa-induced dyskinetic-like movements by interacting with nAChRs in the brain. However, nicotine has the disadvantage that it also acts at α3β4* and α7 nAChRs in the autonomic nervous system, as well as α1β1δγ nAChRs present in skeletal muscle although the concentrations required to activate the muscle receptor are much higher than those for neuronal subtypes (Albuquerque et al., 2009; Baddick and Marks, 2011; Gotti et al., 2009; Millar and Gotti, 2009; Quik et al., 2011). Notably, the primary subtypes in the brain are the α4β2*, α6β2* and α7 receptor populations, as well as a smaller, localized population of α3β4* nAChRs (Albuquerque et al., 2009; Baddick and Marks, 2011; Gotti et al., 2009; Millar and Gotti, 2009; Quik et al., 2011) (the asterisk indicates the possible presence of other subunits in the receptor complex). The β2* nAChRs are the main subtypes in the nigrostriatal pathway, which is damaged in Parkinson’s disease and parkinsonian animal models. Thus, nAChR drugs directed to these populations may provide a more selective treatment approach to reduce L-dopa-induced dyskinetic-like movements.
To investigate this possibility, we had previously used two approaches. In the first, we showed that the CNS selective β2* nAChR agonist A85380 reduced L-dopa-induced AIMs in 6-OHDA lesioned rats (Huang et al., 2011b). Experiments using null mutant mice next demonstrated that the expression of L-dopa-induced AIMs was reduced by ~40% compared in β2 nAChR knockout mice compared to wild type littermates, but that nicotine treatment did not further reduce AIMs in the β2 deficient mice. Taken together these studies support a role for β2* nAChRs in the occurrence of L-dopa-induced AIMs. Subsequent work using α6 nAChR (−/−) mice yielded similar results to those with β2 nAChR (−/−) mice (Quik et al., 2012b), suggesting a role specifically for α6β2* nAChRs.
The objective of the present study was to evaluate the involvement of α4β2* nAChRs in L-dopa-induced AIMs. In striatum, ~20% of these receptors are located on dopamine terminals with the remaining 80% present on other striatal neurons (Gotti et al., 2009; Millar and Gotti, 2009; Quik et al., 2011). In contrast, α6β2* nAChRs are primarily expressed on dopamine neurons. This differential localization provides an approach to investigate the role of postsynaptic α4β2* nAChRs, as these receptors are the predominant ones remaining with severe nigrostriatal dopaminergic damage. We therefore performed experiments in which rats received a severe unilateral 6-hydroxydopamine (6-OHDA) lesion, which led to an almost complete loss of the striatal dopamine transporter and α6β2* nAChRs, although α4β2* nAChRs are left relatively intact. We then tested the effect of nAChR compounds that are selective for β2* over α7, α3β4* and α1β1δγ nAChRs. These compounds led to a 23–32% reduction in L-dopa-induced AIMs in rats with severe nigrostriatal damage. These data support a role for α4β2* nAChRs on non-dopaminergic neurons in the antidyskinetic effect of nicotine. Since α4α5β2 nAChRs are primarily localized to dopaminergic neurons that are lost with nigrostriatal damage (Zoli et al., 2002), drugs targeting α4β2 nAChRs may be most useful for treating L-dopa-induced dyskinesias in patients with severe Parkinson’s disease.
2. Materials and methods
2.1 Animals
Male Sprague-Dawley rats (~200 g) were purchased from Charles River Laboratories, Inc (Gilroy, CA, USA). They were housed 3–4 per cage with free access to food and water. Housing was in a temperature- and humidity-controlled environment under a 12 h light-dark cycle. All procedures were done at SRI in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
2.2. Nigrostriatal lesioning
After several days of acclimation to the laboratory, the rats (230–250 g) were lesioned by unilateral intracranial administration of 6-OHDA (free base; Sigma-Aldrich Co., St. Louis, MO) into the right medial forebrain bundle under isofluorane anesthesia as previously described (Bordia et al., 2008; Bordia et al., 2010; Cenci and Lundblad, 2007). An injection of 2 μl 6-OHDA (3 μg free base/μl dissolved in 0.02% ascorbic acid, 0.9% saline) was stereotaxically injected at the following two coordinates: (1) AP, −4.4; ML, 1.2; DV, 7.8; tooth bar at −2.4; (2) AP, −4.0; ML, 0.75; DV, 8.0; tooth bar at +3.4 relative to the bregma (Bordia et al., 2008; Bordia et al., 2010; Cenci and Lundblad, 2007). 6-OHDA injection into each of the medial forebrain bundle sites was done over a 2 min period, with the needle kept at the injection site for 2 more min before withdrawal. The rats were given buprenorphine (0.02 mg/kg sc) postoperatively.
Two to three weeks after lesioning, rats were tested for amphetamine-induced rotational behavior in an automated behavioral measurement apparatus (ROTOMAX, AccuScan Instruments Inc. Columbus, Ohio, USA). Each rat was placed in a cylindrical glass chamber for 30 min for acclimatization, after which 4.0 mg/kg amphetamine (Sigma Chemical Co., St. Louis, MO) was administered intraperitoneally (ip). Amphetamine-induced rotations were monitored for 90 min, with rats making at least 200 ipsilateral turns/90 min used for further study.
2.3. Drug treatment
The nAChR compounds TC-2696, TI-10165, TC-8831 and TC-10600 (from Targacept, Inc.) were administered by systemic infusion via osmotic minipumps (Alzet model 2006; Durect Corporation, Cupertino, CA, USA), with compound delivery at a rate of 3.6 μl/day. Pumps were pre-filled with either sterilized water or test compound in water (pH 7.0) to provide the indicated doses. The doses of the compounds were selected based on their Ki values for nAChRs, preliminary PK parameters and observed behavioral effects in other CNS behavioral assessments in rodents (unpublished results). The minipumps were subcutaneously implanted along the dorsal aspect of the neck between the shoulder blades under isofluorane anesthesia, according to the manufacturer’s instruction. After implantation, rats were acutely given buprenorphine (0.02 mg/kg sc) for post-operative pain. Sazetidine (generously provided by Dr. K. Kellar, Georgetown Univ) was injected sc 10 min before L-dopa administration and also 8 h later.
2.4. Measurement of L-dopa-induced AIMs
Two weeks after the start of nAChR compound treatment, the rats were given single daily sc injections of L-dopa methyl ester (6 or 8 mg/kg) plus 15 mg/kg benserazide, an aromatic amino acid decarboxylase inhibitor that does not cross the blood-brain barrier and thus inhibits the breakdown of L-dopa in the periphery. There was no significant difference in total AIM scores with 6 or 8 mg/kg L-dopa (see Figs. 4 to 7). We therefore used 6 mg/kg in our later studies. L-dopa-induced AIMs were determined after daily L-dopa dosing, at the time points indicated in the timelines (Figs. 4–8). Behavioral testing was done in the morning starting between 9 and 10 AM. Three different AIMs subtypes were measured including (1) axial dystonia, contralateral twisted posturing of the neck and/or upper body; (2) abnormal orolingual movements, stereotyped jaw movements and/or contralateral tongue protrusion; and (3) abnormal forelimb movements, repetitive rhythmic jerks or dystonic posturing of the contralateral forelimb and/or grabbing movements of the contralateral paw (Bordia et al., 2008; Bordia et al., 2010; Carta et al., 2006; Cenci et al., 1998; Cenci et al., 2002). Rats were scored on a scale from 0 to 4 for each of these three AIM subtypes as follows: 1 = occasional; 2 = frequent; 3 = continuous but interrupted by sensory distraction; and 4 = continuous, severe, not interrupted by sensory distraction. Animals were evaluated for AIMs by a rater blinded to treatment condition for an initial 20 min baseline period, and then for 3 h following L-dopa injection. All the rats developed AIMs to varying extents, with none excluded from the study. Assessment of the different AIM subtypes was done over 20 min sessions, thus yielding a total of 9 sessions of testing per animals. The maximum possible score for each animal was 108 (maximum score per session = 12; number of sessions over a 3 h period = 9).
Fig. 4.

Decline in L-dopa-induced AIMs with TC-2696. The treatment regimen is depicted in the upper panel. All rats were unilaterally lesioned with 6-OHDA as described in Methods. They were then pretreated with TC-2696 (1.0 mg/kg/d) for 2 wk via minipump and subsequently injected with L-dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc). After several wk at 1.0 mg/kg/d TC-2696, the initial minipump was replaced with one releasing 2.8 mg/kg/d. The rats were assessed for axial, oral and forelimb AIMs as indicated in the timeline, with the total AIMs representing the sum of these three components. The daily time course of the total L-dopa-induced AIMs is depicted in the graphs at the right. Values are the mean ± S.E.M. of 9–10 rats per group. Significance of difference from vehicle: *p < 0.05, **p < 0.01.
Fig. 7.

