

Abstract
DNA mismatch repair (MMR) is crucial to ensuring the fidelity of the genome. The inability to correct single base mismatches leads to elevated mutation rates and carcinogenesis. Using metalloinsertors–bulky metal complexes that bind with high specificity to mismatched sites in the DNA duplex–our laboratory has adopted a new chemotherapeutic strategy through the selective targeting of MMR-deficient cells, that is, those that have a propensity for cancerous transformation. Rhodium metalloinsertors display inhibitory effects selectively in cells that are deficient in the MMR machinery, consistent with this strategy. However, a highly sensitive structure–function relationship is emerging with the development of new complexes that highlights the importance of subcellular localization. We have found that small structural modifications, for example a hydroxyl versus a methyl functional group, can yield profound differences in biological function. Despite similar binding affinities and selectivities for DNA mismatches, only one metalloinsertor shows selective inhibition of cellular proliferation in MMR-deficient versus -proficient cells. Studies of whole-cell, nuclear and mitochondrial uptake reveal that this selectivity depends upon targeting DNA mismatches in the cell nucleus.
Keywords: metal DNA, mismatches, DNA repair, intercalator
1. Introduction
The mismatch repair (MMR) machinery recognizes and repairs single base lesions and mismatches that arise from errors in DNA replication [1,2]. Deficiencies in the MMR machinery increase the rate of mutagenesis 50- to 1000-fold, resulting in an enhanced susceptibility to cancer [3,4]. Additionally, many MMR-deficient cancers exhibit resistance to chemotherapeutics such as DNA alkylators and platinating agents [5] as MMR proteins are responsible for recognizing the DNA adducts formed by these agents [6]. As a strategy to target MMR-deficient cancers, we have developed a variety of bulky rhodium complexes that target DNA mismatches through metalloinsertion, a binding mode in which a sterically expansive ligand, such as chrysenequinone diimine (chrysi; figure 1), inserts into the DNA base stack at the site of the mismatch and ejects the thermodynamically destabilized bases. These complexes exhibit 1000-fold selectivity over well-matched DNA and target 80 per cent of all mismatches irrespective of sequence context [7,8,9,10].
Figure 1.
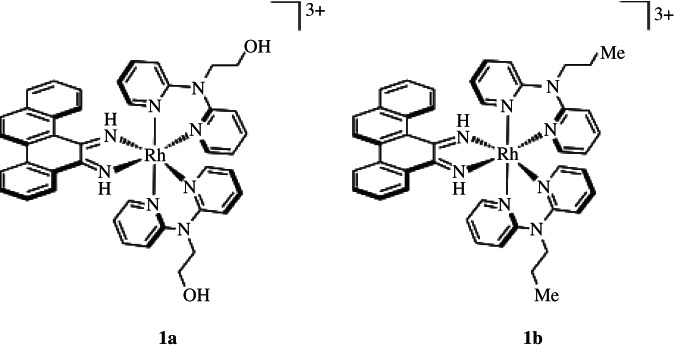
[Rh(L)2chrysi]3+ metalloinsertors. [Rh(DPAE)2chrysi]3+ (1a) contains two ethanol moieties off the central nitrogen atoms, where [Rh(PrDPA)2chrysi]3+ (1b) contains instead two propyl groups.
Metalloinsertion represents a general binding mode for the binding of bulky metal complexes to destabilized mismatches. With intercalative binding, well-matched, hydrogen-bonded base pairs separate, increasing the helical pitch, so that an aromatic heterocyclic ligand can stack within the DNA duplex, essentially like another base pair [11]. For metalloinsertion, the flat aromatic heterocyclic ligand is simply too large to insert easily into the DNA duplex and instead, to accommodate the inserting ligand, the base pairs must separate and be ejected from the helix [12,13]. This ejection only occurs easily at destabilized mismatched sites, and thus the binding affinity for mismatches correlates with the thermodynamic instability of the mismatch, the ease of separation and ejection. Several crystal structures have shown that metalloinsertion occurs from the minor groove side with no increase in helical pitch [12,13,14]. As a result, for the tris(chelate) metalloinsertors, binding within the small minor groove is highly enantioselective for the Δ-isomer.
