

Synopsis
Diabetes mellitus is responsible for nearly 10% of fetal anomalies in diabetic pregnancies. Although aggressive perinatal care and glycemic control are available in developed countries, the birth defect rate in diabetic pregnancies remains much higher than that in the general population. Major cellular activities--i.e., proliferation and apoptosis--and intracellular metabolic conditions--i.e., nitrosative, oxidative, and endoplasmic reticulum stress--have been demonstrated to be associated with diabetic embryopathy using animal models. Translating advances made in animal studies into clinical applications in humans will require collaborative efforts across the basic research, preclinical, and clinical communities.
Keywords: Diabetic embryopathy, birth defects, hyperglycemia, apoptosis, cell proliferation, metabolism, intracellular stress, intervention
Introduction
Diabetes mellitus is a metabolic disease, primarily due to high concentrations of glucose in the circulation (1). Hyperglycemia interrupts normal cellular metabolism and signaling and eventually causes organ dysfunction (2, 3). Nearly two centuries after diabetes was first recognized (4), its association with congenital birth defects and fetal mortality in pregnancy was recognized and referred to as diabetic embryopathy (4, 5). Prior to the introduction of insulin, diabetes-associated fetal and maternal mortality rates were nearly 70% and 40%, respectively (6, 7). Since the administration of insulin to control glycemia in pregnant women, these mortality rates have decreased dramatically to nearly 12% (8–15). In addition to the control of glycemia with insulin, aggressive perinatal care and neonatal management also contributed to the decline of maternal and fetal mortality (10, 16–19).
Unfortunately, the present birth defect rate in diabetic pregnancies (about 10%) is still higher than that in the general population (3%) and appears to be on the rise (7, 12, 13, 15, 20–25). The reasons for this increase in birth defect rates are complex. One reason is that there has been a rapid increase in diabetic patients in the population which includes women of childbearing age (26). It is estimated that approximately 8,000 babies in the United States are born each year with maternal diabetes-associated congenital malformations. The incidence of these malformations also has risen to near-epidemic level in developing countries.
Tackling this issue involves battles at a number of fronts: diagnosing fetal anomalies must be improved; technologies that can recognize developmental malformations as early as the embryogenesis period are needed; and better prenatal and planned pregnancy consultations should be implemented to help reduce diabetes-associated birth defects. These remain ongoing challenges for perinatal care providers.
An important goal in eliminating birth defects is to develop therapeutic interventions that can protect embryos from hyperglycemic insult. This goal can only be achieved by understanding the cellular and molecular mechanisms underlying diabetic embryopathy. Basic research using animal models has contributed a considerable amount of information about the manifestations of fetal abnormalities, but more work is still needed.
Pre-gestational and gestational diabetes
Diabetes mellitus is a chronic disease manifested by hyperglycemia and its associated metabolic factors. This condition is usually diagnosed by measuring the levels of plasma glucose, expressed as mg/dL or mM, and glycosylated hemoglobin A (HbA1c), indexed as percentage of total hemoglobin A (27–29). Manifestation of diabetes can be the result of insulin deficiency (type 1) or insulin resistance (type 2).
Type 1 diabetes, or insulin-dependent diabetes, is caused by autoimmune destruction of insulin-producing β-cells in the pancreas (30, 31).
Type 2 diabetes, or non-insulin-dependent diabetes, is caused by failure in insulin signaling to regulate cellular glucose uptake (32–34).
Diabetes mellitus, either type 1 or type 2, diagnosed in women before pregnancy is referred to as pre-gestational diabetes (20, 35–37). When hyperglycemia is detected after the onset of pregnancy, usually in the third trimester (24–28 weeks), the pregnant woman is considered having gestational diabetes mellitus (GDM) (38–42). According to guidelines from the International Association of Diabetes in Pregnancy Study Group (IADPSG), women who have a fasting plasma glucose ≥ 126 mg/dL and HbA1c ≥ 6.5% are diagnosed as having GDM (43).
Congenital birth defects in infants of diabetic mothers have been found to be associated with pre-gestational diabetes that is uncontrolled in the first trimester of pregnancy (44–48) Although a few cases have suggested a link between GDM and fetal birth defects, this association remains unclear and lacks strong supporting evidence (49, 50). One possible reason for the controversy is that some women with early-onset type 2 diabetes are misdiagnosed as having GDM when first screened in the second trimester (39, 51). Nevertheless, it is well established that GDM can lead to many adverse fetal outcomes, including macrosomia, hypoglycemia, hypocalcemia, and hyperbilirubinemia (51–53).
High glucose as a major teratogenic factor
Human studies have demonstrated a strong link between maternal glycemic level, as indicated by the association of plasma glucose and HbA1c levels (54, 55) with the incidence of congenital malformations in offspring (56–61). Other adverse metabolic factors produced in diabetes mellitus, such as ketone bodies, advanced glycation end products, and branched chain amino acids, may have synergetic effects with glucose on disrupting normal embryonic development (62–66). The putative teratogenic effects of hyperglycemia are supported by studies that demonstrate a reduction in the incidence of birth defects following clinical interventions targeted at achieving euglycemia (67–70). Animal studies also have shown that isolated embryos in culture exposed to high concentrations of glucose develop malformations similar to those seen in human diabetic pregnancies (62, 71–73). These observations indicate that high glucose in either type 1 or type 2 diabetes is a major teratogenic factor that disturbs embryonic development.
The major effort to eliminate adverse outcome in pre-gestational diabetes and GDM complicated pregnancies is to control glycemic levels (20, 74–76). Clinical management of diabetic pregnancy with insulin and oral hypoglycemic medications can achieve the euglycemic standards, recommended by Diabetes Control and Complications Trials and International Federation of Clinical Chemistry and Laboratory Medicine (Table 1) (77–79). However, with respect to pre-gestional diabetes, optimal glycemic control prior to conception appears to be a challenge because:
Table 1.
Recommendations for pre-conception HbA1c targets
| USA | UK | |
|---|---|---|
| DCCT | <7.0% | <6.1% |
| IFCC | <53 mmol/mol | <43 mmol/mol |
DCCT, Diabetes Control and Complications Trials; IFCC, International Federation of Clinical Chemistry and Laboratory Medicine.
most women with diabetes do not seek preconception care,
most have unplanned pregnancies (80).
even in women who plan pregnancies and receive preconception counseling, euglycemia is difficult to achieve and maintain in non-clinical settings.
Therefore, alternative approaches to glucose control to prevent birth defects need to be developed and implemented.
Malformations and Diagnosis
Maternal diabetes-associated fetal anomalies can be seen in any organ system but are most common and severe in the central nervous system (CNS), cardiovascular system (CVS), craniofacial region, and caudal structure (Table 2) (11, 12, 25, 44, 55, 81, 82).
Table 2.
Developmental anomalies in major organ systems in diabetic embryopathy
| Central nervous | Craniofacial | Cardiovascular | Skeletal |
|---|---|---|---|
| Anencephaly | Hemifacial | Conus arteriosus defects | Sacral agenesis |
| Encephalocele | microsomia | Transposition of great vessels | Sacral hypoplasia |
| Exencephaly | Macrostomia | Tetralogy of Fallot | Limb defects |
| Microcephaly | Cleft palate | Ventricular septal defects | Vertebral defects |
| Hydrocephaly | Cleft lip | Pulmonary vavle defects | Caudal regression |
| Holoprosencephaly | Microtia | Patent ductus arteriosus | |
| Spina bifida | Micrognathia | Hypoplastic left heart syndrome | |
| Craniosynostosis | Coarctation of the aorta | ||
| Anotia/Microtia | Right ventricular septal defects | ||
| Eye defects | Atrial septal defects | ||
| Heterotaxia |
Central nervous system
The most common structural defects resulting from diabetic embryopathy occur in the brain region and spinal cord of the CNS () (12, 25, 83–86). Some of these can be recognized as early as in the first trimester using ultrasonography.
Anencephaly is characterized as absence of the cerebral hemispheres with the brain stem and portions of the midbrain intact. It is readily discernible in the first trimester (Figure 1) (76, 87, 88). In the second trimester, it is more evident as poorly formed cranial bones and symmetric absence of the calvarium (76, 87, 89).
Holoprosencephaly, the complete or partial absence of the midline echo within the fetal brain, can be diagnosed as early as 14 weeks of gestation, though diagnosis at the 20-week anomaly scan is more common (90–92).
Excencephaly, the absence of the superior vault and convolution of the brain, can be diagnosed as early as 10 weeks of gestation (Figure 2) (93).
Ultrasonic diagnosis of CNS defects such as spina bifida usually occurs in conjunction with detection of the second trimester maternal blood biomarker α-fetoprotein (81, 94–97).
Figure 1.
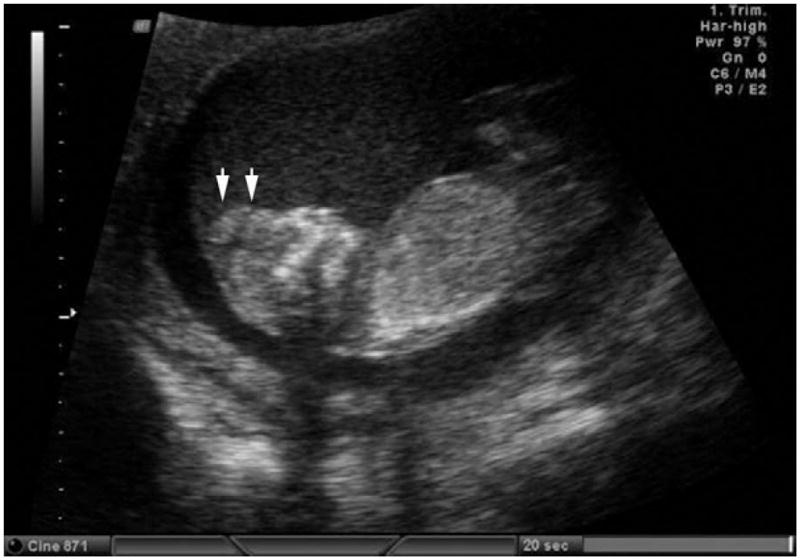
Two-dimensional ultrasonic scanning of anencephaly in the first trimester. Arrows indicate missing forebrain. Modified with permission from; Cameron M, Moran P. Prenatal screening and diagnosis of neural tube defects. Prenat Diagn 2009;29:402–11.
Figure 2.
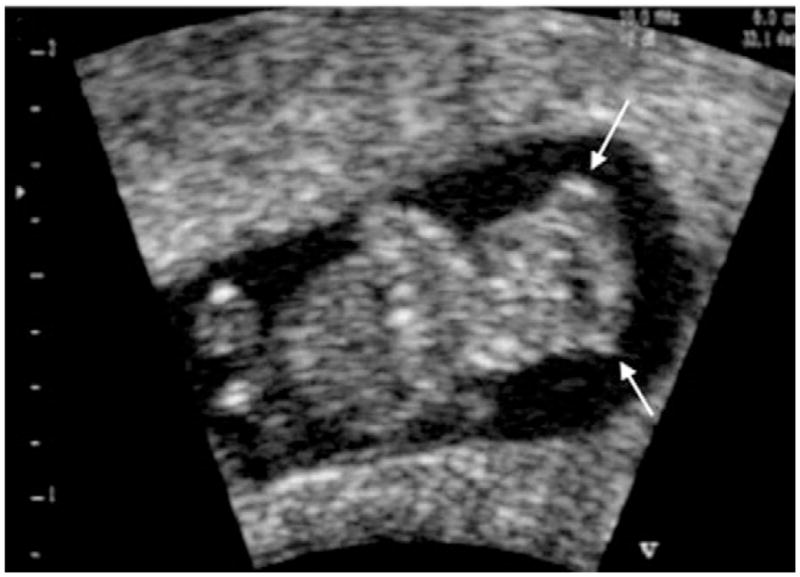
Two-dimensional ultrasonic scanning of exencephaly in the first trimester. The contours of the brain are irregular (arrows). Modified with permission from; Blaas HG, Eik-Nes SH. Sonoembryology and early prenatal diagnosis of neural anomalies. Prenat Diagn 2009;29:312–25.
Craniofacial structures
In newborns of diabetic mothers the most common anomalies in the craniofacial regions are hemifacial microsomia and microtia in newborns of diabetic mothers (82, 98). Cleft palate and lip also occur in relatively high frequency (99–101). Hearing impairment in children has been found to be associated with diabetic pregnancy (82, 98, 102), likely due to developmental defects in the inner and middle ear (102).
Prenatal diagnose of cleft lip and cleft palate relies a combination of coronal and axial two-dimensional ultrasound scans (103). Cleft lip can be easily recognized from coronal scan images, however, when cleft lip extends into the palate, the axial scan of the maxilla can provide the image of the defects (104). Cases of lateral cleft lip/palate often present a so-called maxillary pseudomass visualized in two-dimensional sonographs (105).
Cardiovascular system
Fetal cardiac defects associated with maternal diabetes have been extensively characterized. Anomalies commonly are present in:
-
myocardium-derived structures
atria,
ventricles
interventricular septum.
-
endocardium-derived structures
Sonographic imaging remains the standard method to diagnosis fetal cardiac abnormalities. Technological advances in imaging, especially in high frequency imaging and improvements in resolution, have aided perinatal care providers in recognizing cardiac defects as early as 10 weeks of gestation.