TC-10600 reduced L-dopa-induced AIMs. The treatment regimen with TC-10600 is depicted in the upper panel. All rats were unilaterally lesioned with 6-OHDA, next pretreated with TC-10600 (1.0 mg/kg/d) for 2 wk via minipump and then injected with L-dopa plus benserazide (8 mg/kg plus 15 mg/kg sc). After several wk at 1.0 mg/kg/d TC-10600, the initial minipump was replaced with one releasing 1.8 mg/kg/d. The rats were assessed for axial, oral, and forelimb AIMs as indicated in the timeline, with the total AIMs being the sum of these three components. The daily time course of the total L-dopa-induced AIMs is shown in the graphs at the right. Values are the mean ± S.E.M. of 7–9 rats per group. Significance of difference from vehicle: *p < 0.05.
Fig. 8.

Reduction in L-dopa-induced AIMs with sazetidine. The treatment regimen with sazetidine is depicted in the upper panel. All rats were unilaterally lesioned with 6-OHDA, subsequently injected with L-dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc) for several wk, after which they were treated with sazetidine at the indicated doses (twice daily sc). The rats were assessed for axial, oral and forelimb AIMs as depicted in the timeline, with the total AIMs representing the sum of the three components. The daily time course of the total L-dopa-induced AIMs is depicted in the graphs at the right. Values are the mean ± S.E.M. of 9–10 rats per group. Significance of difference from vehicle: *p < 0.05.
2.5. Limb use asymmetry test
The forelimb asymmetry test was used as an index of motor function after unilateral nigrostriatal damage (Bordia et al., 2008; Bordia et al., 2010; Schallert et al., 2000; Tillerson et al., 2002). Animals were placed in a transparent cage and evaluated over a 5 min period for forepaw placement against the walls of the cage. A mirror was arranged behind the cage to allow the raters to view forelimb movements when the rat was turned away from the rater. Wall exploration was expressed in terms of the percentage of use of the impaired forelimb (contralateral to the lesion) compared to the total number of limb use movements. Raters were blinded with respect to the treatment status of the rats.
2.6. Autoradiography
Rats were killed by CO2 exposure, and the brains quickly removed and frozen in isopentane on dry ice. A series of 8 μm sections from the mid-portion of the striatum (between Bregma 0.2 and −0.30) were cut at −15°C in a cryostat, thaw mounted onto poly-L-lysine coated slides, dried, and stored at −80°C.
Dopamine transporter (DAT) autoradiography was assessed using 125I-RTI-121 (specific activity, 2200 Ci/mmol; PerkinElmer Analytical and Life Sciences, Boston, MA, USA) as described (Bordia et al., 2008). Sections were preincubated twice for 15 min each at room temperature in buffer (50 mM Tris-HCl, pH 7.4, 120 mM NaCl, 5 mM KCl). This was followed by a 2 h incubation at room temperature with 50 pM 125I-RTI-121 in the same buffer also containing 0.025% BSA and 1 μM fluoxetine. Binding was terminated by washing the sections 4 times 15 min each in ice cold buffer, and then once for 10 s in ice cold deionized water. Nonspecific binding was determined in the presence of 100 μM of the dopamine uptake inhibitor nomifensine.
α6β2* nAChR levels were assayed using 125I-α-conotoxinMII (α-CtxMII) (specific activity, 2200 Ci/mmol) (Quik et al., 2003). Sections were preincubated at room temperature for 15 min in binding buffer (20 mM Hepes Buffer, 144 mM NaCl, 1.5 mM KCl, 2.0 mM CaCl2, 1.0 mM MgSO4), plus 1.0 mM phenylmethylsulfonyl fluoride. This was followed by a 1 h incubation at room temperature in binding buffer plus 0.5% BSA, also containing 5 mM EDTA, 5 mM EGTA, 10 μg/ml each of aprotinin, leupeptin, pepstatin A, and 0.5 nM 125I-α-CtxMII. Nicotine (100 μM) was used to determine nonspecific binding. The assay was stopped by washing the slides for 10 min, in ice-cold binding buffer, 2× 10 min in 0.1X buffer at 0°C and two final 5 s washes in ice-cold deionized water.
α4β2* nAChR levels were determined using 125I-epibatidine (specific activity, 2200 Ci/mmol; PerkinElmer Analytical and Life Sciences, Boston, MA, USA) in the presence of 100 nM α-CtxMII to block α6β2* nAChRs (Quik et al., 2003). Slides were pre-incubated at room temperature for 15 min at room temperature in the following buffer; 50 mM Tris, pH 7.5, 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, and 1.0 mM MgCl2. A 40 min incubation was then done at room temperature in the presence of 0.015 nM 125I-epibatidine. Nonspecific binding was evaluated in the presence of nicotine (100 μM). The binding reaction was stopped by washing the sections 2 ×5 min in ice-cold buffer, 1 ×10 s in ice cold deionized water.
For all autoradiographic assays, the dried slides were exposed to Kodak MR film (Eastman Kodak, Rochester, NY) with 3H microscale standards (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) for several days.
2.7. Competition binding to nAChRs in membrane preparations
Binding to α7, α3β4 and muscle nAChRs was done as described using a membrane binding assay (Davies et al., 1999; Lippiello and Fernandes, 1986). Membranes were prepared from SH-SY5Y cells, TE671 cells or CHO cells expressing either human α3β4 or α7 nAChRs (generously provided by Dr. J. Lindstrom, University of Pennsylvania). They were then incubated for 2 hours at 25°C in phosphate buffered saline in the absence of presence of varying concentrations of the compounds (0.001 nM to 100 μM) and 3H-epibatidine (Perkin-Elmer Life Sciences). Incubation was terminated by rapid filtration, and the retained radioactivity was determined by liquid scintillation counting using Perkin-Elmer Trilux Microbeta (Waltham, MA). Nonspecific binding was defined with 10 μM of epibatidine.
2.8. 3H-Dopamine release from striatal synaptosomes
Dopamine release studies were performed using striatal synaptosomes obtained from rat brain as previously described (Bencherif et al., 1998). Striatal tissue from two rats (female, Sprague-Dawley, weighing 150–250 g) was pooled and homogenized in ice-cold 0.32 M sucrose (8 ml) containing 5 mM HEPES, pH 7.4, using a glass/glass homogenizer. The tissue was then centrifuged at 1,000 × g for 10 min. The pellet was discarded and the supernatant was centrifuged at 12,500 × g for 20 min. The resulting pellet was re-suspended in ice-cold perfusion buffer containing monoamine oxidase inhibitors (128 mM NaCl, 1.2 mM KH2PO4, 2.4 mM KCl, 3.2 mM CaCl2, 1.2 mM MgSO4, 25 mM HEPES, 1 mM ascorbic acid, 0.02 mM pargyline HCl and 10 mM glucose, pH 7.4) and centrifuged for 15 min at 23,000 × g. The final pellet was re-suspended in perfusion buffer (2 ml) for immediate use.
The synaptosomal suspension was incubated for 10 min in a 37°C shaking incubator to restore metabolic activity. 3H-Dopamine, specific activity = 28.0 Ci/mmol, NEN Research Products) was added at a final concentration of 0.1 μM and the suspension was incubated at 37°C for another 10 min. Aliquots of perfusion buffer (100 μl) and tissue (100 μl) were loaded into the suprafusion chambers of a Brandel Suprafusion System (series 2500, Gaithersburg, MD). Perfusion buffer (room temperature) was pumped into the chambers at a rate of approximately 0.6 ml/min for a wash period of 8 min. Nicotine (10 μM) or the indicated concentrations of compound were then applied in the perfusion stream for 48 s. Fractions (12 s each) were continuously collected from each chamber throughout the experiment to capture basal release and agonist-induced peak release and to re-establish the baseline after the compound application. Released 3H-dopamine was quantified by scintillation counting. For each chamber, the integrated area of the peak was normalized to its baseline. Release was expressed as a percentage of release obtained with control nicotine. Within each assay, each test compound concentration was replicated using 2 chambers; replicates were averaged. The compound concentration resulting in half maximal activation (EC50) of specific release was determined using GraphPad Prism (La Jolla, CA).
2.9. Data and statistical analysis
The ImageQuant system and software (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) was used for quantification of optical density measures. Data was converted to fmol/mg tissue using standard curves generated from radioactive 3H-standards also exposed to the films. Specific binding was determined by subtracting total binding from non-specific binding. The standards were calibrated for 125I-autoradiography, as described (Artymyshyn et al., 1990), with the optical density readings of the samples always within the linear range of the film. For the competition studies, the Ki was defined as follows: Ki = IC50/[1 + ligand concentration /Kd], where the concentration of 125I- α -CtxMII was 0.5 nM and that of 125I-epibatidine was 0.015 nM; the Kd value for 125I- α -CtxMII binding is 1.0 nM and that of 125I-epibatidine binding 0.1 nM.
All statistics were performed using GraphPad Prism® (GraphPad Software, Inc, San Diego, CA.). Values are the mean ± SEM of the indicated number of rats, and represent data from one to two separate experiments. Differences in rating scores between groups were analyzed using nonparametric tests (Mann-Whitney test). For the time course studies, repeated measures analysis of variance (ANOVA) followed by Bonferroni multiple comparison test was used. A level of 0.05 was considered significant.