Previously, we have demonstrated that, because of this high specificity for DNA mismatches, these rhodium metalloinsertors have unique biological properties [15,16,17]. Their biological activity has been characterized in two isogenic cell lines derived from human colorectal carcinoma (HCT116), one MMR-deficient (HCT116O) and the other MMR-proficient (HCT116N) [18]. Cellular proliferation assays have shown that our rhodium metalloinsertors exhibit anti-proliferative activity preferentially in the MMR-deficient HCT116O line. Moreover, the extent of this cell selectivity is dependent on binding of the complex to a mismatched site: the higher the mismatch binding affinity, the greater the differential inhibition of cellular proliferation in MMR-deficient versus -proficient cells [16]. Recently, complexes prepared with more efficient cellular uptake have also shown a differential cytotoxicity in MMR-deficient versus -proficient cells [17]. The results, therefore, support the strategy of a cell-selective chemotherapeutic strategy based upon DNA mismatch targeting.
In the development of novel metalloinsertors for improved cell-selective anti-proliferative activity, two complexes were discovered to have strikingly different biological activities, despite containing only minor functional group changes to their overall structure. The complexes, depicted in figure 1, are tris(chelate) compounds that consist of two N-functionalized dipyridylamine (DPA) ligands in addition to the inserting chrysi ligand. The modified DPA ligands contain either ethanol or N-propyl moieties, affording [Rh(DPAE)2chrysi]3+ (1a) and [Rh(PrDPA)2chrysi]3+ (1b), respectively. [Rh(DPAE)2chrysi]3+ exhibits exceptional inhibition of growth selectively in MMR-deficient cells, whereas [Rh(PrDPA)2chrysi]3+ displays little detectable cell selectivity; instead, the PrDPA complex inhibits cellular proliferation in both cell lines. Here, we explore the various factors that contribute to this cell-selective biological activity for one complex with no activity for the closely related complex. We find that the selective activity in MMR-deficient cells depends not only upon a high binding affinity for single base mismatches, present for both complexes, but also upon efficient targeting of the complexes to nuclear rather than mitochondrial DNA. Specifically, genomic DNA mismatches are implicated as the target for rhodium metalloinsertors in cellulo, whereas the mitochondrion appears to be an undesirable target. These results underscore subcellular localization as an important factor also in therapeutic design.
2. Experimental
(a). Material and methods
All organic reagents were purchased from Sigma-Aldrich unless otherwise noted. Commercially available chemicals were used as received without further purification. RhCl3 starting material was purchased from Pressure Chemical Co. Media and supplements were purchased from Invitrogen (Carlsbad, CA, USA). BrdU, antibodies and buffers were purchased in kit format from Roche Molecular Biochemical (Mannheim, Germany).
Oligonucleotides were ordered from Integrated DNA Technologies and purified by high-performance liquid chromatography (HPLC) using a C18 reverse-phase column (Varian, Inc.). All HPLC purifications were carried out on a Hewlett-Packard 1100 HPLC system. DNA purity was confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry and quantified by UV–vis using the extinction coefficients at 260 nm estimated for single-stranded DNA. UV–vis characterizations were performed on a Beckmann DU 7400 spectrophotometer.
(b). Ligand synthesis
The ancillary ligands 2-(di(pyridin-2-yl)amino)ethanol (DPAE, 5a) and N-propyl-N-(pyridin-2-yl)pyridin-2-amine (PrDPA, 5b) were synthesized from 2,2′-dipyridylamine (2) according to scheme 1.
Scheme 1.
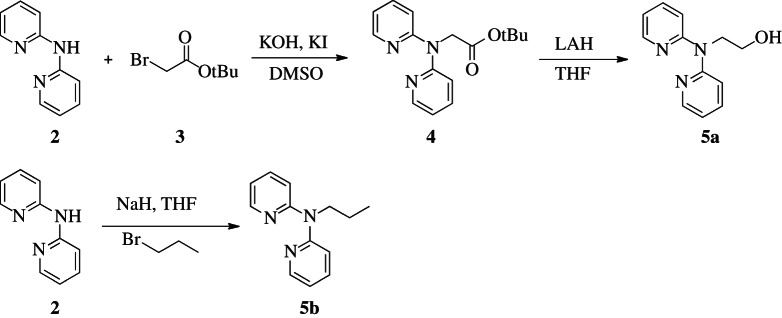
Synthesis of ancillary ligands DPAE (5a) and PrDPA (5b) from dipyridylamine (2).