Most heart anomalies, including double outlet hearts, atrial septal defects, and ventricular septal defects, can be diagnosed using the four-chamber view, a transverse projection through the fetal thorax above the level of the diaphragm (Figure 3) (109). Measurement of the thickness of the ventricular walls in the sonographs can reveal myocardial hypoplasia. Due to the position of the canning plane, the four-chamber view usually misses defects in the structures in the outflow tracts. A so-called base view, with scans superior to the four-chamber view, can reveal abnormalities in the aorta and pulmonary artery. Complex defects such as Tetralogy of Fallot, which involves ventricular septal defects and great vessel defects, may require a combination of four-chambered and base views to diagnose.
Figure 3.
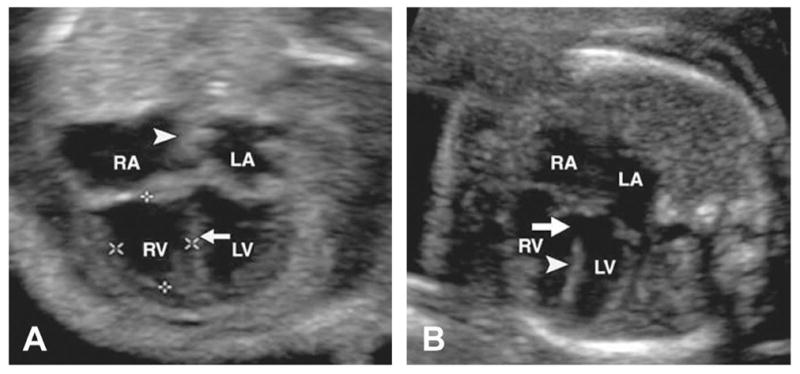
Four-chamber view of fetal hearts. (A) Normal heart. Arrow indicates interventricular septum. Arrowhead indicates interatrial septum. (B) Heart with VSD (arrow) in inlet portion of interventricular septum (arrowhead). LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Modified with permission from; Rajiah P, Mak C, Dubinksy TJ, et al. Ultrasound of fetal cardiac anomalies. AJR Am J Roentgenol 2011;197:W747–60.
Color Doppler provides an added advantage to practitioners because it can detect minor defects in the heart by exhibiting changes in blood flow pattern. The association of increased first trimester nuchal translucency with fetal cardiac defects has been observed; however, more studies are still needed to establish it as a diagnostic marker (110, 111).
Caudal regression
Caudal regression syndrome, also known as caudal dysplasia, is characterized as the absence or hypoplasia of caudal trunk and limbs (112, 113). Caudal regression syndrome can be diagnosed by noting a shortened spine and abnormal lower limbs. This anomaly can be detected in the fetus as early as second trimester.
Mechanisms of Diabetic Embryopathy
Developmental mechanisms
Central Nervous System
Diabetic embryopathy causes embryonic and fetal abnormalities as a result of a disturbance in normal organogenesis. In the CNS, most anomalies occur because of a failure of early neural tube formation, referred to as neural tube defects (NTDs) (114–116). The formation of the neural tube, or neurulation, occurs in human embryos from weeks 3–6 of gestation (114). Neurulation begins with formation of the neural ectoderm, which develops into the neural folds. The neural folds grow dorsal-laterally and eventually fuse at the dorsal midline along the body axis to form the neural tube (117). In diabetic embryopathy, neurulation is perturbed in young embryos, leading to NTDs such as exencephaly or spina bifida (62, 73, 118, 119). Studies using diabetic rodents with poorly controlled hyperglycemia have recapitulated the same CNS defects observed in human fetuses (Figure 4) (120–122).
Figure 4.
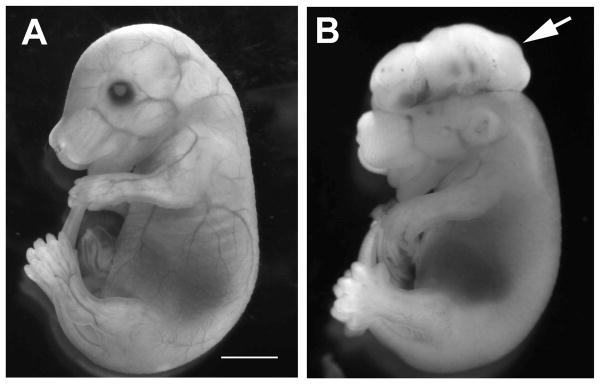
Exencephaly in mouse E15.5 fetus of diabetic pregnancy. (A) Nondiabetic control. (B) Diabetes. Arrow indicates exencephaly. Scale bar=3 mm.
Cardiovascular System
The development of the heart is a complex process, involving multiple tissue types. The atria, ventricles, and interventricular septum are derived from the myocardium, whereas the membranous septa in the atrioventricular channel and the outflow tracts and associated valves are derived from the endocardium (123–126).
The most common abnormality of the ventricles is hypoplastic left heart syndrome (44). It is associated with under development of the myocardium during early cardiogenesis (121). Defects in endocardium-derived structures appear to be associated with endocardial cushions (Figure 5) (121, 127), bulbous structures that develop during early embryogenesis at the atrioventricular junction and the bulbous cortis (outflow tract) as a result of the production of the extracellular matrix (128, 129). During development of the endocardial cushions, endocardial (endothelial) cells differentiate into mesenchymal cells. This process is known as the endothelial-mesenchymal transformation (EMT) (128, 130). These mesenchymal cells migrate into the extracellular matrix to fill the acellular space and promote the growth of the cardiac structures (130). The endocardial cushions develop toward each other and eventually fuse to form a continuous septum (125). After fusion, the endocardial tissues undergo dramatic remodeling to connect with the interventricular and primary atrial septum and form valves (130, 131).
Figure 5.
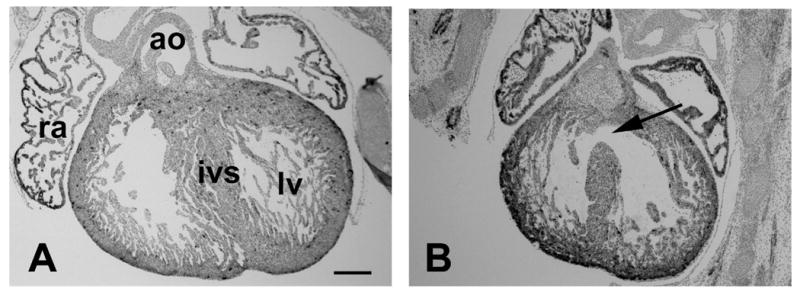
AVSD in mouse E15.5 fetus of diabetic pregnancy. (A) Heart of nondiabetic control. (B) Heart of diabetic group with VSD (arrow). ao, aorta; ivs, interventricular septum; lv, left ventricle; ra, right atrium. Scale bar=200 μm.
In embryos of diabetic pregnancies, early development of the endocardial cushions is inhibited (121). An impact of maternal hyperglycemia on later endocardial cushion remodeling also has been observed (132). Further research is needed to determine the significance of these developmental processes in cardiac malformation in diabetic embryopathy.
Craniofacial
In the craniofacial region, abnormalities in facial structures may be associated with dysmorphogenesis of cartilages (133, 134). These cartilages, such as the Meckel’s cartilage in the mandibular arch, are not only involved in early morphogenesis of the facial processes, but also give rise to many types of bony structures in the craniofacial regions such as the auditory ossicles (135, 136).
Cellular mechanisms
The development of organ systems involves cell proliferation, death, migration, and differentiation. Excessive programmed cell death (apoptosis) has been observed in the dorsal region of the neural tube and is associated with NTDs in diabetic embryopathy (71, 122, 137–142). In the developing heart, cell proliferation at the early stages and apoptosis at the late stages of cardiogenesis appear to be associated with cardiac malformations in the embryos of diabetic pregnancy (121). In addition to cell proliferation, endocardial cell differentiation and migration also are suppressed by maternal hyperglycemia (121, 127).
Development of the cardiac outflow tract requires cell migration from the neural crest in the dorsal region of the neural tube. These neural crest cells contribute significantly to the septation of the outflow segment and the formation of the great vessels (143, 144). Malformations in the outflow segment may be due to impaired neural crest cell migration, which can be caused by increased apoptosis (145, 146).
Craniofacial structure development also requires neural crest cells that migrate from the anterior neural tube during early embryogenesis (147–149). Studies using animal models have shown that maternal diabetes perturbs neural crest cell migration in embryos (133, 150). Increased apoptosis in the mandibular arch has been observed in embryos of diabetic women, though it is uncertain whether those cells are of neural crest-origin (146). The impact of maternal diabetes on the differentiation of the neural crest-origin craniofacial mesenchymal cells remains to be demonstrated.
Molecular mechanisms in promoting apoptosis in diabetic embryopathy
Endoplasmic reticulum stress
When exposed to high glucose, embryonic cells take up glucose via glucose transporters (151, 152). An influx of glucose disturbs intracellular metabolic homeostasis and organelle function. Dysfunction of the endoplasmic reticulum (ER) leads to aberrant protein folding and subsequent accumulation of unfolded and misfolded proteins in its lumen (153–155), which cause ER stress (156–159). Under stress conditions, the ER activates a number of molecular cascades, collectively known as the unfolded protein response (UPR), to increase expression of chaperone protein to resolve protein folding crisis, inhibit protein translation, suppress mitosis, and even trigger apoptosis (Figure 6) (160–162). ER stress has been observed in the embryos from diabetic pregnancies (127, 163, 164). The potential causative role of ER stress in embryonic malformation has been demonstrated with experiments using a chemical chaperone to resolve protein folding crisis (127).
Figure 6.
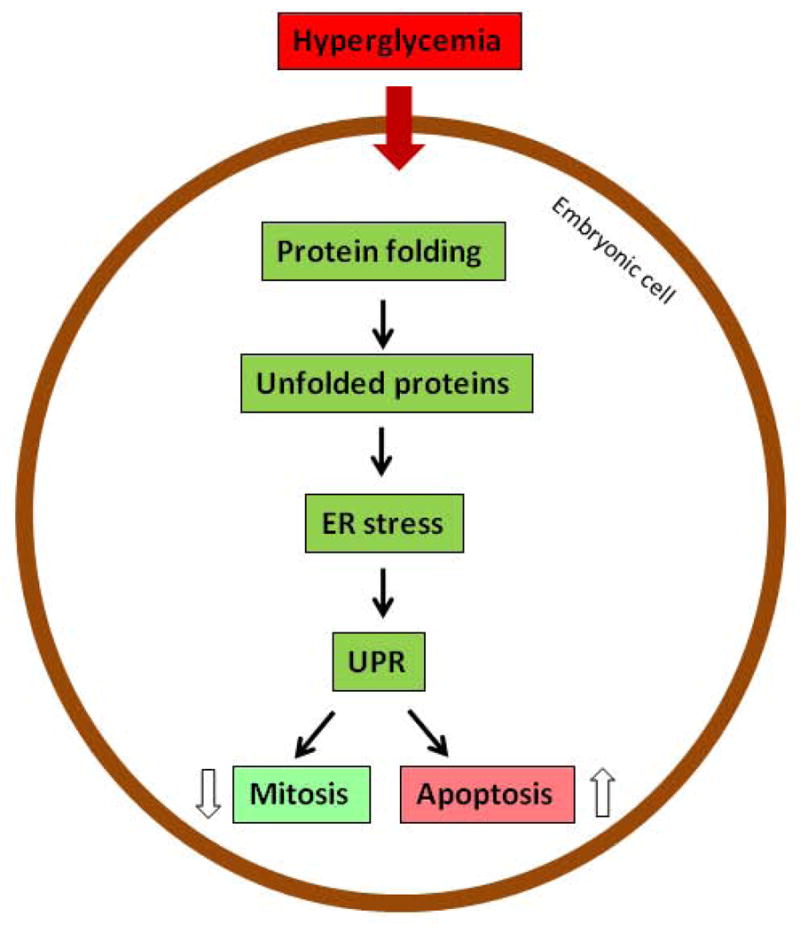
ER stress in diabetic embryopathy. ER, endoplasmic reticulum; UPR, unfolded protein response.
Oxidative stress
High glucose also changes the morphology and function of mitochondria (165). Changes in mitochondria interrupt the electron transport chain, leading to generation of reactive oxygen species (ROS) (166, 167). High glucose also reduces the level of intracellular antioxidants, including glutathione (GSH) and thioredoxin (168–171). The imbalance of ROS and antioxidative buffering results in oxidative stress, which perturbs intracellular signaling (Figure 7) (172, 173). Treating diabetic pregnant animals or embryos cultured in high glucose with antioxidants decreases embryonic malformation rates (174–184). Moreover, embryos treated with antioxidants overexpress a transgene of superoxide dismutase resist maternal hyperglycemic insult (185, 186).
Figure 7.