3. Results
3.1. Characteristics of the severely-lesioned parkinsonian rat model
The results in Table 1 show that there is an almost complete loss of the striatal dopamine transporter, a measure of dopamine nerve terminal integrity, in the lesioned striatum of rats after intracranial 6-OHDA administration. The results shown are the dopamine transporter values for the different sets of rats for each of the treatment regimens. Sections were taken from the mid- portion of the striatum between Bregma 0.2 and −0.30. Our previous data had shown there were no anterior to posterior gradients in transporter losses with lesioning (Quik et al., 2003). Quantitative analyses showed that the dopamine transporter density measurements (Table 1) on the lesioned side were ≤1% of the control side, with essentially no signal uniformly across the striatum. There was some variation in dopamine transporter expression in control rats most likely due to differences in the batches of the radioisotope (125I-RTI-121), in the radioisotope standards and other reagents and chemicals used, as these studies were done over a two to three year period.
TABLE 1.
Effect of severe nigrostriatal damage on the dopamine transporter in rat striatum for each of the different studies. Rats were unilaterally lesioned with 6-OHDA as described in Materials and Methods. 125I-RTI-121 autoradiography was done to measure the striatal dopamine transporter in the different sets of studies. Sazetidine was tested after a 5 wk washout of the rats in the TI-10165 study. Values represent the mean ± SEM of 10 to 12 rats.
| Drug Study | Dopamine transporter (fmol/mg tissue)
|
% Unlesioned | |||
|---|---|---|---|---|---|
| Unlesioned side | Lesioned side | ||||
| TC-2696 | 24.2 | ± 1.26 | 0.12 | ± 0.12 | 0.5 |
| TI-10165 | 11.8 | ± 0.62 | 0.11 | ± 0.08 | 0.9 |
| TC-8831 | 33.7 | ± 0.85 | 0.35 | ± 0.18 | 1.0 |
| TC-10600 | 15.5 | ± 1.20 | 0.04 | ± 0.02 | 0.3 |
A new set of animals was used for each compound to avoid carry over effects from previous treatments, except for sazetidine. Sazetidine was tested after a 5 wk washout of the rats initially treated with TI-10165; AIMs had returned to control levels after this time period (data not shown).
We also measured α4β2* and α6β2* nAChRs to determine the extent of loss of these two receptor populations with lesioning. Large declines were observed in α6β2* nAChRs (97%) (p < 0.001) in the lesioned striatum, as expected since these receptors are present on nigrostriatal dopamine terminals (Quik et al., 2001; Quik et al., 2003). By contrast, α4β2* nAChR were reduced by only 12% (p < 0.05) compared to the unlesioned side (Table 2), as previously shown (Quik et al., 2003). For these studies, we measured receptors in control rats from the TI-10165 study. These data are consistent with earlier findings that the majority of α4β2* nAChRs (75–80%) are present on nondopaminergic neurons in the striatum (Kulak et al., 2002; Quik et al., 2003).
TABLE 2.
Effect of the 6-OHDA lesion on α6β2* nAChRs and α4β2* nAChRs in rat striatum. 125I-Epibatidine binding in the presence of the α6β2* nAChR antagonist α-CtxMII was used to identify α4β2* nAChRs, while 125I-α-CtxMII was used to detect α6β2* nAChRs in striatum. The concentrations of 125I-α-CtxMII and 125I-epibatidine were 0.5 nM and 0.015 nM, respectively. Values represent the mean ± SEM of 8 rats 6-OHDA-lesioned rats not treated with nAChR drugs.
| nAChRs | fmol/mg tissue
|
% Control | |
|---|---|---|---|
| Unlesioned side | Lesioned side | ||
| α6β2* | 3.45 ± 0.13 | 0.10 ± 0.01*** | 2.9 |
| α4β2* | 6.86 ± 0.23 | 6.08 ± 0.03* | 88 |
Significance of difference from unlesioned side:
p < 0.05,
p < 0.001.
As a behavioral measure of parkinsonism, rats were rated for forelimb use asymmetry for a 5 min period. Forelimb use was similar on both the left and right sides in unlesioned rats, with values of 50.7 ± 5.1% and 49.3 ± 5.1 %, respectively. The current 6-OHDA lesioning protocol led to a reduction in % contralateral limb use ranging from 3–16% of the unlesioned side (Table 3). This residual forelimb use, despite an almost complete loss of striatal dopamine terminals, may be due to functional compensation mediated by dopamine terminals on the unlesioned side (Zigmond et al., 1990) and/or by other neurotransmitter systems. L-dopa treatment improved parkinsonism, as assessed using the forepaw placement test, in each of the 3 study groups, with a a significant main effect of L-dopa using two-way ANOVAs in the different groups as follows; TC-2696 (F(1,24) = 6.95, P < 0.05), TI-10165 (F(1,33) = 26.24, P < 0.0001), TC-8831 (F(1,35) = 28.28, P < 0.0001).
TABLE 3.
NAChR compounds do not modify parkinsonism either OFF or ON L-dopa. Vehicle and compound treated unilaterally lesioned rats were rated for forelimb use asymmetry to assess parkinsonism. Impaired forelimb use was measured for a 5 min period before and 60 min after administration of L-dopa, when its effect is maximal. A large reduction in % contralateral limb use is observed on the lesioned side. L-dopa treatment improved % contralateral limb use, while the nAChR compounds had no effect. Parkinsonism was not evaluated for TC-10600 or sazetidine. The values represent the mean ± S.E.M of 5–9 rats per group.
| Drug | L-dopa | % Contralateral limb use
|
|
|---|---|---|---|
| Vehicle | nAChR compound | ||
| TC-2696 | OFF | 3.57 ± 2.9 | 3.27 ± 1.5 |
| ON | 21.0 ± 8.5 | 35.6 ± 13.1 | |
| TI-10165 | OFF | 16.4 ± 3.3 | 16.8 ± 5.2 |
| ON | 39.3 ± 9.2 | 54.7 ± 5.9 | |
| TC-8831 | OFF | 12.3 ± 2.8 | 12.9 ± 3.0 |
| ON | 54.0 ± 12.1 | 51.3 ± 16.5 | |
3.2. Test compound characterization
The structures of the different nAChR compounds tested are shown in Fig. 1. Receptor competition studies were done to evaluate the ability of these compounds to interact at α4β2* and α6β2* nAChRs (Fig. 2 and Table 4). We used 125I-epibatidine binding in the presence of α-CtxMII to measure striatal α4β2* nAChRs, and 125I-α-CtxMII to identify striatal α6β2* nAChRs. All the nAChR compounds interacted with both nAChR subtypes with a similar affinity in the binding studies. Data for nicotine, varenicline and sazetidine are provided for comparison. The affinity of the compounds for α4β2* nAChRs ranged from 0.31 nM for varenicline to 30.2 nM for TI-10165. Thus, the affinity of the compounds for α6β2* nAChRs was generally comparable to that for the α4β2* nAChRs (Table 4).
Fig. 1.

Chemical structures of nAChR compounds included in this study.
Fig. 2.
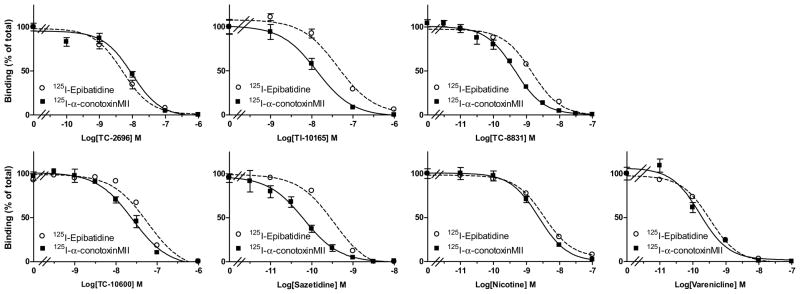
Receptor competition studies with nAChR compounds. 125I-Epibatidine autoradiography (in the presence of the α6β2* nAChR antagonist α-CtxMII) and 125I-α-CtxMII autoradiography were done to evaluate the interaction of the different compounds at α4β2* and α6β2* nAChRs, respectively, in rat striatum. The compounds yielded similar competition curves for α4β2* and α6β2* nAChR binding. Each value represents the mean ± SEM of 8 to 10 rats.
TABLE 4.