(i). Tert-butyl 2-(di(pyridine-2-yl)amino)acetate (4)
Tert-butyl 2-(di(pyridine-2-yl)amino)acetate (4) was prepared according to a modified procedure from the literature [19]. Potassium hydroxide (3.0 g, 53.6 mmol, 4.6 equiv.) was added to a solution of 2,2′-dipyridylamine (2) (2.0 g, 11.7 mmol) in 40 ml DMSO and stirred at room temperature for 16 h. Potassium iodide (200 mg, 1.2 mmol, 0.1 equiv.) and tert-butyl bromoacetate (3) (4 ml, 2.3 equiv.) were added to the mixture, and the reaction was stirred for 2 h at room temperature. The reaction mixture was extracted with diethyl ether (3×50 ml). The organic fractions were combined and dried over magnesium sulfate, and the solvent was removed by rotary evaporation. The crude product was isolated by flash chromatography (SiO2, hexane/ethyl acetate=8:2) to give a yellow oil. Yield: 2.92 g (88%). 1H NMR (CDCl3, 300 MHz): δ 8.33 (ddd, J=5.0, 1.9, 0.9 Hz; 2H), 7.53 (m, 2H), 7.23 (m, 2H), 6.88 (ddd, J=7.2, 5.0, 0.9 Hz; 2H), 4.84 (s, 2H), 1.42 (s, 9H). Electrospray ionization mass spectrometry (ESI-MS) (cation): 286 m/z (M+H+) obsd, 286 m/z calcd.
(ii). 2-(di(pyridine-2-yl)amino)ethanol (5a)
To a slurry of lithium aluminium hydride (LAH; 1.17 g, 30.8 mmol, 3.0 equiv) in tetrahydrofuran (THF; 45 ml) was added 4 (2.9 g, 10.2 mmol) at 0°C under 1 atm argon. The reaction was slowly warmed to room temperature over 4 h. The reaction mixture was then diluted with ethyl ether and cooled to 0°C. The reaction was quenched via careful addition of water (4.0 ml) and then dried with magnesium sulfate. The solvent was removed in vacuo, and the crude product was purified by flash chromatography (SiO2, hexane/ethyl acetate=1:1) to afford DPAE (5a) as a pale yellow oil. Yield: 1.2 g (55%). 1H NMR (DMSO-d6, 300 MHz): δ 8.27 (m, 2H), 7.62 (m, 2H), 7.16 (d, J=8.4 Hz, 2H), 6.93 (m, 2H), 4.92 (t, J=5.4 Hz, 1H), 4.16 (t, J=6.5 Hz, 2H), 3.58 (q, J=6.5 Hz, 2H). ESI-MS (cation): 216.1 m/z (M+H+) obsd, 216 m/z calcd.
(iii). N-propyl-N-(pyridin-2-yl)pyridin-2-amine (5b)
To a slurry of sodium hydride (70 mg, 2.9 mmol) in THF (10 ml) was added 2 (500 mg, 2.9 mmol) in 5 ml THF at 0°C under 1 atm argon. The reaction was purged with argon for 15 min, and 1-bromopropane (468 mg, 3.8 mmol) was added dropwise and warmed to room temperature. The reaction was stirred for an additional 18 h under argon at reflux temperature. The reaction mixture was extracted with dilute sodium bicarbonate, and the aqueous phase was extracted with CH2Cl2 (3×40 ml). The organic fractions were combined and dried over magnesium sulfate, and the solvent was removed in vacuo. 5b was purified via flash chromatography (SiO2, hexane/ethyl acetate=9:1). Yield: 100 mg (25%). 1H NMR (CDCl3, 300 MHz): δ 8.34 (d, J=7.7 Hz, 2H), 7.57–7.45 (m, 2H), 7.06 (d, J=0.7 Hz, 2H), 6.90–6.79 (m, 2H), 4.19–4.07 (m, 2H), 1.79–1.65 (m, 2H), 0.99–0.85 (m, 3H) ppm. ESI-MS (cation): 214.1 m/z (M+H+) obsd, 214 m/z calcd.
(c). Metal complexes
(i). [Rh(NH3)4chrysi]3+ (6)
Rhodium precursor 6 was synthesized from [Rh(NH3)5Cl]2+ according to published protocols [16].