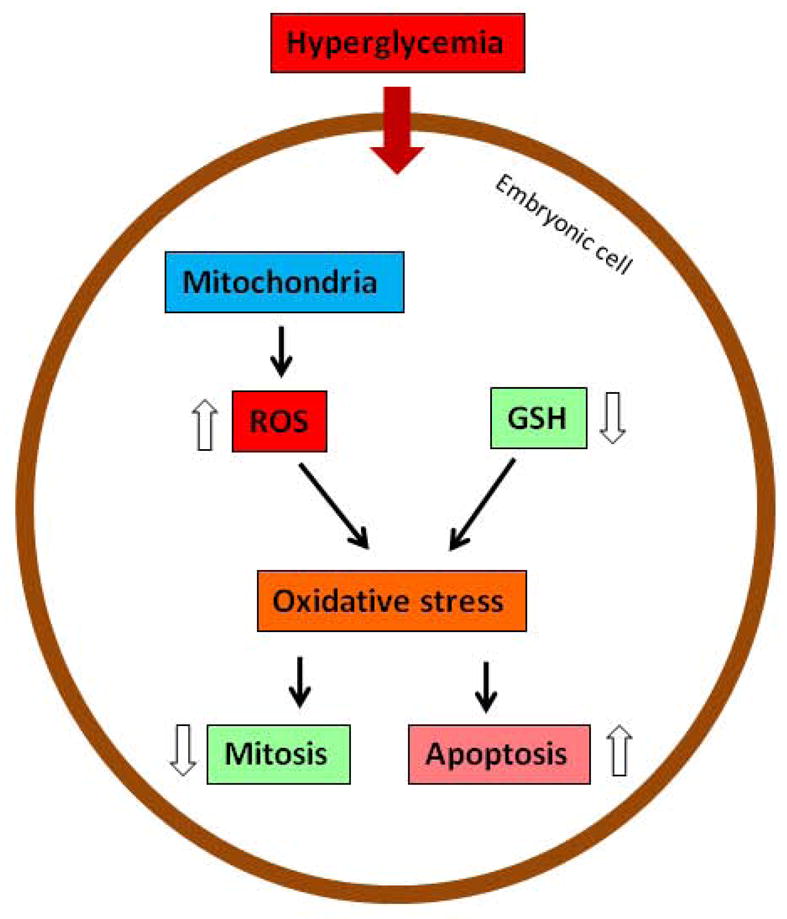
Oxidative stress in diabetic embryopathy. GSH, glutathione; ROS, reactive oxygen species.
Nitrosative stress
Under hyperglycemic conditions, embryonic cells produce high levels of nitric oxide (NO). NO is a second messenger that regulates various intracellular signaling pathways (187, 188) and also can react with ROS to produce more toxic radicals, called reactive nitrogen species (RNS), than NO or ROS alone (189, 190). The high levels of RNS, such as peroxynitrite, generate a condition known as nitrosative stress (Figure 8).
Figure 8.
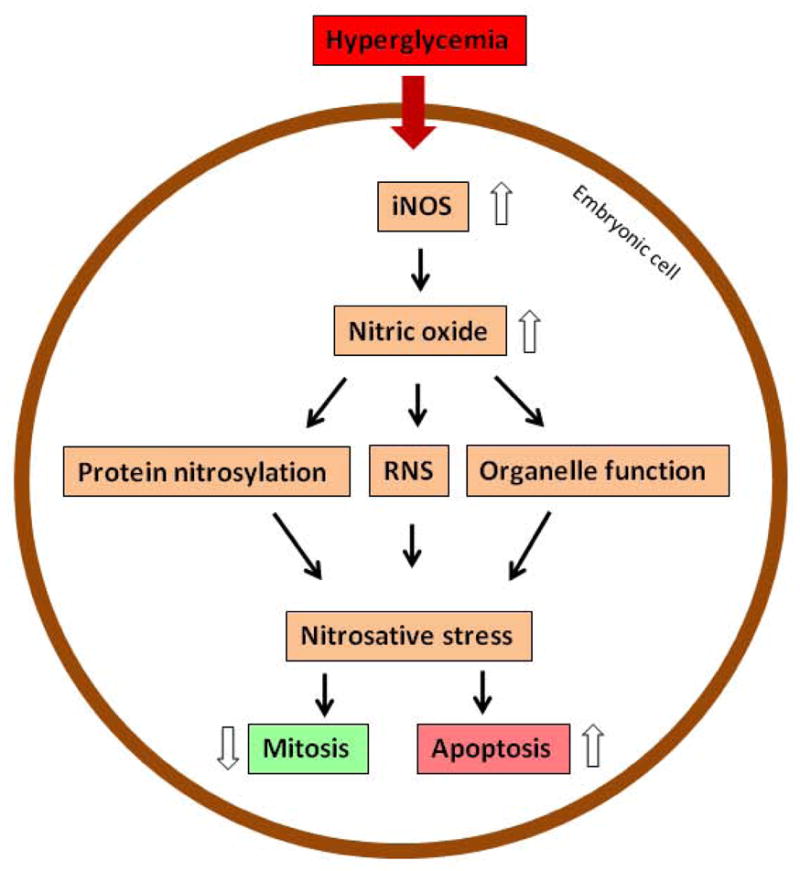
Nitrosative stress in diabetic embryopathy. iNOS, inducible nitric oxide synthase; RNS, reactive nitrogen species.
Synthesis of NO is catalyzed by NO synthase (NOS) enzymes. There are three main forms of NOS enzymes: neuronal (nNOS and NOS1); endothelial (eNOS and NOS3); and inducible (iNOS and NOS2)(191–193). Both nNOS and eNOS are constitutively expressed and do not vigorously respond to extracellular stimulation (194, 195), but iNOS actively responds to extracellular changes with marked upregulation in expression and activity (196–198).
In embryos of diabetic animals, eNOS expression is decreased (199); in contrast, iNOS expression is dramatically increased (200, 201). Experiments using an iNOS knockout model clearly demonstrate that high level of NO is detrimental to the embryo (Figure 8) (120).
Stress response
Phospholipid peroxidation
Cellular stress perturbs intracellular metabolic homeostasis. Phospholipid metabolism is initiated by phospholipases (PLs), including PLA, PLC, and PLD (202, 203). Cytosolic phospholipase A2 (cPLA2) cleaves arachidonic acid from the cell membrane (204–206). In the cytoplasm, arachidonic acid undergoes two major pathways of metabolism: it can be converted into prostaglandin E2 (PGE2) by cyclooxygenase-2 (COX-2) (207, 208), or it can be converted into PGE2-like isoprostanes, such as 8-iso-prostagladin F2 (8-iso-PGF2) and 8-iso-PGF2α, by non-COX-mediated peroxidation involving free radicals (Figure 9) (209, 210).
Figure 9.
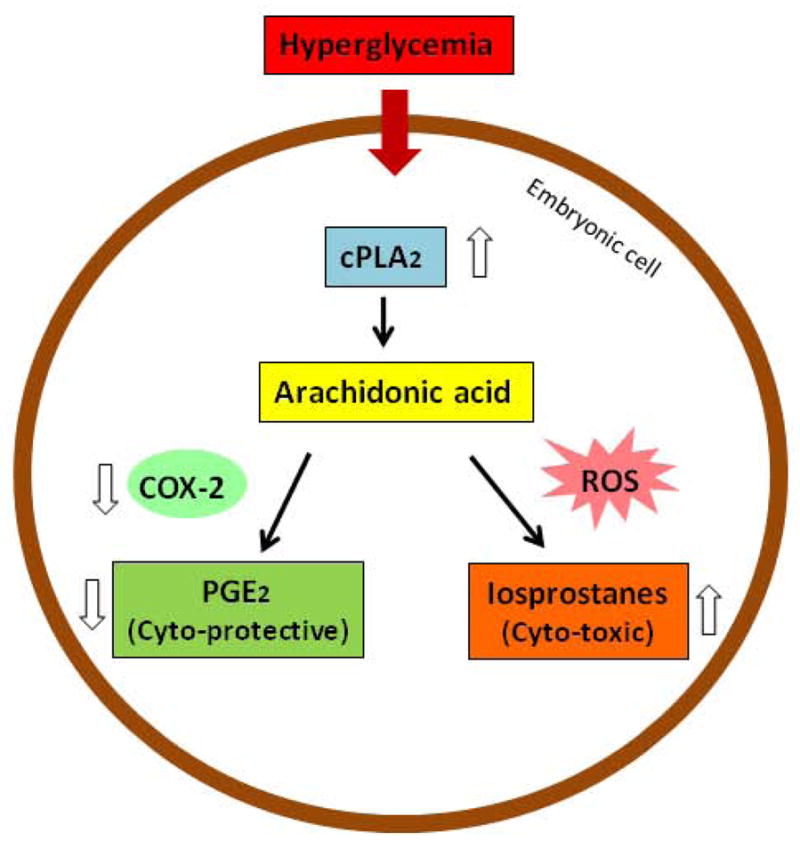
Lipoperoxidation in diabetic embryopathy. COX-2, cyclooxygenase-2; cPLA2, cytosolic phospholipase A2; PGE2, prostaglandin E2; ROS, reactive oxygen species.
In embryos of diabetic animals or embryos exposed to high concentrations of glucose in vitro, the level of PGE2 decreases dramatically (211–215). On the other hand, the level of 8-iso-PGF2 is dramatically elevated in embryos under hyperglycemic conditions as well as in diabetic patients (209, 216). The reduction in PGE2 in the embryo may be due to a decrease in COX-2 activity and expression (211, 217, 218), suggesting that a shift in arachidonic acid metabolism has occurred from producing PGE2 to generating isoprostanes. These PGE2-like isoprostanes have been shown to have damaging effects in animal models of diabetic pregnancy and embryos exposed to high glucose in culture (216, 219), whereas PGE2 protects embryos from the damages due to hyperglycemic conditions (Figure 9) (211, 218, 220).
Protein kinase C family
Under cellular stress conditions, a number of intracellular signaling systems are affected, including the ones regulated by members of the protein kinase C (PKC) family. The PKC family of serine/threonine protein kinases consists of 12 members, which can be divided into the following three groups, based on their activation mechanisms (221, 222): 1) PKCα, β1, β2, and γ require calcium and diacylglycerol (DAG) for activation; 2) PKCδ, ε, η, ν, and θ require only DAG; and 3) PKCμ, ξ, and ι/λ do not require calcium or DAG, but instead require distinct lipid cofactors (i.e., ceramide and phosphatidylinositol-4-phosphate) (221).
In embryos of diabetic animals, each PKC isoform responds differently to hyperglycemia. For example, PKCα, β2, and δ are phosphorylated and activated under hyperglycemic conditions (141). Consequently, inhibiting PKCα, β2, and δ with isoform-specific inhibitors reduces malformation rates in embryos cultured in a high concentration of glucose (141, 223). Additionally, embryos from diabetic pkcδ knockout animal models exhibit significantly lower malformation rate than embryos from diabetic mice having the gene (164). These and other similar studies demonstrate that PKCs play critical roles in diabetic embryopathy.
Mitogen-activated protein kinase family
The mitogen-activated protein kinase (MAPK) family also plays an important role in mediating the effects of maternal hyperglycemia on embryonic development. Members of the MAPK family, including extracellular signal-regulated kinases (ERKs) and c-jun N-terminal kinases/stress-activated protein kinases (JNKs/SAPKs), can usually be activated by oxidative stress. ERKs primarily regulate cell proliferation and survival, whereas JNKs act on pro-apoptotic pathways (224–226). In embryos of diabetic animals, the levels of phosphorylated ERK1 and 2 are significantly reduced (Figure 8) (227, 228). In contrast, JNKs are activated in embryos of diabetic animals (227–230). Embryos treated with a JNK inhibitor while simultaneously incubated in a high concentration of glucose have a lower malformation rate, compared with those without JNK inhibition (231). More convincing evidence of the role of JNKs in diabetic embryopathy comes from the experiments using jnk1 and jnk2 knockout mice. Embryos homozygous for either jnk1 or jnk2 deletion show significantly lower malformation rates compared with a wild-type embryos from diabetic animals (231, 232).
Programmed cell death (Apoptosis)
Apoptosis is precisely regulated by a number of factors, including PKCs, JNKs, and members of the Bcl-2 and caspase families (233). Although the mechanisms by which PKCs and JNKs promote apoptosis in diabetic embryopathy remain to be delineated, much is known about the roles that Bcl-2 and caspase family members play in programmed cell death.
In the Bcl-2 family, some members are pro-apoptotic, such as Bax, Bak, and Bid, whereas other members are anti-apoptotic, such as Bcl-2 and Bcl-xL (234). In the caspase family, some members play a role in executing apoptosis. These members, known as effector or executioner caspases, include casapse-3, -6, and -7 (235–237). Effector caspases can be activated by another group of caspases, known as initiator caspases, including caspase-8, -9, and -10 (235–237).
Bax and Bak form a pore in the outer membrane of mitochondria (238, 239), allowing cytochrome C to be release to cytoplasm to induce apoptosis (240–242). During apoptosis, Bax and Bim expression levels increase in the cell (238, 239). Bax is activated by truncated Bid (tBid), which is produced when Bid is cleaved by serine/threonine proteases such as caspase-8.
In diabetic embryopathy, caspase-8 cleaves Bid into tBid, which stimulates the release of cytochrome C (140). Cytochrome C binds to apoptosis protease-activating factor-1 (Apaf-1), and the resulting complex activates Caspase-9 by forming an apoptosome. Activated Caspase-9 then activates effector caspases, including Caspase-3, 6, and 7, which turn on caspase-activated DNase and other factors, leading to DNA fragmentation and cell death (Figure 10) (240, 241).
Figure 10.