Affinity of nAChR compounds for various nAChR subtypes. Autoradiography was used to measure the Ki of the nAChR drugs for α4β2* and α6β2* nAChRs (columns 2 and 3) using control rat striatal sections. 125I-Epibatidine binding in the presence of the α6β2* nicotinic receptor antagonist α-CtxMII was used to identify α4β2*, while 125I-α-CtxMII was used to detect α6β2* nAChRs. Compounds were tested at concentrations ranging from 10−14 to 10−6 M, depending on the specific compound. Each value represents the mean ± SEM of 8 to 10 rats. To determine the interaction of nAChR compounds at α7, α3β4 and muscle nAChRs membrane binding studies were done (columns 4, 5 and 6). Membranes were prepared from SH-SY5Y cells (α7 nAChRs), TE671 cells (muscle nAChRs) or CHO cells expressing either human α3β4 nAChRs or human α7 nAChRs. Binding to the different nAChR subtypes was done as described in Materials and Methods. Binding data are expressed as percent total specific binding, with replicates for each point averaged and plotted against the log concentration of the compound. The values represent an average ± SEM of a seven-point concentration-response curve run in multiple assays.
| Compound | Ki α4β2* nAChRs (nM) | Ki α6β2* nAChRs (nM) | Kiα7 nAChRs (nM) | Ki α3β4 nAChRs (nM) | Ki Muscle(nM) |
|---|---|---|---|---|---|
| TC-2696 | 5.32 ± 0.23 | 6.54 ± 0.62 | 85,000 ± 14682 | 14,000 ± 5046 | > 100,000 |
| TI-10165 | 30.2 ± 0.8 | 8.60 ± 0.90 | 15,000 ± 5170 | 620 ± 153 | 36,000 ± 15612 |
| TC-8831 | 1.36 ± 0.01 | 0.29 ± 0.03 | 620 ± 95 | 270 ± 63 | 470 ± 86 |
| TC-10600 | 17.4 ± 3.40 | 51.8 ± 2.40 | 790 ± 73 | 1,100 ± 159 | 7,100 ± 1641 |
| Sazetidine | 0.78 ± 0.34 | 0.05 ± 0.01 | 3,300 ± 745 | 30 ± 2 | 180 ± 84 |
| Nicotine | 3.10 ± 0.16 | 1.56 ± 0.09 | 2,800 ± 438 | 350 ± 101 | 3,500 ± 2637 |
| Varenicline | 0.31 ± 0.03 | 0.08 ± 0.01 | 290 ± 34 | 160 ± 84 | 160,000 ± 9206 |
The nAChR compounds were also tested for their ability to interact with α7, α3β4 and muscle-type nAChRs to determine their selectivity for β2* receptors. As shown in (Table 4), the compounds tested were at least 15-fold less potent at the α7, α3β4 and the α1β1 δγ nAChR compared to the β2* subtypes, and in most cases the selectivity was much greater.
Studies were subsequently done to determine the functional characteristics of the different nAChR compounds (Fig. 3). To approach this, we measured total 3H-dopamine release from rat striatal synaptosomes. The data in Table 5 show that potency among the tested compounds ranged from 0.14 μM for TC-8831 to 6.3 μM for TC-2696. The effect of the drugs on α4β2* and α6β2* nAChR-mediated release was not determined.
Fig. 3.

Dose response curves of compound-induced 3H-dopamine release from rat striatal synaptosomes. Release was done as described in Methods at the concentrations depicted on the x-axis. Values are expressed as the % 3H-dopamine release relative to that occurring with 10 μM nicotine and represent the mean ± SEM of 3 to 4 rats.
TABLE 5.
EC50 and Rmax values for compound-evoked 3H-dopamine release from rat striatum. Rat striatal synaptosomes were exposed to the nAChR compounds to evoke 3H-dopamine release. Release is expressed as the % release relative to that occurring with 10 μM nicotine. For the EC50 values, the numbers in parentheses represent the 95% confidence intervals (CI). Rmax values are expressed as mean ± SEM of 3 to 4 assays, with 2 rats per assay.
| Compound | EC50 (CI) μM | Rmax % of 10 μM nicotine |
|---|---|---|
| TC-2696 | 6.3 (1.2 – 31) | 204 ± 43 |
| TI-10165 | 0.81 (0.14 – 4.0) | 78 ± 9.1 |
| TC-8831 | 0.14 (0.10 – 0.26) | 91 ± 3.8 |
| TC-10600 | 1.49 (0.65 – 3.39) | 72 ± 5.7 |
| Sazetidinea | 0.001 | 96 ± 6 |
| Nicotine | 3.0 (1.3 – 7.3) | 100 ± 8.9 |
| Varenicline | 0.05 (0.01 – 0.20) | 41 ± 5.0 |
Taken from (Zwart et al., 2008)
3.3. nAChR compounds reduced L-dopa induced AIMs in severely lesioned rats
The effect of the different nAChR compounds on L-dopa-induced AIMs in rats are depicted in Figs. 4–8 and summarized in Table 6. The data for the first of these (TC-2696) is shown in Fig. 4. As depicted in the timeline, all rats were first lesioned with 6-OHDA and then tested for amphetamine-induced rotations. Five wk after 6-OHDA lesioning, they were implanted with minipumps containing vehicle or TC-2696 (1.0 mg/kg/d). This dose was selected based on previous studies which showed optimal effects on cognitive behavior (unpublished results). L- dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc) treatment was then initiated two weeks after the start of TC-2696. The rats were evaluated for axial, oral and forelimb AIMs, with total AIMs representing the sum of these three components, 1 and 3 wk after L-dopa treatment was started (AIMs I and II). Compared to control rats, there were fewer total L-dopa-induced AIMs in rats pretreated with TC-2696. Ratings of the different AIM subtypes showed that this was primarily due to a reduction in axial AIMs with no significant decline in forelimb and oral AIMs. The effect of this dose of TC-2696 persisted for a further 4 wk of compound administration. The rats were then implanted with new minipumps releasing 2.8 mg/kg/d TC-2696. AIMs (AIMs III and IV) were tested 1 and 3 wk after minipump implantation, with results similar to those at the 1.0 mg/kg/d dose of the compound, except for a significant decline in forelimb AIMs (Fig. 4). The time course studies show that vehicle-treated rats (Fig. 4) develop AIMs maximal by 60–120 min after L-dopa injection, which gradually diminished over time. TC-2696 treatment reduced AIMs with a significant main effect of treatment (F(1,152) = 5.76, P < 0.05) and a significant interaction (F(8,152) = 2.44, P < 0.05) at the 1 mg/kg/d dose (repeated measures ANOVA). There was also a significant main effect of treatment (F(1,152) = 7.95, P < 0.05) and a significant interaction (F(8,152) = 2.56, P < 0.05) with the 2.8 mg/kg/d dose. The rats were tested for parkinsonism using the forepaw asymmetry test 4 wk after the initiation of L-dopa treatment, with no significant effect of the test compound (Table 3).
TABLE 6.
Summary of the effects of nicotinic receptor compounds on L-dopa-induced AIMs in rats with severe dopaminergic lesions. Rats were lesioned and pre-treated with compound for 2 wk. They were then given L-dopa (6 or 8 mg/kg) plus benserazide (15mg/kg) sc for several wk as indicated in the timelines for the different nAChR compounds in Figs 4 to 8. L-dopa-induced AIMs were rated throughout with the values below representing the maximal decrease in total AIM scores over the treatment periods depicted in Figs. 4 to 8. Values are the mean ± SEM of 7–13 rats per group.
| Compound | Dose (mg/kg/day) | Decrease in total AIM scores | Total AIMs score | AIM subtypes significantly affected by nAChR compounds |
|---|---|---|---|---|
| TC-2696 (Fig. 4) | 1.0 – 2.8 | 30% | ~75 | Axial > forelimb |
| TI-10165 (Fig. 5) | 0.1 – 0.3 | 32% | ~55 | Oral |
| TC-8831 (Fig. 6) | 0.3 – 0.75 | 24% | ~55 | Oral = axial = forelimb |
| TC-10600 (Fig. 7) | 1.0 – 1.8 | 32% | ~60 | Oral |
| Sazetidine (Fig. 8) | 0.5 – 2.0 | 23% | ~75 | Oral = forelimb |
| Nicotinea | 0.5 – 2.0 | 35% | ~75 | Oral = forelimb |
| Vareniclineb | 0.5 – 2.0 | 10% | ~60 |
Taken from (Bordia et al., 2010);
Taken from (Huang et al., 2011b).
A new set of rats was then treated with TI-10165 (Fig. 5). Briefly, all rats were unilaterally lesioned with 6-OHDA and then implanted with minipumps containing vehicle or TI-10165 (0.1 mg/kg/d). After 2 wk, L-dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc) was initiated. The rats were evaluated for L-dopa-induced AIMs at the time points indicated in Fig. 5. In rats pretreated with TI-10165, there were fewer total L-dopa-induced AIMs compared to rats receiving vehicle (Fig. 5). Analyses of the different AIM subtypes indicated that this was primarily due to a reduction in oral AIMs, with no significant decline in axial and forelimb AIMs. Assessment of impaired forepaw use showed that there was no significant difference in motor function with TI-10165 with L-dopa-treatment (Table 3). After several wk at 0.1 mg/kg/d TI-10165, the initial minipump was replaced with one releasing 0.3 mg/kg/d. The decline in L-dopa-induced AIMs was similar to that observed with the lower dose. The time course studies show that vehicle-treated rats (Fig. 5) develop AIMs maximally by 60–120 min after L-dopa injection, and that this gradually diminished over time. TC-10165 treatment reduced AIMs with a significant main effect of treatment (F(1,160) = 4.09, P = 0.0567) and no significant interaction at the 0.1 mg/kg/d dose (repeated measures ANOVA). There was no significant main effect of treatment nor a significant interaction with the 0.3 mg/kg/d dose.