(ii). rac-[Rh(DPAE)2chrysi]3+ (1a)
[Rh(NH3)4chrysi]Cl3 (6) (20 mg, 0.038 mmol) and 5a (17.8 mg, 0.082 mmol, excess) were dissolved in a 1:1 mixture of ethanol and water (100 ml) and heated under reflux for 28 h. The solvent was removed in vacuo, and the crude product was purified by HPLC (95:5:0.001 H2O:MeCN:trifluoroacetic acid (TFA)), using a C18 reverse-phase column (Varian, Inc.). The purified product was dried under vacuum and redissolved in a minimal volume of water. The TFA counterion was exchanged for a chloride with a Sephadex QAE-125 ion-exchange resin primed with 1 M MgCl2. Yield: 4.5 mg (13.5%). 1H NMR (DMSO-d6, 300 MHz): δ 13.47 (s, 1H), 13.03 (s, 1H), 9.27 (d, J=8.1 Hz, 1H), 9.02–8.75 (overlapping m, 6H), 8.52–8.27 (overlapping m, 3H), 8.21–7.60 (overlapping m, 8H), 7.41–7.01 (m, 8H), 4.23–4.04 (m, 4H), 3.82 (s, 2H), 3.71–3.54 (m, 4H) ppm; UV–vis (H2O pH 8): 297 nm (47 000 M−1 cm−1), 391 nm (9300 M−1 cm−1). ESI-MS (cation): 787.1 m/z (M – 2H+), 394.2 m/z (M – H2+) obsd, 787 m/z (M – 2H+) calcd.
(iii). rac-[Rh(PrDPA)2chrysi]3+ (1b)
1b was synthesized from 6 (20 mg, 0.038 mmol) and 5b (17 mg, 0.08 mmol) as described for 1a. The resulting product was purified by HPLC (95:5:0.001 H2O:MeCN:TFA) and passed through a Sephadex QAE-125 ion-exchanged resin primed with 1 M MgCl2 to give the chloride salt. Yield: 3 mg (15%). 1H NMR (DMSO-d6, 300 MHz): δ 10.08 (d, J=8.6 Hz, 1H), 8.05 (dd, J=26.7, 8.6 Hz, 4H), 7.58 (dd, J=21.1, 8.5 Hz, 4H), 7.46–7.32 (m, 6H), 7.27–7.11 (m, 3H), 6.90–6.78 (m, 8H), 0.97–0.85 (m, 4H), 0.62 (t, J=7.2 Hz, 4H), 0.02 (t, J=7.3 Hz, 6H) ppm; UV–vis (H2O pH 8): 295 nm (51 000 M−1 cm−1), 388 nm (13 000 M−1 cm−1). ESI-MS (cation): 783.1 m/z (M – 2H+), 392.4 m/z (M – H2+) obsd, 783 m/z calcd.
(d). Octanol/water partition coefficient (log P)
Solid [Rh(DPAE)2chrysi]3+, [Rh(PrDPA)2chrysi]3+ and [Rh(DIP)2chrysi]3+ were dissolved in 10 ml 1-octanol-saturated H2O. Aliquots (2 ml) of each sample were taken in triplicate, mixed with an equal volume of H2O-saturated 1-octanol, and vortexed for 10 s. The samples were incubated at room temperature for 4 h and centrifuged for 5 min at 3000 r.p.m. to allow for the separation of the two phases. The concentrations of rhodium in the aqueous and organic phases were determined by UV–vis; to account for the change in the molar absorptivity of rhodium in 1-octanol, [Rh]oct was defined as [Rh]stock−[Rh]aq. Log P is defined as 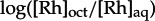 .
.
(e). Cell culture
HCT116N and HCT116O cells were grown in RPMI-1640 medium containing 10 per cent foetal bovine serum, 2 mM l-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 U ml−1 penicillin, 10 μg ml−1 streptomycin and 400 μg ml−1 Geneticin (G418). Cells were grown in tissue culture flasks (Corning Costar, Acton, MA, USA) at 37°C and under 5 per cent CO2 humidified atmosphere.
(f). Cellular proliferation ELISAs
Enzyme-linked immunosorbent assays (ELISAs) were performed with HCT116N and HCT116O cells as described in the literature. Cells were incubated with varying concentrations of 1a or 1b for the durations specified, then grown in rhodium-free medium for the remainder of the 72 h period. After 48 h, BrdU was added, and, at 72 h, BrdU incorporation was quantified by antibody assay [20]. Cellular proliferation was expressed as the ratio of BrdU incorporation into treated cells versus that of untreated cells, and standard errors were calculated from five replicates.