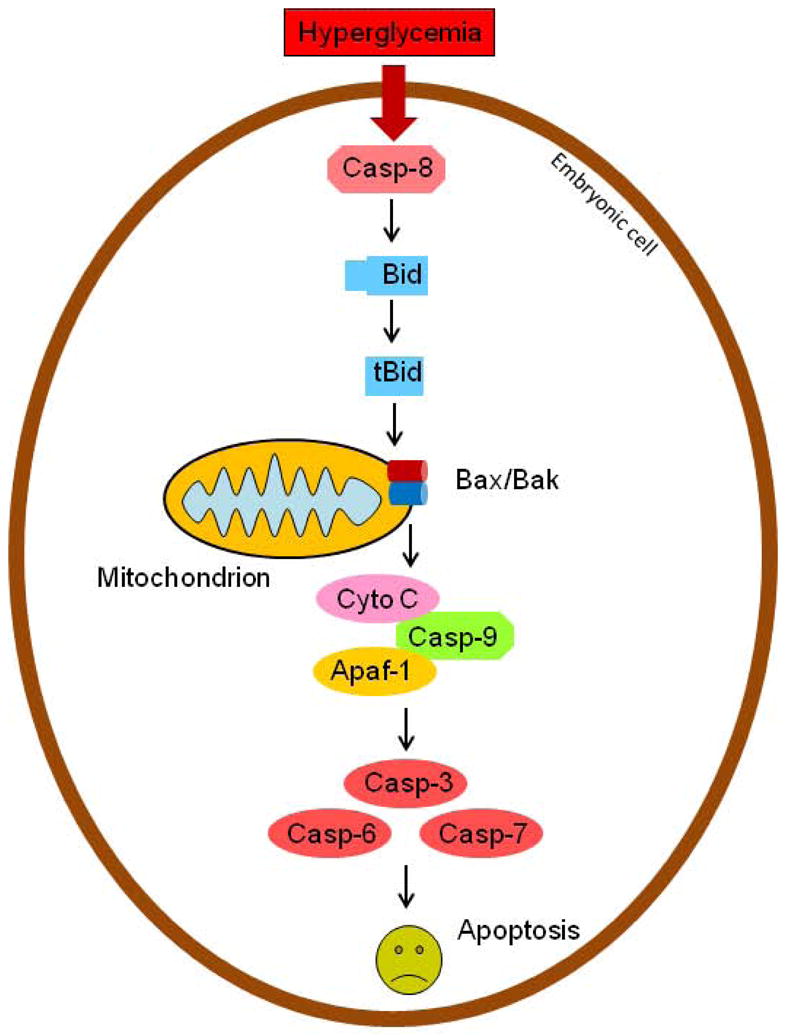
Caspase-8-regulated apoptotic pathway in diabetic embryopathy. Apaf-1, apoptotic protease-activating factors-1; Casp, caspase; Cyto, cytochrome;
In animal studies, suppression of apoptosis using caspase inhibitors can reduce embryonic malformations in the embryos cultured in high glucose (140). The results imply potential interventional strategies to block apoptosis in embryos in diabetic pregnancies.
Interventions and Clinical Challenges
Research conducted in animal models of diabetic pregnancy has revealed some of the major molecular changes and subsequent intracellular metabolic conditions that occur in embryos in response to hyperglycemia. This information has guided efforts to explore interventional approaches to reduce diabetes-induced embryonic malformations (122, 174, 183, 184, 227, 243–247).
Targeting lipid metabolism
One anomaly that occurs in diabetic embryopathy is aberrant phospholipid metabolism, especially lipoperoxidation. Dietary supplementation with arachidonic acid or myo-inositol in diabetic pregnant animals has been shown to reduce embryonic malformations due to lipoperoxidation (183, 184, 244). Treatment of diabetic pregnancy animals with polyunsaturated fatty acids also have been shown to exert similar helpful effects on embryonic development (227, 248).
Antioxidative strategies
Targeting oxidative stress to treat diabetic embryopathy, using lipoic acid, ergothioneine, vitamin C, and vitamin E, has been tested in diabetic pregnant animals and shown to decrease embryonic malformations (174, 181, 183, 184, 243, 249). N-acetylcysteine (NAC), which has been used as an antioxidant in clinical practices to treat acetaminophen poisoning (250, 251), has been observed to reduce malformations in animal embryos in culture exposed to high glucose (252–254); however, its effect in human diabetic pregnancy remains to be demonstrated.
Folic acid, or water-soluble vitamin B9, has long been known to reduce NTDs in humans (255, 256). The effect of folic acid in reducing embryonic malformations associated with maternal diabetes has been explored in animal models. Treating cultured rodent embryos with folic acid can reduce neural tube dysmorphogenesis induced by high concentrations of glucose (246). In in vivo experiments, diabetic pregnant rats injected with folic acid also exhibit decreased NTDs in their embryos, compared with diabetic pregnant rats not given the supplement (122, 246, 257).
Combination approaches
The combination of antioxidants and phospholipids that target multiple molecular pathways could have more potent effects in reducing fetal abnormalities compared with monotherapy. Cocktails of vitamin E, arachidonic acid, and myo-inositol have been explored in diabetic animal models and have been shown to decrease embryonic malformations (183, 184). The efficacy of antioxidants to prevent fetal defects in human diabetic pregnancy has not been examined. Antioxidant approaches tested to treat similar diseases, such as preeclampsia, have produced unsatisfactory results (258–262). These results have tampered the enthusiasm in applying similar strategies to treat diabetic embryopathy, but exploration of other strategies is warranted.
Recently, alleviating nitrosative stress via iNOS inhibitors has been tested in diabetic embryopathy (163). Oral treatment of diabetic pregnant animals with an iNOS inhibitor decreased embryonic malformation rates (163). Additional studies are needed to test therapeutic agents or dietary supplements that inhibit iNOS in order to translate these basic research findings into clinical applications for human disease.
Clinical challenges
Diabetic embryopathy involves complex molecular interactions. Extensive understanding of its mechanisms is essential for identifying therapeutic targets and developing effective interventions. Many challenges in developing these approaches lie ahead. First, interventions must be safe for the embryo and mother. The ideal approaches include dietary supplementation of non-toxic agents. Second, because developmental malformations occur early on in the first trimester of pregnancy, the dietary supplements must be easily accessible for women to take before conception or soon thereafter. Third, any therapeutic agents administered via oral treatments or dietary supplements must be able to pass the maternal-fetal barrier to exert potent effects on the developing embryo.
To overcome these challenges, it not only needs scientific research to decipher the molecular mechanisms underlying embryonic malformations and develop prevention strategies, but also requires education of the public to raise awareness about the risk of birth defects in diabetic pregnancy. Preconception counseling and pregnancy planning have been shown to be correlated with reduction in adverse outcomes of pregnancies (263–266). However, more cooperative efforts between perinatal care providers and patients are needed to achieve the goal of eliminating birth defects.
Concluding remarks
Diabetic embryopathy is a global public health issue. Although pregestational screening for maternal diabetes, perinatal care, and postnatal management in developed countries are available to most pregnant women, the rate of birth defects in infants of diabetic mothers remains high. In developing countries, the rates of birth defects and even mortality are high because of unavailable and inadequate care for pregnant women. With the worldwide increase in obesity and type 2 diabetes, diagnosis and management of diabetic pregnancy is a big challenge for the medical community. The increasing public health burden of diabetes in women of childbearing age makes it important to develop interventional approaches to prevent embryonic malformations.
Further understanding of the mechanisms underlying diabetic embryopathy will provide crucial information for developing effective interventions. Clinical approaches must be safe to administer in early pregnancy and accessible to women prior to and soon after conception to be highly effective because diabetes in early pregnancy often goes undetected and many women have unplanned pregnancies. Major cellular activities--i.e., proliferation and apoptosis--and intracellular metabolic conditions--i.e., nitrosative, ER, and oxidative stress--have been demonstrated to be associated with diabetic embryopathy using animal models. Translating advances made in animal studies into clinical applications in humans will require collaborative efforts across the basic research, preclinical, and clinical communities.
Key points.
Diabetes mellitus in early pregnancy increases the risk of birth defects in infants.
High glucose induces intracellular stress, increases programmed cell death (apoptosis), and decreases cell proliferation in the embryo.
Reduction of the risk includes pre-conceptional and early gestational glycemic control.
Interventions via dietary supplementation remain to be developed.
Pre-conceptional counseling and planned pregnancy are encouraged for physicians and patients.
Contributor Information
Zhiyong Zhao, Email: zzhao@fpi.umaryland.edu, Department of Obstetrics, Gynecology and Reproductive Sciences, University of Maryland School of Medicine, 655 West Baltimore Street, BRB 11-041, Baltimore, Maryland, 21201. Phone (410) 706-8401
E. Albert Reece, Email: deanmed@som.umaryland.edu, Department of Obstetrics, Gynecology and Reproductive Sciences, University of Maryland School of Medicine, 655 West Baltimore Street, BRB 14-029, Baltimore, Maryland, 21201. Phone (410) 706-7410
References
- 1.American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35 (Suppl 1):S64–71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porte D, Jr, Schwartz MW. Diabetes complications: why is glucose potentially toxic? Science. 1996;272:699–700. doi: 10.1126/science.272.5262.699. [DOI] [PubMed] [Google Scholar]
- 3.King GL. The role of hyperglycaemia and hyperinsulinaemia in causing vascular dysfunction in diabetes. Ann Med. 1996;28:427–32. doi: 10.3109/07853899608999103. [DOI] [PubMed] [Google Scholar]
- 4.Barnett DM, Krall LP. The history of Diabetes. In: Kahn BB, King GL, Moses AC, Weir GC, Jacobson AMJSR, editors. Joslin’s Diabetes Mellitus. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 1–17. [Google Scholar]
- 5.LeCorche E. Du Diabetic dans se rapports avec la vie uterine menstruation et al grusesse. Ann Gynecol. 1885;24:257. [Google Scholar]
- 6.Duncan JM. On puerperal diabetes. Trans Obstet Soc Lond. 1882;24:256. [Google Scholar]
- 7.Joslin EP. Treatment of Diabetes Mellitus. Philadelphia: Lea and Febiger; 1923. Number of pages. [Google Scholar]
- 8.Pedersen J. The Pregnant Diabetic and Her Newborn: Problems and Management. Baltimore: Williams and Wilkins; 1977. Number of pages. [Google Scholar]
- 9.Olofsson P, Liedholm H, Sartor G, et al. Diabetes and pregnancy. A 21-year Swedish material. Acta Obstet Gynecol Scand Suppl. 1984;122:3–62. [PubMed] [Google Scholar]
- 10.Kitzmiller JL, Gavin LA, Gin GD, et al. Preconception care of diabetes. Glycemic control prevents congenital anomalies. Jama. 1991;265:731–6. [PubMed] [Google Scholar]
- 11.Kucera J. Rate and type of congenital anomalies among offspring of diabetic women. J Reprod Med. 1971;7:73–82. [PubMed] [Google Scholar]
- 12.Soler NG, Walsh CH, Malins JM. Congenital malformations in infants of diabetic mothers. Q J Med. 1976;45:303–13. [PubMed] [Google Scholar]
- 13.Miodovnik M, Mimouni F, Dignan PS, et al. Major malformations in infants of IDDM women. Vasculopathy and early first-trimester poor glycemic control. Diabetes Care. 1988;11:713–8. doi: 10.2337/diacare.11.9.713. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen JF, Molsted-Pedersen L, Mortensen HB. Fetal growth delay and maternal hemoglobin A1c in early diabetic pregnancy. Obstet Gynecol. 1984;64:351–2. [PubMed] [Google Scholar]
- 15.Rubin A, Murphy DP. Studies in human reproduction. III. The frequency of congenital malformations in the offspring of nondiabetic and diabetic individuals. J Pediatr. 1958;53:579–85. doi: 10.1016/s0022-3476(58)80148-1. [DOI] [PubMed] [Google Scholar]
- 16.Damm P, Molsted-Pedersen L. Significant decrease in congenital malformations in newborn infants of an unselected population of diabetic women. Am J Obstet Gynecol. 1989;161:1163–7. doi: 10.1016/0002-9378(89)90656-x. [DOI] [PubMed] [Google Scholar]
- 17.Hanson U, Persson B, Thunell S. Relationship between haemoglobin A1C in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia. 1990;33:100–4. doi: 10.1007/BF00401047. [DOI] [PubMed] [Google Scholar]
- 18.Fuhrmann K, Reiher H, Semmler K, et al. Prevention of congenital malformations in infants of insulin-dependent diabetic mothers. Diabetes Care. 1983;6:219–23. doi: 10.2337/diacare.6.3.219. [DOI] [PubMed] [Google Scholar]
- 19.Greene MF, Hare JW, Cloherty JP, et al. First-trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology. 1989;39:225–31. doi: 10.1002/tera.1420390303. [DOI] [PubMed] [Google Scholar]
- 20.Reece EA, Homko CJ. Assessment and management of pregnancies complicated by pregestational and gestational diabetes mellitus. J Assoc Acad Minor Phys. 1994;5:87–97. [PubMed] [Google Scholar]
- 21.Kalter H, Warkany J. Medical progress. Congenital malformations: etiologic factors and their role in prevention (first of two parts) N Engl J Med. 1983;308:424–31. doi: 10.1056/NEJM198302243080804. [DOI] [PubMed] [Google Scholar]
- 22.Tsang RC, Ballard J, Braun C. The infant of the diabetic mother: today and tomorrow. Clin Obstet Gynecol. 1981;24:125–47. doi: 10.1097/00003081-198103000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Hadden DR. Diabetes in pregnancy 1985. Diabetologia. 1986;29:1–9. doi: 10.1007/BF02427272. [DOI] [PubMed] [Google Scholar]
- 24.Ballard JL, Holroyde J, Tsang RC, et al. High malformation rates and decreased mortality in infants of diabetic mothers managed after the first trimester of pregnancy (1956–1978) Am J Obstet Gynecol. 1984;148:1111–8. doi: 10.1016/0002-9378(84)90637-9. [DOI] [PubMed] [Google Scholar]