Fig. 5.

Reduction in L-dopa-induced AIMs with TI-10165. The treatment regimen with TI-10165 is depicted in the upper panel. All rats were unilaterally lesioned with 6-OHDA, pretreated with TI-10165 (0.1 mg/kg/d) for 2 wk via minipump and subsequently injected with L-dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc). After several wk at 0.1 mg/kg/d TI-10165, the initial minipump was replaced with one releasing 0.3 mg/kg/d. The rats were assessed for axial, oral and forelimb AIMs as indicated in the timeline, with the total AIMs representing the sum of these three components. The daily time course of the total L-dopa-induced AIMs is depicted in the graphs at the right. Values are the mean ± S.E.M. of 9–10 rats per group. Significance of difference from vehicle: *p < 0.05.
We next tested the effect of TC-8831 against L-dopa-induced AIMs (Fig. 6). Five wk after lesioning, rats were pretreated with TC-8831 via minipump at a dose of 0.75 mg/kg/d. Daily L-dopa plus benserazide (6.0 mg/kg plus 15 mg/kg sc) treatment was initiated 2 wk later. The rats were subsequently assessed for total, axial, oral and forelimb AIMs over the course of several wk as indicated in Fig. 6. TC-8831 reduced total AIMs (Fig. 6) and all L-dopa-induced AIM components. The L-dopa treatment was continued but the initial minipump containing TC-8831 was replaced with one releasing a lower dose of TC-8831 (0.3 mg/kg/d) to determine if the effect was maintained at a lower dose. AIMs were again tested 2 and 4 wk after implantation. At the lower dose, there was a significant decrease in all L-dopa-induced AIM components. The time course studies show that vehicle-treated rats develop maximal AIMs by 60–120 min after L-dopa injection, with a decline to baseline by 3 h. While there was a trend for a decrease in AIMs with TC-8831, there was no significant main effect of treatment although there was a significant interaction (F(8,168) = 2.20, P < 0.05) at the 0.75 mg/kg/d dose (repeated measures ANOVA). There was a significant main effect of treatment (F(1,176) = 4.44, P < 0.05) and a significant interaction (F(8,176) = 3.23, P < 0.01) with the 0.3 mg/kg/d dose. TC-8831 did not affect parkinsonism (Table 3).
Fig. 6.

TC-8831 decreased L-dopa-induced AIMs. The treatment regimen, depicted in the upper panel, shows that all rats were unilaterally lesioned with 6-OHDA. This was followed by pretreatment with TC-8831 (0.75 mg/kg/d) for 2 wk via minipump and subsequent injection with L-dopa plus benserazide (6.0 mg/kg plus 15 mg/kg sc). After several wk at 0.75 mg/kg/d TC-8831, the initial minipump was replaced with one releasing 0.3 mg/kg/d. Rats were rated for axial, oral and forelimb AIMs as indicated in the timeline, with the total AIMs being the sum of these three components. The daily time course of total L-dopa-induced AIMs is provided in the graphs at the right. Values are the mean ± S.E.M. of 10–13 rats per group. Significance of difference from vehicle: *p < 0.05, **p < 0.01, ***p < 0.001.
The effect of TC-10600 is depicted in Fig. 7. Unilaterally lesioned rats were pretreated with TC-10600 (1.0 mg/kg/d) for 2 wk via minipump, followed by initiation of daily injection with L-dopa plus benserazide (8 mg/kg plus 15 mg/kg sc). There was no significant decline in total L-dopa-induced AIMs at the 1.0 mg/kg/d dose. The initial minipump containing TC-10600 was then replaced with one releasing a higher dose (1.8 mg/kg/d), with the L-dopa/benserazide treatment remaining throughout. A significant decline in total and oral L-dopa-induced AIMs was observed at the higher dose of 1.8 mg/kg/d (P < 0.05). There was also a trend for a decline in AIMs in the daily time course studies with TC-10600. However, there was no significant main effect of treatment and no significant interaction with 1.0 mg/kg/d. Similarly, there was no significant main effect of treatment nor significant interaction with the 1.8 mg/kg/d dose. Parkinsonism was not evaluated in this group, thus it is possible that antidyskinetic effect was due to motor suppression.
Sazetidine was also tested for its ability to reduce L-dopa-induced AIMs (Fig. 8). For these studies we used the groups of rats that had been treated with TI-10165 and that had subsequently undergone a washout period for 5 wk. They were injected with L-dopa plus benserazide (8.0 mg/kg plus 15 mg/kg sc) throughout the washout to maintain AIMs. They were subsequently injected sc with sazetidine at the indicated doses 10 min before L-dopa plus benserazide and also 8 h later. There was no effect of the 0.5 mg/kg dose on L-dopa-induced AIMS. There was a decline in total, oral and forelimb AIMs with the 1.0 mg/kg dose (P < 0.05). Results were similar with the 2.0 mg/kg dose (data not shown). There were also trends for a decline in the time course studies. However, at the 1.0 mg/kg/d dose there was no significant main effect of treatment and no significant interaction. Parkinsonism was not tested.
These combined data show that β2* selective nAChR compounds reduced L-dopa-induced AIMs in rats with severe nigrostriatal damage (see Table 6 for summary).
4. Discussion
The present studies were conducted to understand the role of non-dopaminergic α4β2* nAChRs in the nicotine-mediated reduction in L-dopa-induced AIMs in severely parkinsonian animals (Bordia et al., 2008; Bordia et al., 2010; Quik et al., 2007). This is important because numerous non-dopaminergic systems contribute to the occurrence of these abnormal involuntary movements (Brotchie and Jenner, 2011; Carta et al., 2008a; Cenci and Konradi, 2010; Jenner, 2008; Morin et al., 2010). In addition, there are multiple CNS nAChRs with which nicotine interacts to modulate function of non-dopaminergic neurotransmitter systems. Identification of the subtypes relevant to L-dopa-induced AIMs will help in the development of targeted therapies that have an improved side effect profile compared to that of nicotine. In the present study we focused on the nAChRs in the striatum, since an extensive literature indicates that this brain region plays a key role in the generation of L-dopa-induced AIMs (Brotchie and Jenner, 2011; Carta and Bezard, 2011; Cenci et al., 2011; Fisone and Bezard, 2011; Iravani et al., 2012).
Candidate nAChRs in the striatum that may be involved in the nAChR-mediated reduction in L-dopa-induced AIMs include the α4β2*, α6β2* and α7 nAChR populations (Millar and Gotti, 2009; Quik and Wonnacott, 2011). Studies using genetically modified nAChR mice have shown that β2* nAChRs play an essential role in the development and maintenance of levodopa-induced dyskinesias. Significantly fewer L-dopa-induced AIMs were observed in β2 knockout mice compared to their wild-type littermates, while nicotine treatment elicited no further reduction in AIMs in β2 knockout mice compared to their wild-type littermates (Huang et al., 2011a). Subsequent studies with mice lacking α6 nAChRs, yielded similar results as with β2 knockout mice. These latter data suggest that the α6β2* nAChR population may be critical for the development of L-dopa-induced AIMs and for nicotine’s anti-dyskinetic effects (Quik et al., 2012b). There are several α6β2* nAChR subtypes in striatum including α6 α4β2β3, α6β2β3 and αβ2 subtypes (Grady et al., 2010; Letchworth and Whiteaker, 2011; Millar and Gotti, 2009; Quik and Wonnacott, 2011). Continued studies are necessary to determine which populations are most important for the nicotine-mediated reduction in L-dopa-induced AIMs.
In addition to α6β2* nAChRs, another subtype that may be involved in the nAChR-mediated decline in L-dopa-induced AIMs is the α4β2* population present in striatum. The α4β2* nAChR are more widely expressed in striatum than the α6β2* nAChRs, which are present primarily on striatal dopaminergic neurons (Gotti et al., 2009; Millar and Gotti, 2009; Quik et al., 2011). By contrast, only 20% of α4β2* nAChRs are located on dopaminergic terminals while the remaining 80% are on other striatal neuronal populations, possibly GABAergic or glutamatergic neurons (Gotti et al., 2009; Millar and Gotti, 2009; Quik et al., 2011). Because of this differential distribution of α6β2* and α4β2*, one approach to understand the role of α4β2* nAChRs is to lesion dopaminergic neurons. The α6β2* nAChRs would be lost with extensive nigrostriatal damage, while the majority of the α4β2* receptors would remain. The present studies show that compounds that interact with both α4β2* and α6β2* nAChRs still reduce L-dopa-induced AIMs with severe nigrostriatal damage. These data would suggest that α4β2* nAChRs on non-dopaminergic neurons are also involved in the occurrence of L-dopa-induced AIMs. Previous nAChR antibody immunoprecipitation studies by Zoli and coworkers had shown that striatal nAChRs expressing the α5 subunit are lost with severe nigrostriatal damage (Zoli et al., 2002). The only nAChR expressing this subunit is the α4 α5β2 subtype. Since the α4 α5β2 subtype is lost with severe lesioning, these findings suggest that the primary nAChR population remaining in striatum is the α4β2 nAChR (Zoli et al., 2002). These combined studies suggest that drugs acting at α4β2 nAChRs may help reduce L-dopa-induced dyskinesias with severe nigrostriatal damage as occurs in late stage Parkinson’s disease. Our earlier work using α6 knockout mice had shown that α6β2* nAChRs were also involved in the nicotine-mediated decline in L-dopa-induced AIMs primarily in mice with moderate nigrostriatal damage. These data, coupled with the present results, suggest that an interaction at α6β2* nAChRs may be more important with a mild-moderate lesion while α4β2* nAChRs play a greater role with more severe lesioning.