(g). Cellular proliferation with MTT
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) experiments were performed with HCT116N and HCT116O cells as described in the literature [21]. HCT116N and HCT116O cells were inoculated with 0–40 μM 1a or 1b and plated in 96-well plates at 50 000 cells per well. Cells were incubated for 24 h at 37°C under humidified atmosphere. After the incubation period, MTT was added, and the cells were incubated for an additional 4 h. The resulting formazan crystals were solubilized over a period of 24 h at 37°C, 5 per cent CO2. Formazan formation was quantified via electronic absorption at 550–600 nm with a reference wavelength of 690 nm. Cell viability is expressed as a function of formazan formation and normalized to that of untreated cells. Standard errors were calculated from five replicates.
(h). Binding competition titrations
A 29-mer DNA hairpin containing a CC mismatch (*5′-GGCAGGCATGGCTTTTTGCCATCCCTGCC-3′) was labelled with 32P at the 5′-end according to established procedures [22]. A 1:1 mixture of labelled and unlabelled DNA was prepared in buffer (100 mM NaCl, 20 mM NaPi, pH 7.1) to a final concentration of 2 μM. The hairpin was annealed by heating to 90°C for 10 min and slowly cooled to room temperature. To prepare samples for gel electrophoresis, 5 μL of a 4 μM solution of [Rh(bpy)2chrysi]Cl3 (which photocleaves the DNA backbone at the site of a mismatch or abasic site upon irradiation [8,9,10]) and varying concentrations of 1a or 1b (5 μl) were added to 2 μM annealed DNA hairpin (10 μl). A light control (10 μl DNA, 10 μl H2O), a dark control (10 μl DNA, 5 μl [Rh(bpy)2chrysi]3+, 5 μl 1a or 1b, no irradiation) and a positive control (10 μl DNA, 5 μl [Rh(bpy)2chrysi]3+, 5 μl H2O) were also prepared. Samples were vortexed and, except for the dark control, irradiated on an Oriel (Darmstadt, Germany) 1000 W Hg/Xe solar simulator (340–440 nm) for 15 min. Samples were then incubated at 37°C for 20 min, dried, then electrophoresed through a 20 per cent denaturing polyacrylamide gel. The gel was exposed on a phosphor screen, phosphorimaged, and the amounts of DNA cleavage were quantified using ImageQuant.
To determine the KB values of 1a and 1b, competition gels were run in triplicate for each complex, and the percentage of DNA cleavage at each concentration was averaged and plotted as a function of log [Rh]. The data were fitted to a sigmoidal curve using OriginPro v. 8.1. KB values were determined by calculating the concentration of rhodium at the inflection points of the curve and solving simultaneous equilibria involving DNA, [Rh(bpy)2chrysi]3+ and 1a or 1b in Mathematica 8.0. The dissociation constant KD is defined as 1/KB.
(i). Whole-cell rhodium accumulation
HCT116O cells were plated in six-well plates at 1.0×106 cells per well (3 ml medium), and allowed 24 h to adhere. The cells were then incubated with 10 μM 1a or 1b for an additional 24 h. Cells were lysed with 1 per cent sodium dodecyl sulfate (SDS) and sonicated. Samples were aliquoted (0.75 ml) and diluted with 2 per cent HNO3 (0.75 ml), and the cellular rhodium content was quantified on an HP-4500 inductively coupled plasma mass spectrometry (ICP-MS) unit. The remainder of the cell lysates were analysed for protein content via bicinchoninic acid (BCA) assay [23]. Rhodium counts were normalized to cellular protein content, and standard errors were calculated from three replicates.