- 25.Mills JL. Malformations in infants of diabetic mothers. Teratology. 1982;25:385–94. doi: 10.1002/tera.1420250316. [DOI] [PubMed] [Google Scholar]
- 26.Harris MI, Flegal KM, Cowie CC, et al. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes Care. 1998;21:518–24. doi: 10.2337/diacare.21.4.518. [DOI] [PubMed] [Google Scholar]
- 27.Peacock I. Glycosylated haemoglobin: measurement and clinical use. J Clin Pathol. 1984;37:841–51. doi: 10.1136/jcp.37.8.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai LC. Global standardisation of HbA1c. Malays J Pathol. 2008;30:67–71. [PubMed] [Google Scholar]
- 29.Consensus C. Consensus statement on the worldwide standardization of the hemoglobin A1C measurement: the American Diabetes Association, European Association for the Study of Diabetes, International Federation of Clinical Chemistry and Laboratory Medicine, and the International Diabetes Federation. Diabetes Care. 2007;30:2399–400. doi: 10.2337/dc07-9925. [DOI] [PubMed] [Google Scholar]
- 30.Eisenbarth GS. Type 1 Diabetes Mellitus. In: Kahn CR, Weir GC, King GL, Jacobson AM, Moses AC, Smith RJ, editors. Diabetes Mellitus. Philladelphia: Lippincott Williams & Wilkins; 2005. pp. 399–424. [Google Scholar]
- 31.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–7. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 32.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nature reviews. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 33.Meeuwisse-Pasterkamp SH, van der Klauw MM, Wolffenbuttel BH. Type 2 diabetes mellitus: prevention of macrovascular complications. Expert review of cardiovascular therapy. 2008;6:323–41. doi: 10.1586/14779072.6.3.323. [DOI] [PubMed] [Google Scholar]
- 34.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 35.Meyer BA, Palmer SM. Pregestational diabetes. Semin Perinatol. 1990;14:12–23. [PubMed] [Google Scholar]
- 36.Dunne FP. Pregestational Diabetes Mellitus and Pregnancy. Trends Endocrinol Metab. 1999;10:179–82. doi: 10.1016/s1043-2760(98)00147-7. [DOI] [PubMed] [Google Scholar]
- 37.Forsbach-Sanchez G, Tamez-Perez HE, Vazquez-Lara J. Diabetes and pregnancy. Arch Med Res. 2005;36:291–9. doi: 10.1016/j.arcmed.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Ratner RE, Passaro MD. Gestational diabetes mellitus. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus : A Fundamental and Clinical Text. Philadelphia: Lippincott Williams & Wilkins; 2004. pp. 1291–300. [Google Scholar]
- 39.Reece EA, Leguizamon G, Wiznitzer A. Gestational diabetes: the need for a common ground. Lancet. 2009;373:1789–97. doi: 10.1016/S0140-6736(09)60515-8. [DOI] [PubMed] [Google Scholar]
- 40.Ullrich I, Yeater R. Gestational diabetes. W V Med J. 1995;91:148–51. [PubMed] [Google Scholar]
- 41.Persson B, Hanson U. Neonatal morbidities in gestational diabetes mellitus. Diabetes Care. 1998;21 (Suppl 2):B79–84. [PubMed] [Google Scholar]
- 42.Lucas MJ. Diabetes complicating pregnancy. Obstet Gynecol Clin North Am. 2001;28:513–36. doi: 10.1016/s0889-8545(05)70215-1. [DOI] [PubMed] [Google Scholar]
- 43.Metzger BE, et al. International Association of D Pregnancy Study Groups Consensus P. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33:676–82. doi: 10.2337/dc09-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Correa A, Gilboa SM, Besser LM, et al. Diabetes mellitus and birth defects. Am J Obstet Gynecol. 2008;199:237 e1–9. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reece EA. Diabetes-induced birth defects: what do we know? What can we do? Curr Diab Rep. 2012;12:24–32. doi: 10.1007/s11892-011-0251-6. [DOI] [PubMed] [Google Scholar]
- 46.Becerra JE, Khoury MJ, Cordero JF, et al. Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics. 1990;85:1–9. [PubMed] [Google Scholar]
- 47.Loffredo CA, Wilson PD, Ferencz C. Maternal diabetes: an independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology. 2001;64:98–106. doi: 10.1002/tera.1051. [DOI] [PubMed] [Google Scholar]
- 48.Ramos-Arroyo MA, Rodriguez-Pinilla E, Cordero JF. Maternal diabetes: the risk for specific birth defects. EurJ Epidemiol. 1992;8:503–08. doi: 10.1007/BF00146367. [DOI] [PubMed] [Google Scholar]
- 49.Farrell T, Neale L, Cundy T. Congenital anomalies in the offspring of women with type 1, type 2 and gestational diabetes. Diabet Med. 2002;19:322–6. doi: 10.1046/j.1464-5491.2002.00700.x. [DOI] [PubMed] [Google Scholar]
- 50.Sheffield JS, Butler-Koster EL, Casey BM, et al. Maternal diabetes mellitus and infant malformations. Obstet Gynecol. 2002;100:925–30. doi: 10.1016/s0029-7844(02)02242-1. [DOI] [PubMed] [Google Scholar]
- 51.Reece EA. The fetal and maternal consequences of gestational diabetes mellitus. J Matern Fetal Neonatal Med. 2010;23:199–203. doi: 10.3109/14767050903550659. [DOI] [PubMed] [Google Scholar]
- 52.Garcia Carrapato MR. The offspring of gestational diabetes. J Perinat Med. 2003;31:5–11. doi: 10.1515/JPM.2003.001. [DOI] [PubMed] [Google Scholar]
- 53.Jones CW. Gestational diabetes and its impact on the neonate. Neonatal Netw. 2001;20:17–23. doi: 10.1891/0730-0832.20.6.17. [DOI] [PubMed] [Google Scholar]
- 54.Shields LE, Gan EA, Murphy HF, et al. The prognostic value of hemoglobin A1c in predicting fetal heart disease in diabetic pregnancies. Obstet Gynecol. 1993;81:954–7. [PubMed] [Google Scholar]
- 55.Miller E, Hare JW, Cloherty JP, et al. Elevated maternal hemoglobin A1c in early pregnancy and major congenital anomalies in infants of diabetic mothers. N Engl J Med. 1981;304:1331–4. doi: 10.1056/NEJM198105283042204. [DOI] [PubMed] [Google Scholar]
- 56.Greene MF, Hare JW, Cloherty JP, et al. First-trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology. 1989;39:225–31. doi: 10.1002/tera.1420390303. [DOI] [PubMed] [Google Scholar]
- 57.Rose BI, Graff S, Spencer R, et al. Major congenital anomalies in infants and glycosylated hemoglobin levels in insulin-requiring diabetic mothers. J Perinatol. 1988;8:309–11. [PubMed] [Google Scholar]
- 58.Lucas MJ, Leveno KJ, Williams ML, et al. Early pregnancy glycosylated hemoglobin, severity of diabetes, and fetal malformations. Am J ObstetGynecol. 1989;161:426–31. doi: 10.1016/0002-9378(89)90536-x. [DOI] [PubMed] [Google Scholar]
- 59.Key TC, Giuffrida R, Moore TR. Predictive value of early pregnancy glycohemoglobin in the insulin- treated diabetic patient. Am J ObstetGynecol. 1987;156:1096–100. doi: 10.1016/0002-9378(87)90117-7. [DOI] [PubMed] [Google Scholar]
- 60.Miller E, Hare JW, Cloherty JP, et al. Elevated maternal hemoglobin A1c in early pregnancy and major congenital anomalies in infants of diabetic mothers. N Engl J Med. 1981;304:1331–34. doi: 10.1056/NEJM198105283042204. [DOI] [PubMed] [Google Scholar]
- 61.Ylinen K, Aula P, Stenman UH, et al. Risk of minor and major fetal malformations in diabetics with high haemoglobin A1c values in early pregnancy. BrMed J (Clin ResEd) 1984;289:345–46. doi: 10.1136/bmj.289.6441.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eriksson UJ, Borg LA, Cederberg J, et al. Pathogenesis of diabetes-induced congenital malformations. Ups J Med Sci. 2000;105:53–84. doi: 10.1517/03009734000000055. [DOI] [PubMed] [Google Scholar]
- 63.Horton WE, Jr, Sadler TW. Effects of maternal diabetes on early embryogenesis. Alterations in morphogenesis produced by the ketone body, B-hydroxybutyrate. Diabetes. 1983;32:610–6. doi: 10.2337/diab.32.7.610. [DOI] [PubMed] [Google Scholar]
- 64.Hunter ES, 3rd, Sadler TW, Wynn RE. A potential mechanism of DL-beta-hydroxybutyrate-induced malformations in mouse embryos. Am J Physiol. 1987;253:E72–80. doi: 10.1152/ajpendo.1987.253.1.E72. [DOI] [PubMed] [Google Scholar]
- 65.Hunter ES, 3rd, Sadler TW. D-(−)-beta-hydroxybutyrate-induced effects on mouse embryos in vitro. Teratology. 1987;36:259–64. doi: 10.1002/tera.1420360214. [DOI] [PubMed] [Google Scholar]
- 66.Moore DC, Stanisstreet M, Clarke CA. Morphological and physiological effects of beta-hydroxybutyrate on rat embryos grown in vitro at different stages. Teratology. 1989;40:237–51. doi: 10.1002/tera.1420400305. [DOI] [PubMed] [Google Scholar]
- 67.Eriksson UJ, Cederberg J, Wentzel P. Congenital malformations in offspring of diabetic mothers--animal and human studies. Rev Endocr Metab Disord. 2003;4:79–93. doi: 10.1023/a:1021879504372. [DOI] [PubMed] [Google Scholar]
- 68.Persson B. Prevention of fetal malformation with antioxidants in diabetic pregnancy. PediatrRes. 2001;49:742–43. doi: 10.1203/00006450-200106000-00004. [DOI] [PubMed] [Google Scholar]
- 69.Fuhrmann K, Reiher H, Semmler K, et al. The effect of intensified conventional insulin therapy before and during pregnancy on the malformation rate in offspring of diabetic mothers. Exp Clin Endocrinol. 1984;83:173–77. doi: 10.1055/s-0029-1210327. [DOI] [PubMed] [Google Scholar]
- 70.Kitzmiller JL, Gavin LA, Gin GD, et al. Preconception care of diabetes. Glycemic control prevents congenital anomalies. JAMA. 1991;265:731–36. [PubMed] [Google Scholar]
- 71.Gareskog M, Cederberg J, Eriksson UJ, et al. Maternal diabetes in vivo and high glucose concentration in vitro increases apoptosis in rat embryos. Reprod Toxicol. 2007;23:63–74. doi: 10.1016/j.reprotox.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 72.Akazawa S. Diabetic embryopathy: studies using a rat embryo culture system and an animal model. Congenit Anom (Kyoto) 2005;45:73–9. doi: 10.1111/j.1741-4520.2005.00070.x. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Z, Reece EA. Experimental mechanisms of diabetic embryopathy and strategies for developing therapeutic interventions. J Soc Gynecol Investig. 2005;12:549–57. doi: 10.1016/j.jsgi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 74.Lee-Parritz A. New technologies for the management of pregestational diabetes mellitus. Obstet Gynecol Surv. 2012;67:167–75. doi: 10.1097/OGX.0b013e31824bb538. [DOI] [PubMed] [Google Scholar]
- 75.Ballas J, Moore TR, Ramos GA. Management of diabetes in pregnancy. Curr Diab Rep. 2012;12:33–42. doi: 10.1007/s11892-011-0249-0. [DOI] [PubMed] [Google Scholar]
- 76.Reece EA, Friedman AM, Copel JA, et al. Prenatal diagnosis and management of deviant fetal growth and congenital malformations. In: Reece EA, Coustan DR, editors. Diabetes Mellitus in Pregnancy. New York: Churchill Livingstone; 1995. pp. 219–49. [Google Scholar]
- 77.Guideline Development G. Management of diabetes from preconception to the postnatal period: summary of NICE guidance. BMJ. 2008;336:714–7. doi: 10.1136/bmj.39505.641273.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ali S, Dornhorst A. Diabetes in pregnancy: health risks and management. Postgrad Med J. 2011;87:417–27. doi: 10.1136/pgmj.2010.109157. [DOI] [PubMed] [Google Scholar]
- 79.Kitzmiller JL, Block JM, Brown FM, et al. Managing preexisting diabetes for pregnancy: summary of evidence and consensus recommendations for care. Diabetes Care. 2008;31:1060–79. doi: 10.2337/dc08-9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holing EV, Beyer CS, Brown ZA, et al. Why don’t women with diabetes plan their pregnancies? Diabetes Care. 1998;21:889–95. doi: 10.2337/diacare.21.6.889. [DOI] [PubMed] [Google Scholar]
- 81.Reece EA, Hobbins JC. Diabetic embryopathy: pathogenesis, prenatal diagnosis and prevention. Obstet Gynecol Surv. 1986;41:325–35. [PubMed] [Google Scholar]
- 82.Ewart-Toland A, Yankowitz J, Winder A, et al. Oculoauriculovertebral abnormalities in children of diabetic mothers. Am J Med Genet. 2000;90:303–9. [PubMed] [Google Scholar]
- 83.Zacharias JF, Jenkins JH, Marion JP. The incidence of neural tube defects in the fetus and neonate of the insulin-dependent diabetic woman. Am J Obstet Gynecol. 1984;150:797–8. doi: 10.1016/0002-9378(84)90702-6. [DOI] [PubMed] [Google Scholar]
- 84.Barr M, Jr, Hanson JW, Currey K, et al. Holoprosencephaly in infants of diabetic mothers. J Pediatr. 1983;102:565–8. doi: 10.1016/s0022-3476(83)80185-1. [DOI] [PubMed] [Google Scholar]
- 85.Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16:261–72. doi: 10.1002/tera.1420160304. [DOI] [PubMed] [Google Scholar]