The current results show that α4β2* nAChRs mediate a 20–30% reduction in L-dopa-induced AIMs. Our previous work with β2 and α6 knockout mice indicated that the maximum reduction in AIMs by gene deletion or nicotine administration was 40–60% (Huang et al., 2011a; Quik et al., 2012a). In addition, we had shown that nicotine appeared to lead to a maximal 50–60% decline in L-dopa-induced dyskinesias in rats and monkeys (Bordia et al., 2008; Bordia et al., 2010; Quik et al., 2007; Quik et al., 2013). Altogether, this suggests there may be a floor effect such that the maximum nicotine-mediated reduction in dyskinesias ranges around 40–60%, depending on the specific treatment conditions and species.
A point of note is that our previous studies had shown that the agonist varenicline did not significantly reduce L-dopa-induced AIMs in severely lesioned rats although there was a non-significant 10% decline in their appearance (Huang et al., 2011b). A possible explanation for its reduced effectiveness may relate to the fact that varenicline is a partial agonist and thus may not adequately activate the relevant nAChR populations involved in the nicotine-mediated reduction in L-dopa-induced AIMs (Reperant et al., 2009). Alternatively or as well, the ability of varenicline to activate other neurotransmitter receptors (5-HT) may interfere with its effectiveness in reducing L-dopa induced AIMs (Lummis et al., 2011).
The current data show that nAChR compounds reduced L-dopa-induced AIMs in rats with extensive nigrostriatal damage. However, in a recent nonhuman primate study, nicotine did not significantly attenuate L-dopa-induced dyskinesias in monkeys with severe nigrostriatal damage at doses that successfully reduced dyskinesias in animals with a more moderate lesion (Quik et al., 2013). These discrepant effects in the two parkinsonian animal models may be due to species differences in, for instance, receptor subtype expression. Mode of administration also varied between the studies, with drugs given to the rats via minipump but to nonhuman primates via the drinking water.
Another nAChR subtype in the striatum is the α7 nAChR, although it is present only at a very low density in most species except mice (Millar and Gotti, 2009; Quik and Wonnacott, 2011). α7 nAChRs are located on cortical glutamatergic afferents that indirectly regulate dopamine release (Kaiser and Wonnacott, 2000). They may therefore represent another potential site for regulating L-dopa-induced dyskinesias; however, their role remains to be elucidated. The compounds in the current study had minimal interaction with α7 nAChRs.
In the present studies we focus on striatal nAChRs because both α4β2* and α6β2* nAChRs are highly expressed in the striatum, a region very important to Parkinson’s disease pathology and because these nAChRs are associated with changes in locomotor activity (Quik and Wonnacott, 2011). However, it should be noted that α4β2* nAChRs are not only present in striatum but also highly expressed in the thalamus, multiple cortical areas, hippocampus, globus pallidus, cerebellum, and other brain regions (Grady et al., 2010; Millar and Gotti, 2009; Quik and Wonnacott, 2011). Thus, although our focus has been on the striatum it is possible that other brain regions are involved in the α4β2* nAChR-mediated improvement in L-dopa-induced AIMs. This includes the cortex, thalamus and cerebellum which are all implicated in motor control (Haber and Calzavara, 2009; Manto et al., 2012; Penhune and Steele, 2012).
The mechanism(s) whereby an interaction at α4β2* nAChRs improves L-dopa-induced AIMs remains to be identified. Our results show that TC-2696, TI-10165, TC-8831 and TC-10600, like nicotine and varenicline, stimulate striatal dopamine release indicating that they have full or partial agonist properties, at least at β2* nAChRs on striatal dopaminergic terminals. Sazetidine also has nAChR agonist properties and stimulates dopamine release (Zwart et al., 2008). It is well known that agonists lead to nAChR desensitization, which results in an effective block of receptor function (Buccafusco et al., 2009; McCarthy et al., 2011; Picciotto et al., 2008). In fact, it has been suggested that nAChR agonists such as varenicline and sazetidine may act via nAChR desensitization or inactivation (Hussmann et al., 2012; Rezvani et al., 2012; Rollema et al., 2010; Zwart et al., 2008). Such a mechanism for the nAChR-mediated reduction in L-dopa-induced AIMs is supported by results showing that the nAChR antagonist mecamylamine also attenuated AIMs in parkinsonian rats (Bordia et al., 2010).
Although all the nAChR compounds tested, except varenicline (Huang et al., 2011b), significantly reduced L-dopa-induced AIMs in rodents with severe dopaminergic lesions, they variably modulated the different AIM components. All of the drugs effectively reduced oral AIMs, except for TC-2696 that best reduced axial AIMs. In addition, some of the drugs most effectively reduced oral AIMs, while others were more like nicotine and reduced both oral and forelimb AIMs in a similar fashion. These varying effects on some AIM components and not others may relate to the differential interaction of the different drug with α4β2* and α6β2* nAChRs. For example, the drugs that only reduce oral AIMs (TI-10165 and TC10600) have the lowest affinities for α4β2* nAChRs. Thus, a greater interaction at α4β2* nAChRs may be necessary for the nAChR-mediated decline in forelimb AIMs. Another point of note is that the nAChR-mediated reduction in forelimb AIMs appears to occur primarily with the drugs that have a high affinity for α6β2* nAChRs, such as TC-8831, sazetidine and nicotine. In addition, the compounds may have subtle effects on other neurotransmitter receptors, although they may interact at such receptors with a much lower potency. Numerous neurotransmitter systems have been implicated in the development of L-dopa-induced AIMs. These include the serotonergic (Carta and Bezard, 2011; Carta et al., 2008b; Eskow et al., 2009; Eskow et al., 2007; Huot et al., 2011; Iravani and Jenner, 2011) and glutamatergic systems (Blandini and Armentero, 2012; Rylander et al., 2010; Sgambato-Faure and Cenci, 2012), and also the noradrenergic, opiate, cannabinoid, adenosine and/or histaminergic ones (Iravani and Jenner, 2011).
Notably the nAChR compounds tested reduced the occurrence of L-dopa-induced AIMs but did not significantly affect the L-dopa-mediated improvement in parkinsonism. This differential effect of nAChRs compounds on these two motor components is probably due to the fact that the neuronal circuits through which L-dopa improves the bradykinesia, tremor, and rigidity observed in Parkinson’s disease are unique from those through which it causes dyskinesias. In fact, the effect of L-dopa on parkinsonism is thought to occur via the indirect dopaminergic pathway, while the development of L-dopa-induced dyskinesias may predominantly involve the direct pathway (Gerfen and Surmeier, 2011; Guigoni et al., 2005; Obeso et al., 2008).
With respect to time course, several weeks of treatment were generally required for the nAChR compounds to reduce L-dopa-induced AIMs. This observation suggests that long term molecular alterations underlie the beneficial effect of these compounds. Although the intracellular mechanisms remain to be elucidated, alterations in calcium entry and signaling appear to be a first step (Hosur and Loring, 2011; Kawamata and Shimohama, 2011; Mudo et al., 2007; Picciotto and Zoli, 2008; Quik and Wonnacott, 2011; Shimohama, 2009; Ward et al., 2008). nAChR-mediated increases in calcium can then trigger any one of numerous downstream signaling pathways such as protein kinase A, extracellular signal-regulated mitogen-activated protein kinase (ERK/MAPK), calmodulin, phosphatidylinositol 3-kinase (PI3K)/Akt-or protein kinase B-dependent signaling, JAK2/STAT3, and others. Activation of these diverse signalling cascades may then modulate caspases, CREB and/or other mechanisms (Hosur and Loring, 2011; Kawamata and Shimohama, 2011; Mudo et al., 2007; Picciotto and Zoli, 2008; Quik and Wonnacott, 2011; Shimohama, 2009; Ward et al., 2008). Interestingly, tolerance did not develop to the beneficial effect of the nAChR compounds with up to 3 months of treatment. This is in agreement with our previous findings, which show that the effects of nicotine persisted with at least 4 months of treatment at which time the study was ended (Bordia et al., 2008; Bordia et al., 2010). These data suggest that the molecular mechanisms that are involved appear to endure with time.