(ii). Mitochondrial rhodium accumulation
HCT116O cells were plated in 75 cm2 culture flasks at 2.0×107 cells per plate and incubated at 37°C, 5 per cent CO2, for 24 h. Rhodium was added to 10 μM and cells were grown for an additional 24 h. The cells were then harvested by trypsinization and centrifuged for 5 min at 1200 r.p.m. The supernatants were decanted, and the cell pellets were resuspended in 1 ml cold phosphate-buffered saline (PBS) (pH 7.2). The cells were centrifuged again for 5 min at 1200 r.p.m. The supernatants were discarded, and the resultant pellets were resuspended in 0.5 ml mitochondrial extraction buffer (200 mM mannitol, 68 mM sucrose, 50 mM PIPES (piperazine-1,2-bis[2-ethanesulfonic acid]), 50 mM KCl, 5 mM ethylene glycol tetraacetic acid, 2 mM MgCl2; 1 mM dithiothreitol and protease inhibitors were added immediately before use). The samples were incubated on ice for 20 min, and the suspensions were homogenized via passage through a needle and syringe (35×). The homogenized cells were then centrifuged for 5 min at 750 r.p.m. The supernatants were collected and spun again at 14 000g for 10 min. The supernatants were decanted, and the resulting mitochondrial pellet was suspended in 0.8 ml H2O via probe sonication. All samples were diluted 1× with 2 per cent HNO3. Aliquots (20 μl) were used in a BCA assay to determine mitochondrial protein content, which was carried out according to standard protocol. Rh counts from ICP-MS were converted to ppb and normalized to mitochondrial protein content (ng Rh per mg protein). As the mitochondria were isolated from whole cells, the rhodium content determined is strictly mitochondrial and, therefore, cannot be directly compared with total cellular rhodium accumulation. It should be noted that the Rh counts obtained are a lower-bound estimate, given the possibility of rhodium diffusion during organelle isolation. However, the experiments were performed in triplicate and were repeated by different experimenters at different times with a high level of reproducibility. The purity of mitochondrial fractions was ascertained by western blot [24].
(k). Nuclear rhodium accumulation
HCT116O cells were plated in 75 cm2 culture flasks at 1.5×107 cells per plate and incubated at 37°C, 5 per cent CO2, for 24 h. Rhodium was then added to 10 μM and cells were grown for an additional 24 h. The cells were trypsinized according to standard protocol, and the cell pellets were washed with 3 ml 1×PBS (pH 7.2) and spun at 1200 r.p.m. for 5 min. The supernatant was discarded, and the pellets were resuspended in 1 ml 1×PBS and divided into 2×0.5 ml aliquots (nuclear and whole cell). The samples were spun at 450g for 5 min at 4°C. The supernatants were decanted and the whole-cell pellets were dissolved in 1 ml Milli-Q water. The nuclear pellets were dissolved in 1 ml hypotonic buffer (20 mM Tris-HCl, pH 7.4; 10 mM NaCl, 3 mM MgCl2) and incubated on ice for 15 min. After 15 min, 50 μl of NP-40 detergent was added and the samples were vortexed for 10 s. Samples were then spun at 3000g for 10 min at 4°C. The supernatants were discarded, and the nuclear pellets were dissolved in 1 ml Milli-Q water via sonication. All samples were diluted 1× with 2 per cent HNO3. A total of 20 μl aliquots were used in a BCA assay to determine nuclear protein content, which was carried out according to standard protocol. Rh counts from ICP-MS were converted to ppb and normalized to nuclear protein content (ng Rh mg−1 protein). Experiments were performed in biological triplicate, and standard errors were calculated from six replicates.
3. Results and discussion
(a). Synthesis and characterization of metal complexes
The complexes studied were prepared in a straightforward manner. The ancillary ligands DPAE 5a and PrDPA 5b, were synthesized from 2,2'-dipyridylamine [19], and the rhodium precursor, [Rh(NH3)4chrysi]3+(6), was synthesized from [Rh(NH3)5Cl]2+ [16]. The rac-tris(chelate) complexes (1a, 1b) were prepared by reacting 6 with either 5a or 5b (2.1 equiv.) in a 1:1 mixture of ethanol and water at reflux temperature (scheme 2). The octanol/water partition coefficients (log P) were determined to be −1.5 and −1.0 for [Rh(DPAE)2chrysi]3+ and [Rh(PrDPA)2chrysi]3+, respectively, illustrating that simple functional group manipulations can appreciably alter the lipophilicity of a complex. These log P values for [Rh(DPAE)2chrysi]3+ and [Rh(PrDPA)2chrysi]3+ may be compared with that of [Rh(DIP)2chrysi]3+, a highly lipophilic complex (log P=1.3) but with no cell-selective activity, given its poor binding to mismatches.
Scheme 2.