- 86.Rusnak SL, Driscoll SG. Congenital Spinal Anomalies In Infants Of Diabetic Mothers. Pediatrics. 1965;35:989–95. [PubMed] [Google Scholar]
- 87.Aubry MC, Aubry JP, Dommergues M. Sonographic prenatal diagnosis of central nervous system abnormalities. Childs Nerv Syst. 2003;19:391–402. doi: 10.1007/s00381-003-0768-3. [DOI] [PubMed] [Google Scholar]
- 88.Cameron M, Moran P. Prenatal screening and diagnosis of neural tube defects. Prenat Diagn. 2009;29:402–11. doi: 10.1002/pd.2250. [DOI] [PubMed] [Google Scholar]
- 89.Drugan A, Weissman A, Evans MI. Screening for neural tube defects. Clin Perinatol. 2001;28:279–87. vii. doi: 10.1016/s0095-5108(05)70083-x. [DOI] [PubMed] [Google Scholar]
- 90.Turner CD, Silva S, Jeanty P. Prenatal diagnosis of alobar holoprosencephaly at 10 weeks of gestation. Ultrasound Obstet Gynecol. 1999;13:360–2. doi: 10.1046/j.1469-0705.1999.13050360.x. [DOI] [PubMed] [Google Scholar]
- 91.Peebles DM. Holoprosencephaly. Prenat Diagn. 1998;18:477–80. [PubMed] [Google Scholar]
- 92.Wong HS, Tang MH, Yan KW, et al. Histological findings in a case of alobar holoprosencephaly diagnosed at 10 weeks of pregnancy. Prenat Diagn. 1999;19:859–62. doi: 10.1002/(sici)1097-0223(199909)19:9<859::aid-pd633>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 93.Blaas HG, Eik-Nes SH. Sonoembryology and early prenatal diagnosis of neural anomalies. Prenat Diagn. 2009;29:312–25. doi: 10.1002/pd.2170. [DOI] [PubMed] [Google Scholar]
- 94.Cuckle HS. Screening for neural tube defects. Ciba Found Symp. 1994;181:253–66. doi: 10.1002/9780470514559.ch15. discussion 66–9. [DOI] [PubMed] [Google Scholar]
- 95.Pergament E. Alpha-fetoprotein and the prenatal diagnosis of neural tube defects. Obstet Gynecol Annu. 1977;6:173–89. [PubMed] [Google Scholar]
- 96.Haddow JE, Knight GJ, Kloza EM. Alpha-fetoprotein screening in pregnancy-a test whose time has come. Ariz Med. 1982;39:436–9. [PubMed] [Google Scholar]
- 97.Haddow JE, Miller WA. Prenatal diagnosis of open neural tube defects. Methods Cell Biol. 1982;26:67–94. doi: 10.1016/s0091-679x(08)61364-3. [DOI] [PubMed] [Google Scholar]
- 98.Wang R, Martinez-Frias ML, Graham JM., Jr Infants of diabetic mothers are at increased risk for the oculo-auriculo-vertebral sequence: A case-based and case-control approach. J Pediatr. 2002;141:611–7. doi: 10.1067/mpd.2002.128891. [DOI] [PubMed] [Google Scholar]
- 99.Aberg A, Westbom L, Kallen B. Congenital malformations among infants whose mothers had gestational diabetes or preexisting diabetes. Early Hum Dev. 2001;61:85–95. doi: 10.1016/s0378-3782(00)00125-0. [DOI] [PubMed] [Google Scholar]
- 100.Garcia-Patterson A, Erdozain L, Ginovart G, et al. In human gestational diabetes mellitus congenital malformations are related to pre-pregnancy body mass index and to severity of diabetes. Diabetologia. 2004;47:509–14. doi: 10.1007/s00125-004-1337-3. [DOI] [PubMed] [Google Scholar]
- 101.Spilson SV, Kim HJ, Chung KC. Association between maternal diabetes mellitus and newborn oral cleft. Ann Plast Surg. 2001;47:477–81. doi: 10.1097/00000637-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 102.Gratz ES, Pollack MA, Zimmerman RD. Congenital facial palsy and ipsilateral deafness: association with maternal diabetes mellitus. Int J Pediatr Otorhinolaryngol. 1981;3:335–41. doi: 10.1016/0165-5876(81)90058-6. [DOI] [PubMed] [Google Scholar]
- 103.Pilu G, Reece EA, Romero R, et al. Prenatal diagnosis of craniofacial malformations with ultrasonography. Am J Obstet Gynecol. 1986;155:45–50. doi: 10.1016/0002-9378(86)90075-x. [DOI] [PubMed] [Google Scholar]
- 104.Ghi T, Perolo A, Banzi C, et al. Two-dimensional ultrasound is accurate in the diagnosis of fetal craniofacial malformation. Ultrasound Obstet Gynecol. 2002;19:543–51. doi: 10.1046/j.1469-0705.2002.00721.x. [DOI] [PubMed] [Google Scholar]
- 105.Nyberg DA, Hegge FN, Kramer D, et al. Premaxillary protrusion: a sonographic clue to bilateral cleft lip and palate. J Ultrasound Med. 1993;12:331–5. doi: 10.7863/jum.1993.12.6.331. [DOI] [PubMed] [Google Scholar]
- 106.Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart. 2003;89:1217–20. doi: 10.1136/heart.89.10.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ferencz C, Rubin JD, McCarter RJ, et al. Maternal diabetes and cardiovascular malformations: predominance of double outlet right ventricle and truncus arteriosus. Teratology. 1990;41:319–26. doi: 10.1002/tera.1420410309. [DOI] [PubMed] [Google Scholar]
- 108.Rowland TW, Hubbell JP, Jr, Nadas AS. Congenital heart disease in infants of diabetic mothers. J Pediatr. 1973;83:815–20. doi: 10.1016/s0022-3476(73)80374-9. [DOI] [PubMed] [Google Scholar]
- 109.Rajiah P, Mak C, Dubinksy TJ, et al. Ultrasound of fetal cardiac anomalies. AJR Am J Roentgenol. 2011;197:W747–60. doi: 10.2214/AJR.10.7287. [DOI] [PubMed] [Google Scholar]
- 110.Axt-Fliedner R, Gembruch U. Nuchal translucency and fetal cardiac malformations. Ultraschall in der Medizin. 2010;31:144–50. doi: 10.1055/s-0028-1109702. [DOI] [PubMed] [Google Scholar]
- 111.Clur SA, Ottenkamp J, Bilardo CM. The nuchal translucency and the fetal heart: a literature review. Prenatal diagnosis. 2009;29:739–48. doi: 10.1002/pd.2281. [DOI] [PubMed] [Google Scholar]
- 112.Assemany SR, Muzzo S, Gardner LI. Syndrome of phocomelic diabetic embryopathy (caudal dysplasia) Am J Dis Child. 1972;123:489–91. doi: 10.1001/archpedi.1972.02110110117014. [DOI] [PubMed] [Google Scholar]
- 113.Welch JP, Aterman K. The syndrome of caudal dysplasia: a review, including etiologic considerations and evidence of heterogeneity. Pediatr Pathol. 1984;2:313–27. doi: 10.3109/15513818409022263. [DOI] [PubMed] [Google Scholar]
- 114.Sadler TW. Embryology of neural tube development. American journal of medical genetics. 2005;135C:2–8. doi: 10.1002/ajmg.c.30049. [DOI] [PubMed] [Google Scholar]
- 115.Kaufman BA. Neural tube defects. Pediatr Clin North Am. 2004;51:389–419. doi: 10.1016/S0031-3955(03)00207-4. [DOI] [PubMed] [Google Scholar]
- 116.Hollyday M. Neurogenesis in the vertebrate neural tube. Int J Dev Neurosci. 2001;19:161–73. doi: 10.1016/s0736-5748(00)00093-9. [DOI] [PubMed] [Google Scholar]
- 117.Altmann CR, Brivanlou AH. Neural patterning in the vertebrate embryo. International review of cytology. 2001;203:447–82. doi: 10.1016/s0074-7696(01)03013-3. [DOI] [PubMed] [Google Scholar]
- 118.Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. Am J Med Genet C Semin Med Genet. 2005;135C:77–87. doi: 10.1002/ajmg.c.30056. [DOI] [PubMed] [Google Scholar]
- 119.Chappell JH, Jr, Wang XD, Loeken MR. Diabetes and apoptosis: neural crest cells and neural tube. Apoptosis. 2009;14:1472–83. doi: 10.1007/s10495-009-0338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sugimura Y, Murase T, Oyama K, et al. Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes. Diabetologia. 2009;52:962–71. doi: 10.1007/s00125-009-1312-0. [DOI] [PubMed] [Google Scholar]
- 121.Zhao Z. Cardiac malformations and alteration of TGFbeta signaling system in diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2010;89:97–105. doi: 10.1002/bdrb.20225. [DOI] [PubMed] [Google Scholar]
- 122.Oyama K, Sugimura Y, Murase T, et al. Folic acid prevents congenital malformations in the offspring of diabetic mice. Endocr J. 2009;56:29–37. doi: 10.1507/endocrj.k08e-180. [DOI] [PubMed] [Google Scholar]
- 123.Wessels A, Markman MW, Vermeulen JL, et al. The development of the atrioventricular junction in the human heart. Circ Res. 1996;78:110–7. doi: 10.1161/01.res.78.1.110. [DOI] [PubMed] [Google Scholar]
- 124.Moorman A, Webb S, Brown NA, et al. Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart. 2003;89:806–14. doi: 10.1136/heart.89.7.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Anderson RH, Webb S, Brown NA, et al. Development of the heart: (2) Septation of the atriums and ventricles. Heart. 2003;89:949–58. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anderson RH, Webb S, Brown NA, et al. Development of the heart: (3) formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart. 2003;89:1110–8. doi: 10.1136/heart.89.9.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao Z. Endoplasmic reticulum stress in maternal diabetes-induced cardiac malformations during critical cardiogenesis period. Birth Defects Res B Dev Reprod Toxicol. 2012;95:1–6. doi: 10.1002/bdrb.20330. [DOI] [PubMed] [Google Scholar]
- 128.Markwald R, Eisenberg C, Eisenberg L, et al. Epithelial-mesenchymal transformations in early avian heart development. Acta Anat. 1996;156:173–86. doi: 10.1159/000147845. [DOI] [PubMed] [Google Scholar]
- 129.Mjaatvedt GH, Yamamura H, Wessels A, et al. Mechanisms of segmentation, septation, and remodeling of the tubular heart: endocardial cushion fate and cardiac looping. In: Harvey RP, Rosenthal N, editors. Heart Development. New York: Academic Press; 1999. pp. 159–77. [Google Scholar]
- 130.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 131.Zhao Z, Rivkees SA. Programmed cell death in the developing heart: regulation by BMP4 and FGF2. Dev Dyn. 2000;217:388–400. doi: 10.1002/(SICI)1097-0177(200004)217:4<388::AID-DVDY6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 132.Kumar SD, Dheen ST, Tay SS. Maternal diabetes induces congenital heart defects in mice by altering the expression of genes involved in cardiovascular development. Cardiovascular diabetology. 2007;6:34. doi: 10.1186/1475-2840-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Siman CM, Gittenberger-De Groot AC, Wisse B, et al. Malformations in offspring of diabetic rats: morphometric analysis of neural crest-derived organs and effects of maternal vitamin E treatment. Teratology. 2000;61:355–67. doi: 10.1002/(SICI)1096-9926(200005)61:5<355::AID-TERA7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 134.Tanigawa K, Kawaguchi M, Tanaka O, et al. Skeletal malformations in rat offspring. Long-term effect of maternal insulin-induced hypoglycemia during organogenesis. Diabetes. 1991;40:1115–21. doi: 10.2337/diab.40.9.1115. [DOI] [PubMed] [Google Scholar]
- 135.Bowden RE. Development of the middle and external ear in man. Proc R Soc Med. 1977;70:807–15. doi: 10.1177/003591577707001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mallo M. Formation of the middle ear: recent progress on the developmental and molecular mechanisms. Dev Biol. 2001;231:410–9. doi: 10.1006/dbio.2001.0154. [DOI] [PubMed] [Google Scholar]
- 137.Sun F, Kawasaki E, Akazawa S, et al. Apoptosis and its pathway in early post-implantation embryos of diabetic rats. Diabetes Res Clin Pract. 2005;67:110–8. doi: 10.1016/j.diabres.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 138.Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–97. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- 139.Fine EL, Horal M, Chang TI, et al. Evidence that elevated glucose causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes. 1999;48:2454–62. doi: 10.2337/diabetes.48.12.2454. [DOI] [PubMed] [Google Scholar]
- 140.Zhao Z, Yang P, Eckert RL, et al. Caspase-8: a key role in the pathogenesis of diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2009;86:72–7. doi: 10.1002/bdrb.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhao Z, Wu Y-K, Reece EA. Demonstration of the essential role of protein kinase C isoforms in hyperglycemia-induced embryonic malformations. Reproductive Sciences. 2008;15:349–56. doi: 10.1177/1933719108316986. [DOI] [PubMed] [Google Scholar]
- 142.Singh CK, Kumar A, Hitchcock DB, et al. Resveratrol prevents embryonic oxidative stress and apoptosis associated with diabetic embryopathy and improves glucose and lipid profile of diabetic dam. Mol Nutr Food Res. 2011;55:1186–96. doi: 10.1002/mnfr.201000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brown CB, Baldwin HS. Neural crest contribution to the cardiovascular system. Adv Exp Med Biol. 2006;589:134–54. doi: 10.1007/978-0-387-46954-6_8. [DOI] [PubMed] [Google Scholar]