In summary, the present results show that β2* nAChR compounds reduced L-dopa-induced AIMs in rats with severe 6-OHDA nigrostriatal damage. The primary nAChRs in the striatum are the α4β2* and α6β2* subtypes. However, the striatal α6β2* nAChRs are almost completely eliminated in severely lesioned rats since they are localized on dopaminergic neurons destroyed with 6-OHDA lesioning. These data suggest that α4β2* nAChRs are also involved in the nAChR-mediated decline in L-dopa-induced AIMs, at least with severe nigrostriatal damage. Thus, drugs targeting α4β2* nAChRs may be useful for reducing L-dopa-induced dyskinesias in late stage Parkinson’s disease.
HIGHLIGHTS.
Previous work showed that nicotine decreased L-dopa-induced dyskinesias via α6β2* nAChRs
Here, we show β2* nAChR drugs reduce L-dopa-induced dyskinesias with severe nigrostriatal damage
α4β2* nAChRs are primarily present in striatum after severe nigrostriatal damage
Thus striatal α4β2* nAChRs are also involved in L-dopa-induced dyskinesias
α4β2 nAChR drugs may be useful for reducing dyskinesias in late stage Parkinson’s disease
Acknowledgments
This work was supported by grants from the Michael J Fox Foundation and the National Institutes of Health [NS59910 and NS65851 to MQ and GM103801 and GM48677 to JMM] and by Targacept. We thank Maya Hrachova for excellent technical assistance. We are grateful to Dr. Kenneth Kellar, Georgetown University School Medicine for the sazetidine used in these studies, and to Dr. Jon Lindstrom, University of Pennsylvania, for the cells expressing different nAChR subtypes.
Abbreviations
- AIMs
abnormal involuntary movements
- ANOVA
analysis of variance
- nAChRs
nicotinic receptors
- CNS
central nervous system
- α-conotoxinMII
α-CtxMII
- 6-OHDA
6-hydroxydopamine
- 125I-RTI-121
125I-3β-(4-iodophenyl)tropane-2 β-carboxylic acid isopropyl ester
- sazetidine-A
6-(5-(((S)-azetidin-2-yl)methoxy)pyridine-3-yl)hex-5-yn-1-ol
- *
the asterisk indicates the possible presence of other nicotinic subunits in the receptor complex, with α4β2* nAChRs including α4β2 and α4α5β2 subtypes, and α6β2* including α6α4β2β3, α6β2β3 and α6β2 subtypes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlskog JE. Parkinson’s disease: is the initial treatment established? Curr Neurol Neurosci Rep. 2003;3:289–295. doi: 10.1007/s11910-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymyshyn R, Smith A, Wolfe BB. The use of 3H standards in 125I autoradiography. J Neurosci Methods. 1990;32:185–192. doi: 10.1016/0165-0270(90)90139-7. [DOI] [PubMed] [Google Scholar]
- Baddick CG, Marks MJ. An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem Pharmacol. 2011;82:828–841. doi: 10.1016/j.bcp.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencherif M, Schmitt JD, Bhatti BS, Crooks P, Caldwell WS, Lovette ME, Fowler K, Reeves L, Lippiello PM. The heterocyclic substituted pyridine derivative (+/−)-2-(−3-pyridinyl)-1-azabicyclo[2.2.2]octane (RJR-2429): a selective ligand at nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 1998;284:886–894. [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero MT. New pharmacological avenues for the treatment of L-DOPA-induced dyskinesias in Parkinson’s disease: targeting glutamate and adenosine receptors. Expert Opin Investig Drugs. 2012;21:153–168. doi: 10.1517/13543784.2012.651457. [DOI] [PubMed] [Google Scholar]
- Bordia T, Campos C, Huang L, Quik M. Continuous and intermittent nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesias in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2008;327:239–247. doi: 10.1124/jpet.108.140897. [DOI] [PubMed] [Google Scholar]
- Bordia T, Campos C, McIntosh JM, Quik M. Nicotinic receptor-mediated reduction in L-dopa-induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther. 2010;333:929–938. doi: 10.1124/jpet.109.162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie J, Jenner P. New approaches to therapy. Int Rev Neurobiol. 2011;98:123–150. doi: 10.1016/B978-0-12-381328-2.00005-5. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Beach JW, Terry AV. Desensitization of nicotinic acetylcholine receptors as a strategy for drug development. J Pharmacol Exp Ther. 2009;328:364–370. doi: 10.1124/jpet.108.145292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Bezard E. Contribution of pre-synaptic mechanisms to l-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–251. doi: 10.1016/j.neuroscience.2011.07.070. [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Munoz A, Kirik D, Bjorklund A. Involvement of the serotonin system in l-dopa-induced dyskinesias. Parkinsonism Relat Disord. 2008a;14(Suppl 2):S154–158. doi: 10.1016/j.parkreldis.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Munoz A, Kirik D, Bjorklund A. Serotonin-dopamine interaction in the induction and maintenance of L-DOPA-induced dyskinesias. Prog Brain Res. 2008b;172:465–478. doi: 10.1016/S0079-6123(08)00922-9. [DOI] [PubMed] [Google Scholar]
- Carta M, Lindgren HS, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J Neurochem. 2006;96:1718–1727. doi: 10.1111/j.1471-4159.2006.03696.x. [DOI] [PubMed] [Google Scholar]
- Cenci M, Lindgren H. Advances in understanding l-DOPA-induced dyskinesia. Curr Opin Neurobiol. 2007;17:665–671. doi: 10.1016/j.conb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Konradi C. Maladaptive striatal plasticity in l-DOPA-induced dyskinesia. Prog Brain Res. 2010;183C:209–233. doi: 10.1016/S0079-6123(10)83011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007;Chapter 9(Unit 9):25. doi: 10.1002/0471142301.ns0925s41. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Ohlin KE, Odin P. Current options and future possibilities for the treatment of dyskinesia and motor fluctuations in Parkinson’s disease. CNS Neurol Disord Drug Targets. 2011;10:670–684. doi: 10.2174/187152711797247885. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: how relevant is the rat? Nat Rev Neurosci. 2002;3:574–579. doi: 10.1038/nrn877. [DOI] [PubMed] [Google Scholar]
- Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38:679–690. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- Eskow KL, Dupre KB, Barnum CJ, Dickinson SO, Park JY, Bishop C. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse. 2009;63:610–620. doi: 10.1002/syn.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskow KL, Gupta V, Alam S, Park JY, Bishop C. The partial 5-HT(1A) agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves l-DOPA efficacy. Pharmacol Biochem Behav. 2007;87:306–314. doi: 10.1016/j.pbb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Fisone G, Bezard E. Molecular mechanisms of l-DOPA-induced dyskinesia. Int Rev Neurobiol. 2011;98:95–122. doi: 10.1016/B978-0-12-381328-2.00004-3. [DOI] [PubMed] [Google Scholar]
- Fox SH, Chuang R, Brotchie JM. Parkinson’s disease--opportunities for novel therapeutics to reduce the problems of levodopa therapy. Prog Brain Res. 2008;172:479–494. doi: 10.1016/S0079-6123(08)00923-0. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, McIntosh JM, Marks MJ, Collins AC. Mouse striatal dopamine nerve terminals express alpha4alpha5beta2 and two stoichiometric forms of alpha4beta2*-nicotinic acetylcholine receptors. J Mol Neurosci. 2010;40:91–95. doi: 10.1007/s12031-009-9263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigoni C, Li Q, Aubert I, Dovero S, Bioulac BH, Bloch B, Crossman AR, Gross CE, Bezard E. Involvement of sensorimotor, limbic, and associative basal ganglia domains in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2005;25:2102–2107. doi: 10.1523/JNEUROSCI.5059-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Calzavara R. The cortico-basal ganglia integrative network: the role of the thalamus. Brain Res Bull. 2009;78:69–74. doi: 10.1016/j.brainresbull.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosur V, Loring RH. alpha4beta2 nicotinic receptors partially mediate anti-inflammatory effects through Janus kinase 2-signal transducer and activator of transcription 3 but not calcium or cAMP signaling. Mol Pharmacol. 2011;79:167–174. doi: 10.1124/mol.110.066381. [DOI] [PubMed] [Google Scholar]
- Huang L, Grady SR, Quik M. Nicotine Reduces L-Dopa-Induced Dyskinesias by Acting at {beta}2 Nicotinic Receptors. J Pharmacol Exp Ther. 2011a;338:932–941. doi: 10.1124/jpet.111.182949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LZ, Campos C, Ly J, Carroll FI, Quik M. Nicotinic receptor agonists decrease L-dopa-induced dyskinesias most effectively in moderately lesioned parkinsonian rats. Neuropharmacology. 2011b;60:861–868. doi: 10.1016/j.neuropharm.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot P, Fox SH, Newman-Tancredi A, Brotchie JM. Anatomically selective serotonergic type 1A and serotonergic type 2A therapies for Parkinson’s disease: an approach to reducing dyskinesia without exacerbating parkinsonism? J Pharmacol Exp Ther. 2011;339:2–8. doi: 10.1124/jpet.111.184093. [DOI] [PubMed] [Google Scholar]