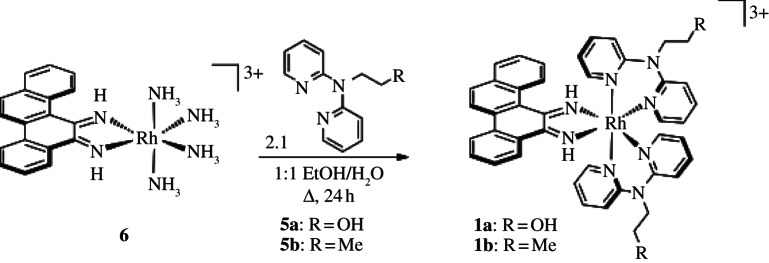
Synthesis of rac-[Rh(L)2chrysi]3+.
(b). Biological studies in MMR-deficient versus -proficient cell lines
We first tested for the selective effects on cellular proliferation of rac-[Rh(DPAE)2chrysi]3+ and rac-[Rh(PrDPA)2chrysi]3+ using the ELISA in the isogenic HCT116 cell lines testing for BrdU incorporation [20]. HCT116N and HCT116O cells were incubated with varying concentrations of each complex, and the proliferation of each cell line was measured over time as a function of incorporation of the thymidine analogue BrdU [20]. The differential activity of rhodium treatment is defined as the difference between the normalized percentages of BrdU incorporation for the two cell lines.
As seen in figure 2a, [Rh(DPAE)2chrysi]3+ exhibits differential inhibition of growth in the MMR-deficient cell line as early as 6 h. At 10 μM, the differential inhibition of the complex is 63±7%. This activity is quite high and early compared with other metalloinsertors tested [16]. By contrast, and remarkably, [Rh(PrDPA)2chrysi]3+ displays little detectable selectivity for MMR-deficient cells; after 72 h, cell growth appears to be inhibited similarly in both cell lines (figure 2b).
Figure 2.
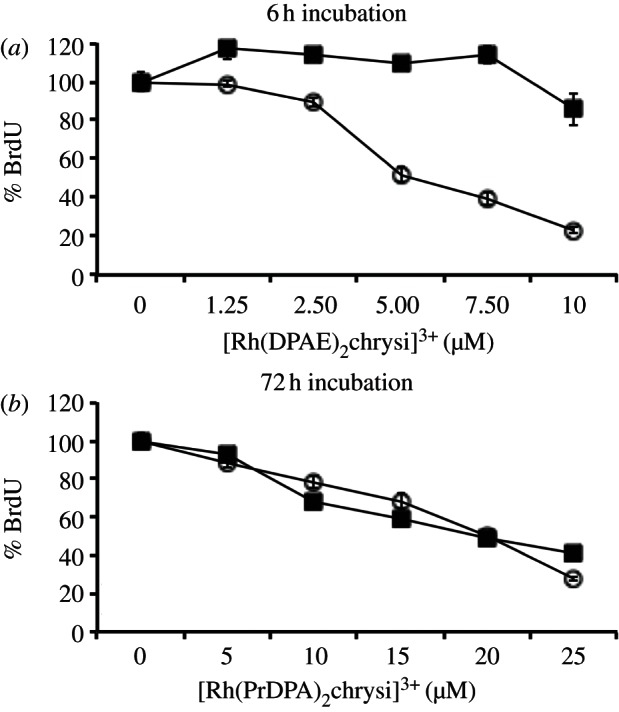
Inhibitory effects of (a) [Rh(DPAE)2chrysi]3+ and (b) [Rh(PrDPA)2chrysi]3+ on MMR-deficient HCT116O (open circles) and MMR-proficient HCT116N (filled squares) cells over time. [Rh(DPAE)2chrysi]3+ exhibits excellent, cell-selective activity at just 6 h, whereas [Rh(PrDPA)2chrysi]3+ imparts non-selective inhibitory effects after 72 h. BrdU incorporation is normalized to that of untreated cells.
We next assayed for cytotoxicity using the MTT assay for mitochondrial function. Mitochondrial enzymes reduce MTT reagent to formazan, which can be quantified by its characteristic absorbance at 570 nm. The absorbance is typically an indicator of the percentage of viable cells present in the medium; however, it more directly reflects the metabolic activity of the cells, and specifically mitochondrion function [25,26,27]. HCT116N and HCT116O cells were treated with 0–40 μM rhodium and incubated for 24 h, after which the cells were exposed to MTT reagent for 4 h. The formazan crystals were solubilized in acidified SDS and quantified using electronic absorption spectroscopy. The percentage of viable cells in a given sample is expressed as a function of the absorbance of formazan at 570 nm. We were interested in particular in comparing the two Rh complexes directly. Neither showed significant differential effects in cytotoxicity at 24 h. However, while we observe that cells treated with rac-[Rh(DPAE)2chrysi]3+ exhibit little cytotoxic effect at 24 h (figure 3), cells treated with [Rh(PrDPA)2chrysi]3+ show some loss in viability, indicative of a change in metabolic activity. This effect for [Rh(PrDPA)2chrysi]3+, however, is not found to be cell selective.