- 144.Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–15. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 145.Morgan SC, Relaix F, Sandell LL, et al. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008;82:453–63. doi: 10.1002/bdra.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wentzel P, Gareskog M, Eriksson UJ. Decreased cardiac glutathione peroxidase levels and enhanced mandibular apoptosis in malformed embryos of diabetic rats. Diabetes. 2008;57:3344–52. doi: 10.2337/db08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Graham A, Begbie J, McGonnell I. Significance of the cranial neural crest. Dev Dyn. 2004;229:5–13. doi: 10.1002/dvdy.10442. [DOI] [PubMed] [Google Scholar]
- 148.Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–75. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- 149.Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nature reviews. 2003;4:806–18. doi: 10.1038/nrn1221. [DOI] [PubMed] [Google Scholar]
- 150.Cederberg J, Picard JJ, Eriksson UJ. Maternal diabetes in the rat impairs the formation of neural-crest derived cranial nerve ganglia in the offspring. Diabetologia. 2003;46:1245–51. doi: 10.1007/s00125-003-1100-1. [DOI] [PubMed] [Google Scholar]
- 151.Li R, Thorens B, Loeken MR. Expression of the gene encoding the high-K (m) glucose transporter 2 by the early postimplantation mouse embryo is essential for neural tube defects associated with diabetic embryopathy. Diabetologia. 2007;50:682–9. doi: 10.1007/s00125-006-0579-7. [DOI] [PubMed] [Google Scholar]
- 152.Matsumoto K, Akazawa S, Ishibashi M, et al. Abundant expression of GLUT1 and GLUT3 in rat embryo during the early organogenesis period. Biochem Biophys Res Commun. 1995;209:95–102. doi: 10.1006/bbrc.1995.1475. [DOI] [PubMed] [Google Scholar]
- 153.Stevens FJ, Argon Y. Protein folding in the ER. Semin Cell Dev Biol. 1999;10:443–54. doi: 10.1006/scdb.1999.0315. [DOI] [PubMed] [Google Scholar]
- 154.Benyair R, Ron E, Lederkremer GZ. Protein quality control, retention, and degradation at the endoplasmic reticulum. International review of cell and molecular biology. 2011;292:197–280. doi: 10.1016/B978-0-12-386033-0.00005-0. [DOI] [PubMed] [Google Scholar]
- 155.Groenendyk J, Sreenivasaiah PK, Kim do H, et al. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–97. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- 156.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 157.Banhegyi G, Baumeister P, Benedetti A, et al. Endoplasmic reticulum stress. Ann N Y Acad Sci. 2007;1113:58–71. doi: 10.1196/annals.1391.007. [DOI] [PubMed] [Google Scholar]
- 158.Fonseca SG, Burcin M, Gromada J, et al. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr Opin Pharmacol. 2009;9:763–70. doi: 10.1016/j.coph.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–94. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–31. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 163.Zhao Z, Eckert RL, Reece EA. Reduction in embryonic malformations and alleviation of endoplasmic reticulum stress by nitric oxide synthase inhibition in diabetic embryopathy. Reprod Sci. 2012;19:823–31. doi: 10.1177/1933719111434543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cao Y, Zhao Z, Eckert RL, et al. The essential role of protein kinase Cdelta in diabetes-induced neural tube defects. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet. 2012;25:2020–4. doi: 10.3109/14767058.2012.677963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang X, Borg LA, Eriksson UJ. Altered mitochondrial morphology of rat embryos in diabetic pregnancy. Anat Rec. 1995;241:255–67. doi: 10.1002/ar.1092410212. [DOI] [PubMed] [Google Scholar]
- 166.Genova ML, Pich MM, Biondi A, et al. Mitochondrial production of oxygen radical species and the role of Coenzyme Q as an antioxidant. Exp Biol Med (Maywood) 2003;228:506–13. doi: 10.1177/15353702-0322805-14. [DOI] [PubMed] [Google Scholar]
- 167.Raha S, Robinson BH. Mitochondria, oxygen free radicals, and apoptosis. Am J Med Genet. 2001;106:62–70. doi: 10.1002/ajmg.1398. [DOI] [PubMed] [Google Scholar]
- 168.Ishibashi M, Akazawa S, Sakamaki H, et al. Oxygen-induced embryopathy and the significance of glutathione-dependent antioxidant system in the rat embryo during early organogenesis. Free Radic Biol Med. 1997;22:447–54. doi: 10.1016/s0891-5849(96)00338-3. [DOI] [PubMed] [Google Scholar]
- 169.Li R, Chase M, Jung SK, et al. Hypoxic Stress In Diabetic Pregnancy Contributes To Impaired Embryo Gene Expression And Defective Development By Inducing Oxidative Stress. Am J Physiol Endocrinol Metab. 2005 doi: 10.1152/ajpendo.00441.2004. [DOI] [PubMed] [Google Scholar]
- 170.Menegola E, Broccia ML, Prati M, et al. Glutathione status in diabetes-induced embryopathies. Biol Neonate. 1996;69:293–7. doi: 10.1159/000244323. [DOI] [PubMed] [Google Scholar]
- 171.Yan J, Hales BF. Depletion of Glutathione Induces 4-Hydroxynonenal Protein Adducts and Hydroxyurea Teratogenicity in the Organogenesis Stage Mouse Embryo. Journal of Pharmacology and Experimental Therapeutics. 2006;319:613–21. doi: 10.1124/jpet.106.109850. [DOI] [PubMed] [Google Scholar]
- 172.Djordjevic VB. Free radicals in cell biology. Int Rev Cytol. 2004;237:57–89. doi: 10.1016/S0074-7696(04)37002-6. [DOI] [PubMed] [Google Scholar]
- 173.Ueda S, Masutani H, Nakamura H, et al. Redox control of cell death. Antioxid Redox Signal. 2002;4:405–14. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- 174.Sivan E, Reece EA, Wu YK, et al. Dietary vitamin E prophylaxis and diabetic embryopathy: morphologic and biochemical analysis. Am J Obstet Gynecol. 1996;175:793–9. doi: 10.1016/s0002-9378(96)80001-9. [DOI] [PubMed] [Google Scholar]
- 175.Viana M, Herrera E, Bonet B. Teratogenic effects of diabetes mellitus in the rat. Prevention by vitamin E Diabetologia. 1996;39:1041–6. doi: 10.1007/BF00400652. [DOI] [PubMed] [Google Scholar]
- 176.Siman CM, Eriksson UJ. Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes. 1997;46:1054–61. doi: 10.2337/diab.46.6.1054. [DOI] [PubMed] [Google Scholar]
- 177.Siman CM, Eriksson UJ. Vitamin C supplementation of the maternal diet reduces the rate of malformation in the offspring of diabetic rats. Diabetologia. 1997;40:1416–24. doi: 10.1007/s001250050844. [DOI] [PubMed] [Google Scholar]
- 178.Yang X, Borg LA, Siman CM, et al. Maternal antioxidant treatments prevent diabetes-induced alterations of mitochondrial morphology in rat embryos. Anat Rec. 1998;251:303–15. doi: 10.1002/(SICI)1097-0185(199807)251:3<303::AID-AR5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 179.Viana M, Aruoma OI, Herrera E, et al. Oxidative damage in pregnant diabetic rats and their embryos. Free Radic Biol Med. 2000;29:1115–21. doi: 10.1016/s0891-5849(00)00397-x. [DOI] [PubMed] [Google Scholar]
- 180.Zaken V, Kohen R, Ornoy A. Vitamins C and E improve rat embryonic antioxidant defense mechanism in diabetic culture medium. Teratology. 2001;64:33–44. doi: 10.1002/tera.1045. [DOI] [PubMed] [Google Scholar]
- 181.Guijarro MV, Indart A, Aruoma OI, et al. Effects of ergothioneine on diabetic embryopathy in pregnant rats. Food Chem Toxicol. 2002;40:1751–5. doi: 10.1016/s0278-6915(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 182.Gareskog M, Eriksson UJ, Wentzel P. Combined supplementation of folic acid and vitamin E diminishes diabetes-induced embryotoxicity in rats. Birth defects research Part A, Clinical and molecular teratology. 2006;76:483–90. doi: 10.1002/bdra.20278. [DOI] [PubMed] [Google Scholar]
- 183.Reece EA, Wu YK, Zhao Z, et al. Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am J Obstet Gynecol. 2006;194:580–5. doi: 10.1016/j.ajog.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 184.Reece EA, Wu YK. Prevention of diabetic embryopathy in offspring of diabetic rats with use of a cocktail of deficient substrates and an antioxidant. Am J Obstet Gynecol. 1997;176:790–7. doi: 10.1016/s0002-9378(97)70602-1. discussion 97–8. [DOI] [PubMed] [Google Scholar]
- 185.Li X, Weng H, Reece EA, et al. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms: substrate activation and consequent lipid peroxidation in diabetic embryopathy. Am J Obstet Gynecol. 2011;205:84 e1–6. doi: 10.1016/j.ajog.2011.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hagay ZJ, Weiss Y, Zusman I, et al. Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol. 1995;173:1036–41. doi: 10.1016/0002-9378(95)91323-8. [DOI] [PubMed] [Google Scholar]
- 187.Jawerbaum A, Gonzalez E. The role of alterations in arachidonic acid metabolism and nitric oxide homeostasis in rat models of diabetes during early pregnancy. Curr Pharm Des. 2005;11:1327–42. doi: 10.2174/1381612053507503. [DOI] [PubMed] [Google Scholar]
- 188.Jawerbaum A, Higa R, White V, et al. Peroxynitrites and impaired modulation of nitric oxide concentrations in embryos from diabetic rats during early organogenesis. Reproduction. 2005;130:695–703. doi: 10.1530/rep.1.00699. [DOI] [PubMed] [Google Scholar]
- 189.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 191.Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298 (Pt 2):249–58. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Torreilles J. Nitric oxide: one of the more conserved and widespread signaling molecules. Front Biosci. 2001;6:D1161–72. doi: 10.2741/torreill. [DOI] [PubMed] [Google Scholar]
- 193.Groves JT, Wang CC. Nitric oxide synthase: models and mechanisms. Curr Opin Chem Biol. 2000;4:687–95. doi: 10.1016/s1367-5931(00)00146-0. [DOI] [PubMed] [Google Scholar]
- 194.Umar S, van der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem. 2010;333:191–201. doi: 10.1007/s11010-009-0219-x. [DOI] [PubMed] [Google Scholar]
- 195.Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–30. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 196.Hesslinger C, Strub A, Boer R, et al. Inhibition of inducible nitric oxide synthase in respiratory diseases. Biochem Soc Trans. 2009;37:886–91. doi: 10.1042/BST0370886. [DOI] [PubMed] [Google Scholar]
- 197.Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–64. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 198.Kroncke KD, Suschek CV, Kolb-Bachofen V. Implications of inducible nitric oxide synthase expression and enzyme activity. Antioxid Redox Signal. 2000;2:585–605. doi: 10.1089/15230860050192341. [DOI] [PubMed] [Google Scholar]
- 199.Kumar SD, Yong S-K, Dheen ST, et al. Cardiac Malformations Are Associated with Altered Expression of Vascular Endothelial Growth Factor and Endothelial Nitric Oxide Synthase Genes in Embryos of Diabetic Mice. Experimental Biology and Medicine. 2008;233:1421–32. doi: 10.3181/0806-RM-186. [DOI] [PubMed] [Google Scholar]
- 200.Jawerbaum A, Gonzalez ET, Novaro V, et al. Increased prostaglandin E generation and enhanced nitric oxide synthase activity in the non-insulin-dependent diabetic embryo during organogenesis. Reprod Fertil Dev. 1998;10:191–6. doi: 10.1071/r97077. [DOI] [PubMed] [Google Scholar]
- 201.Yang P, Cao Y, Li H. Hyperglycemia induces inducible nitric oxide synthase gene expression and consequent nitrosative stress via c-Jun N-terminal kinase activation. Am J Obstet Gynecol. 2010;203:185 e5–11. doi: 10.1016/j.ajog.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Waite M. Phospholipases, enzymes that share a substrate class. Advances in experimental medicine and biology. 1990;279:1–22. doi: 10.1007/978-1-4613-0651-1_1. [DOI] [PubMed] [Google Scholar]
- 203.Kaiser E, Chiba P, Zaky K. Phospholipases in biology and medicine. Clinical biochemistry. 1990;23:349–70. doi: 10.1016/0009-9120(90)90051-u. [DOI] [PubMed] [Google Scholar]
- 204.Blobe GC, Khan WA, Hannun YA. Protein kinase C: cellular target of the second messenger arachidonic acid? Prostaglandins Leukot Essent Fatty Acids. 1995;52:129–35. doi: 10.1016/0952-3278(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 205.Bonventre JV. Phospholipase A2 and signal transduction. Journal of the American Society of Nephrology : JASN. 1992;3:128–50. doi: 10.1681/ASN.V32128. [DOI] [PubMed] [Google Scholar]
- 206.Clark JD, Schievella AR, Nalefski EA, et al. Cytosolic phospholipase A2. Journal of lipid mediators and cell signalling. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 207.Liang X, Wu L, Wang Q, et al. Function of COX-2 and prostaglandins in neurological disease. J Mol Neurosci. 2007;33:94–9. doi: 10.1007/s12031-007-0058-8. [DOI] [PubMed] [Google Scholar]