- Hussmann GP, Turner JR, Lomazzo E, Venkatesh R, Cousins V, Xiao Y, Yasuda RP, Wolfe BB, Perry DC, Rezvani AH, Levin ED, Blendy JA, Kellar KJ. Chronic sazetidine-A at behaviorally active doses does not increase nicotinic cholinergic receptors in rodent brain. J Pharmacol Exp Ther. 2012;343:441–450. doi: 10.1124/jpet.112.198085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iravani MM, Jenner P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J Neural Transm. 2011;118:1661–1690. doi: 10.1007/s00702-011-0698-2. [DOI] [PubMed] [Google Scholar]
- Iravani MM, McCreary AC, Jenner P. Striatal plasticity in Parkinson’s disease and L-DOPA induced dyskinesia. Parkinsonism Relat Disord. 2012;18(Suppl 1):S123–125. doi: 10.1016/S1353-8020(11)70038-4. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S. alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- Kawamata J, Shimohama S. Stimulating nicotinic receptors trigger multiple pathways attenuating cytotoxicity in models of Alzheimer’s and Parkinson’s diseases. J Alzheimers Dis. 2011;24(Suppl 2):95–109. doi: 10.3233/JAD-2011-110173. [DOI] [PubMed] [Google Scholar]
- Kulak JM, Musachio JL, McIntosh JM, Quik M. Declines in different beta2* nicotinic receptor populations in monkey striatum after nigrostriatal damage. J Pharmacol Exp Ther. 2002;303:633–639. doi: 10.1124/jpet.102.039347. [DOI] [PubMed] [Google Scholar]
- Letchworth SR, Whiteaker P. Progress and challenges in the study of alpha6-containing nicotinic acetylcholine receptors. Biochem Pharmacol. 2011;82:862–872. doi: 10.1016/j.bcp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippiello PM, Fernandes KG. The binding of L-[3H]nicotine to a single class of high affinity sites in rat brain membranes. Mol Pharmacol. 1986;29:448–454. [PubMed] [Google Scholar]
- Lummis SC, Thompson AJ, Bencherif M, Lester HA. Varenicline is a potent agonist of the human 5-hydroxytryptamine3 receptor. J Pharmacol Exp Ther. 2011;339:125–131. doi: 10.1124/jpet.111.185306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto M, Bower JM, Conforto AB, Delgado-Garcia JM, da Guarda SN, Gerwig M, Habas C, Hagura N, Ivry RB, Marien P, Molinari M, Naito E, Nowak DA, Oulad Ben Taib N, Pelisson D, Tesche CD, Tilikete C, Timmann D. Consensus paper: roles of the cerebellum in motor control--the diversity of ideas on cerebellar involvement in movement. Cerebellum. 2012;11:457–487. doi: 10.1007/s12311-011-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MJ, Zhang H, Neff NH, Hadjiconstantinou M. Desensitization of delta-opioid receptors in nucleus accumbens during nicotine withdrawal. Psychopharmacology (Berl) 2011;213:735–744. doi: 10.1007/s00213-010-2028-z. [DOI] [PubMed] [Google Scholar]
- Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, Weiner DM, Tison F, Bezard E. Priorities in Parkinson’s disease research. Nat Rev Drug Discov. 2011;10:377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Morin N, Gregoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T. Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology. 2010;58:981–986. doi: 10.1016/j.neuropharm.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Mudo G, Belluardo N, Fuxe K. Nicotinic receptor agonists as neuroprotective/neurotrophic drugs. Progress in molecular mechanisms. J Neural Transm. 2007;114:135–147. doi: 10.1007/s00702-006-0561-z. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG. Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 2000;23:S2–7. doi: 10.1016/s1471-1931(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, Rodriguez M. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord. 2008;23(Suppl 3):S548–559. doi: 10.1002/mds.22062. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. Missing pieces in the Parkinson’s disease puzzle. Nat Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- Penhune VB, Steele CJ. Parallel contributions of cerebellar, striatal and M1 mechanisms to motor sequence learning. Behav Brain Res. 2012;226:579–591. doi: 10.1016/j.bbr.2011.09.044. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M. Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Front Biosci. 2008;13:492–504. doi: 10.2741/2695. [DOI] [PubMed] [Google Scholar]
- Quik M, Cox H, Parameswaran N, O’Leary K, Langston JW, Di Monte D. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Ann Neurol. 2007;62:588–596. doi: 10.1002/ana.21203. [DOI] [PubMed] [Google Scholar]
- Quik M, Mallela A, Chin M, McIntosh JM, Perez XA, Bordia T. Nicotine-mediated improvement in l-dopa-induced dyskinesias in MPTP-lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol Dis. 2013;50:30–41. doi: 10.1016/j.nbd.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Park KM, Hrachova M, Mallela A, Huang LZ, McIntosh JM, Grady SR. Role for alpha6 nicotinic receptors in l-dopa-induced dyskinesias in parkinsonian mice. Neuropharmacology. 2012a;63:450–459. doi: 10.1016/j.neuropharm.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Park KM, Hrachova M, Mallela A, Huang LZ, McIntosh JM, Grady SR. Role for alpha6 nicotinic receptors in l-dopa-induced dyskinesias in parkinsonian mice. Neuropharmacology. 2012b;63:450–459. doi: 10.1016/j.neuropharm.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Perez XA, Grady SR. Role of alpha6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem Pharmacol. 2011;82:873–882. doi: 10.1016/j.bcp.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, Kulak JM, McIntosh JM. Vulnerability of 125I-alpha-conotoxin MII binding sites to nigrostriatal damage in monkey. J Neurosci. 2001;21:5494–5500. doi: 10.1523/JNEUROSCI.21-15-05494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Sum JD, Whiteaker P, McCallum SE, Marks MJ, Musachio J, McIntosh JM, Collins AC, Grady SR. Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol. 2003;63:1169–1179. doi: 10.1124/mol.63.5.1169. [DOI] [PubMed] [Google Scholar]
- Quik M, Wonnacott S. {alpha}6{beta}2* and {alpha}4{beta}2* Nicotinic Acetylcholine Receptors As Drug Targets for Parkinson’s Disease. Pharmacol Rev. 2011;63:938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascol O, Lozano A, Stern M, Poewe W. Milestones in Parkinson’s disease therapeutics. Mov Disord. 2011;26:1072–1082. doi: 10.1002/mds.23714. [DOI] [PubMed] [Google Scholar]
- Reperant C, Pons S, Dufour E, Rollema H, Gardier AM, Maskos U. Effect of the alpha(4)beta(2)(*) nicotinic acetylcholine receptor partial agonist varenicline on dopamine release in beta2 knock-out mice with selective re-expression of the beta2 subunit in the ventral tegmental area. Neuropharmacology. 2009;58:346–350. doi: 10.1016/j.neuropharm.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Timofeeva O, Sexton HG, DeCuir D, Xiao Y, Gordon CJ, Kellar KJ, Levin ED. Effects of sazetidine-A, a selective alpha4beta2* nicotinic receptor desensitizing agent, on body temperature regulation in mice and rats. Eur J Pharmacol. 2012;682:110–117. doi: 10.1016/j.ejphar.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H, Shrikhande A, Ward KM, Tingley FD, 3rd, Coe JW, O’Neill BT, Tseng E, Wang EQ, Mather RJ, Hurst RS, Williams KE, de Vries M, Cremers T, Bertrand S, Bertrand D. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol. 2010;160:334–345. doi: 10.1111/j.1476-5381.2010.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylander D, Iderberg H, Li Q, Dekundy A, Zhang J, Li H, Baishen R, Danysz W, Bezard E, Cenci MA. A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis. 2010;39:352–361. doi: 10.1016/j.nbd.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 2011;26:1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96:69–86. doi: 10.1016/j.pneurobio.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Shimohama S. Nicotinic receptor-mediated neuroprotection in neurodegenerative disease models. Biol Pharm Bull. 2009;32:332–336. doi: 10.1248/bpb.32.332. [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Cohen AD, Caudle WM, Zigmond MJ, Schallert T, Miller GW. Forced nonuse in unilateral parkinsonian rats exacerbates injury. J Neurosci. 2002;22:6790–6799. doi: 10.1523/JNEUROSCI.22-15-06790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Lallemand F, de Witte P, Dexter DT. Neurochemical pathways involved in the protective effects of nicotine and ethanol in preventing the development of Parkinson’s disease: Potential targets for the development of new therapeutic agents. Prog Neurobiol. 2008;85:135–147. doi: 10.1016/j.pneurobio.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, Broad LM, Williams AC, Zhang D, Ding C, Heinz BA, Sher E. Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Mol Pharmacol. 2008;73:1838–1843. doi: 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]