- 208.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50 (Suppl):S29–34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Morrow JD, Hill KE, Burk RF, et al. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc NatlAcadSci USA. 1990;87:9383–87. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Morrow JD. The isoprostanes: their quantification as an index of oxidant stress status in vivo. Drug Metab Rev. 2000;32:377–85. doi: 10.1081/dmr-100102340. [DOI] [PubMed] [Google Scholar]
- 211.Wentzel P, Welsh N, Eriksson UJ. Developmental damage, increased lipid peroxidation, diminished cyclooxygenase-2 gene expression, and lowered prostaglandin E2 levels in rat embryos exposed to a diabetic environment. Diabetes. 1999;48:813–20. doi: 10.2337/diabetes.48.4.813. [DOI] [PubMed] [Google Scholar]
- 212.Wentzel P, Eriksson UJ. A diabetes-like environment increases malformation rate and diminishes prostaglandin E(2) in rat embryos: Reversal by administration of vitamin E and folic acid. Birth Defects Res A Clin Mol Teratol. 2005 doi: 10.1002/bdra.20145. [DOI] [PubMed] [Google Scholar]
- 213.Piddington R, Joyce J, Dhanasekaran P, et al. Diabetes mellitus affects prostaglandin E2 levels in mouse embryos during neurulation. Diabetologia. 1996;39:915–20. doi: 10.1007/BF00403910. [DOI] [PubMed] [Google Scholar]
- 214.Jawerbaum A, Gonzalez ET, Sinner D, et al. Diminished PGE2 content, enhanced PGE2 release and defects in 3H-PGE2 transport in embryos from overtly diabetic rats. Reprod Fertil Dev. 2000;12:141–7. doi: 10.1071/rd00036. [DOI] [PubMed] [Google Scholar]
- 215.Jawerbaum A, Sinner D, White V, et al. Modulation of PGE2 generation in the diabetic embryo: effect of nitric oxide and superoxide dismutase. Prostaglandins Leukot Essent Fatty Acids. 2001;64:127–33. doi: 10.1054/plef.2001.0251. [DOI] [PubMed] [Google Scholar]
- 216.Wentzel P, Eriksson UJ. 8-Iso-PGF(2alpha) administration generates dysmorphogenesis and increased lipid peroxidation in rat embryos in vitro. Teratology. 2002;66:164–8. doi: 10.1002/tera.10068. [DOI] [PubMed] [Google Scholar]
- 217.El-Bassiouni EA, Helmy MH, Abou Rawash N, et al. Embryopathy in experimental diabetic gestation: assessment of PGE2 level, gene expression of cyclooxygenases and apoptosis. Br J Biomed Sci. 2005;62:161–5. doi: 10.1080/09674845.2005.11732704. [DOI] [PubMed] [Google Scholar]
- 218.Wentzel P, Eriksson UJ. Antioxidants diminish developmental damage induced by high glucose and cyclooxygenase inhibitors in rat embryos in vitro. Diabetes. 1998;47:677–84. doi: 10.2337/diabetes.47.4.677. [DOI] [PubMed] [Google Scholar]
- 219.Jawerbaum A, Rosello Catafau J, Gonzalez ET, et al. Glucose metabolism, triglyceride and glycogen levels, as well as eicosanoid production in isolated uterine strips and in embryos in a rat model of non-insulin-dependent diabetes mellitus during pregnancy. Prostaglandins. 1994;47:81–96. doi: 10.1016/0090-6980(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 220.Goto MP, Goldman AS, Uhing MR. PGE2 prevents anomalies induced by hyperglycemia or diabetic serum in mouse embryos. Diabetes. 1992;41:1644–50. doi: 10.2337/diab.41.12.1644. [DOI] [PubMed] [Google Scholar]
- 221.Dempsey EC, Newton AC, Mochly-Rosen D, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 222.Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. Journal of biochemistry. 2002;132:663–8. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- 223.Cao Y, Zhao Z, Eckert RL, et al. Protein kinase Cbeta2 inhibition reduces hyperglycemia-induced neural tube defects through suppression of a caspase 8-triggered apoptotic pathway. Am J Obstet Gynecol. 2011;204:226 e1–5. doi: 10.1016/j.ajog.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 225.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging cell. 2002;1:112–6. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 226.Corson LB, Yamanaka Y, Lai KM, et al. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–37. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- 227.Reece EA, Wu YK, Wiznitzer A, et al. Dietary polyunsaturated fatty acid prevents malformations in offspring of diabetic rats. Am J Obstet Gynecol. 1996;175:818–23. doi: 10.1016/s0002-9378(96)80005-6. [DOI] [PubMed] [Google Scholar]
- 228.Reece EA, Ma XD, Wu YK, et al. Aberrant patterns of cellular communication in diabetes-induced embryopathy. I. Membrane signalling. J Matern Fetal Neonatal Med. 2002;11:249–53. doi: 10.1080/jmf.11.4.249.253. [DOI] [PubMed] [Google Scholar]
- 229.Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol. 2008;198:130 e1–7. doi: 10.1016/j.ajog.2007.06.070. [DOI] [PubMed] [Google Scholar]
- 230.Yang P, Zhao Z, Reece EA. Blockade of c-Jun N-terminal kinase activation abrogates hyperglycemia-induced yolk sac vasculopathy in vitro. Am J Obstet Gynecol. 2008;198:321 e1–7. doi: 10.1016/j.ajog.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 231.Yang P, Zhao Z, Reece EA. Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochem Biophys Res Commun. 2007;357:749–54. doi: 10.1016/j.bbrc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 232.Li X, Xu C, Yang P. c-Jun NH2-Terminal Kinase 1/2 and Endoplasmic Reticulum Stress as Interdependent and Reciprocal Causation in Diabetic Embryopathy. Diabetes. 2012 doi: 10.2337/db12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Molecular and cellular biochemistry. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 234.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 235.Kumar S. Caspase function in programmed cell death. Cell death and differentiation. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 236.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nature reviews. 2008;9:231–41. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 237.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nature reviews. 2008;9:378–90. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 238.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–25. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol. 2006;18:685–9. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 240.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 241.Ferraro E, Corvaro M, Cecconi F. Physiological and pathological roles of Apaf1 and the apoptosome. J Cell Mol Med. 2003;7:21–34. doi: 10.1111/j.1582-4934.2003.tb00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature reviews. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 243.Wiznitzer A, Ayalon N, Hershkovitz R, et al. Lipoic acid prevention of neural tube defects in offspring of rats with streptozocin-induced diabetes. Am J Obstet Gynecol. 1999;180:188–93. doi: 10.1016/s0002-9378(99)70173-0. [DOI] [PubMed] [Google Scholar]
- 244.Khandelwal M, Reece EA, Wu YK, et al. Dietary myo-inositol therapy in hyperglycemia-induced embryopathy. Teratology. 1998;57:79–84. doi: 10.1002/(SICI)1096-9926(199802)57:2<79::AID-TERA6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 245.Reece EA, Khandelwal M, Wu YK, et al. Dietary intake of myo-inositol and neural tube defects in offspring of diabetic rats. Am J Obstet Gynecol. 1997;176:536–9. doi: 10.1016/s0002-9378(97)70543-x. [DOI] [PubMed] [Google Scholar]
- 246.Wentzel P, Gareskog M, Eriksson UJ. Folic Acid Supplementation Diminishes Diabetes- and Glucose-Induced Dysmorphogenesis in Rat Embryos In Vivo and In Vitro. Diabetes. 2005;54:546–53. doi: 10.2337/diabetes.54.2.546. [DOI] [PubMed] [Google Scholar]
- 247.Zabihi S, Eriksson UJ, Wentzel P. Folic acid supplementation affects ROS scavenging enzymes, enhances Vegf-A, and diminishes apoptotic state in yolk sacs of embryos of diabetic rats. Reprod Toxicol. 2007;23:486–98. doi: 10.1016/j.reprotox.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 248.Pinter E, Reece EA, Ogburn PL, Jr, et al. Fatty acid content of yolk sac and embryo in hyperglycemia-induced embryopathy and effect of arachidonic acid supplementation. Am J Obstet Gynecol. 1988;159:1484–90. doi: 10.1016/0002-9378(88)90579-0. [DOI] [PubMed] [Google Scholar]
- 249.Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17:888–95. doi: 10.1016/s0899-9007(01)00658-x. [DOI] [PubMed] [Google Scholar]
- 250.Zed PJ, Krenzelok EP. Treatment of acetaminophen overdose. Am J Health Syst Pharm. 1999;56:1081–91. doi: 10.1093/ajhp/56.11.1081. quiz 91–3. [DOI] [PubMed] [Google Scholar]
- 251.Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm. 2006;63:1821–7. doi: 10.2146/ajhp060050. [DOI] [PubMed] [Google Scholar]
- 252.Gareskog M, Wentzel P. N-Acetylcysteine and {alpha}-cyano-4-hydroxycinnamic acid alter protein kinase C (PKC)-{delta} and PKC-{zeta} and diminish dysmorphogenesis in rat embryos cultured with high glucose in vitro. J Endocrinol. 2007;192:207–14. doi: 10.1677/joe.1.06966. [DOI] [PubMed] [Google Scholar]
- 253.Wentzel P, Ejdesjo A, Eriksson UJ. Maternal diabetes in vivo and high glucose in vitro diminish GAPDH activity in rat embryos. Diabetes. 2003;52:1222–8. doi: 10.2337/diabetes.52.5.1222. [DOI] [PubMed] [Google Scholar]
- 254.Wentzel P, Wentzel CR, Gareskog MB, et al. Induction of embryonic dysmorphogenesis by high glucose concentration, disturbed inositol metabolism, and inhibited protein kinase C activity. Teratology. 2001;63:193–201. doi: 10.1002/tera.1034. [DOI] [PubMed] [Google Scholar]
- 255.Honarbakhsh S, Schachter M. Vitamins and cardiovascular disease. Br J Nutr. 2009;101:1113–31. doi: 10.1017/S000711450809123X. [DOI] [PubMed] [Google Scholar]
- 256.Van Patten CL, de Boer JG, Tomlinson Guns ES. Diet and dietary supplement intervention trials for the prevention of prostate cancer recurrence: a review of the randomized controlled trial evidence. J Urol. 2008;180:2314–21. doi: 10.1016/j.juro.2008.08.078. discussion 721–2. [DOI] [PubMed] [Google Scholar]
- 257.Higa R, Kurtz M, Mazzucco MB, et al. Folic acid and safflower oil supplementation interacts and protects embryos from maternal diabetes-induced damage. Mol Hum Reprod. 2012;18:253–64. doi: 10.1093/molehr/gar080. [DOI] [PubMed] [Google Scholar]
- 258.Villar J, Purwar M, Merialdi M, et al. World Health Organisation multicentre randomised trial of supplementation with vitamins C and E among pregnant women at high risk for pre-eclampsia in populations of low nutritional status from developing countries. Br J Obstet Gynecol. 2009;116:780–8. doi: 10.1111/j.1471-0528.2009.02158.x. [DOI] [PubMed] [Google Scholar]
- 259.Robinson I, de Serna DG, Gutierrez A, et al. Vitamin E in humans: an explanation of clinical trial failure. Endocr Pract. 2006;12:576–82. doi: 10.4158/EP.12.5.576. [DOI] [PubMed] [Google Scholar]
- 260.Polyzos NP, Mauri D, Tsappi M, et al. Combined vitamin C and E supplementation during pregnancy for preeclampsia prevention: a systematic review. Obstet Gynecol Surv. 2007;62:202–6. doi: 10.1097/01.ogx.0000256787.04807.da. [DOI] [PubMed] [Google Scholar]
- 261.Briasoulis A, Tousoulis D, Antoniades C, et al. The oxidative stress menace to coronary vasculature: any place for antioxidants? Curr Pharm Des. 2009;15:3078–90. doi: 10.2174/138161209789058057. [DOI] [PubMed] [Google Scholar]
- 262.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 263.Lipscombe LL, McLaughlin HM, Wu W, et al. Pregnancy planning in women with pregestational diabetes. J Matern Fetal Neonatal Med. 2011;24:1095–101. doi: 10.3109/14767058.2010.545929. [DOI] [PubMed] [Google Scholar]
- 264.Gabbe SG, Graves CR. Management of diabetes mellitus complicating pregnancy. Obstet Gynecol. 2003;102:857–68. doi: 10.1016/j.obstetgynecol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 265.Janz NK, Herman WH, Becker MP, et al. Diabetes and pregnancy. Factors associated with seeking pre-conception care. Diabetes Care. 1995;18:157–65. doi: 10.2337/diacare.18.2.157. [DOI] [PubMed] [Google Scholar]
- 266.Willhoite MB, Bennert HW, Jr, Palomaki GE, et al. The impact of preconception counseling on pregnancy outcomes. The experience of the Maine Diabetes in Pregnancy Program. Diabetes Care. 1993;16:450–5. doi: 10.2337/diacare.16.2.450. [DOI] [PubMed] [Google Scholar]