Figure 3.
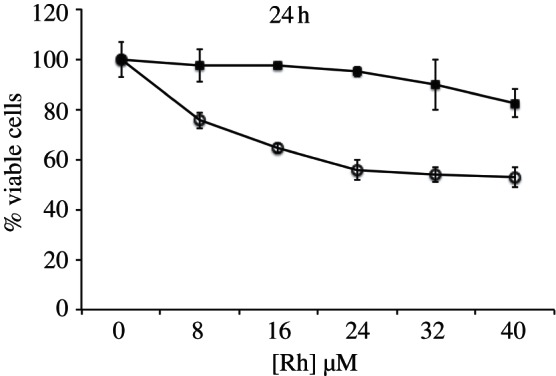
Cell viability of HCT116O cells treated with either [Rh(DPAE)2chrysi]3+ (filled squares) or [Rh(PrDPA)2chrysi]3+ (open circles) over a 24 h period, as determined by MTT assay. The percentage of cell viability is normalized to that of untreated cells.
(c). DNA binding affinity
Previously, a correlation between DNA binding affinity and inhibitory effects on MMR-deficient cells was established [16]. In general, complexes that bind DNA mismatches with the highest affinity were found to have the greatest differential activity in cellulo, with the most effective complexes showing KB=107–108 M−1 for a CC mismatch. We thus sought to examine whether a difference in binding affinity might account for the differences seen in biological activities.
Since [Rh(DPAE)2chrysi]3+ and [Rh(PrDPA)2chrysi]3+ do not promote DNA photocleavage, DNA binding affinities were measured on a 29-mer hairpin containing a CC mismatch in a competition assay through photocleavage by [Rh(bpy)2chrysi]3+ [22]. For a CC mismatch, we find KB=6.8×106 M−1 and 2.5×106 M−1 for [Rh(DPAE)2chrysi]3+ and [Rh(PrDPA)2chrysi]3+, respectively (figure 4). The binding affinity of [Rh(DPAE)2chrysi]3+ for a CC mismatch is, therefore, only slightly greater than that of [Rh(PrDPA)2chrysi]3+. Both complexes show affinities well within the range where differential effects on biological activities have been seen [16]. Thus, binding affinity alone cannot account for the difference in biological activity between the two complexes.
Figure 4.
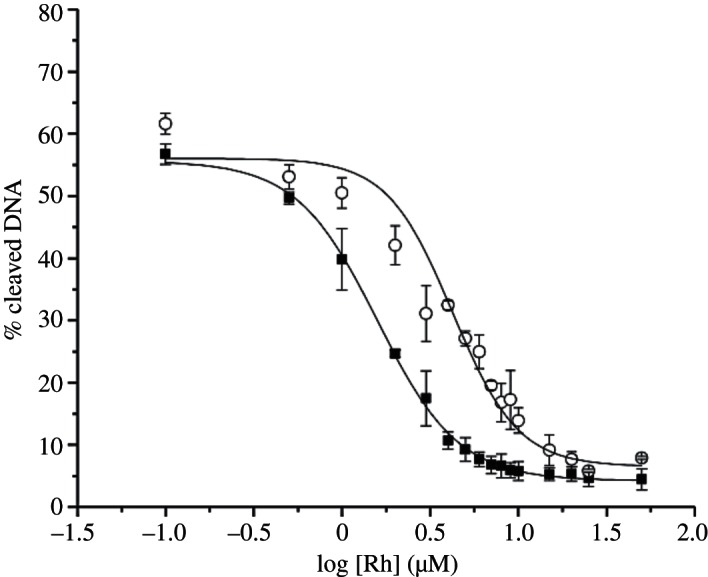
Sigmoidal curve fits for competition titrations with [Rh(DPAE)2chrysi]3+ (filled squares) and [Rh(PrDPA)2chrysi]3+ (open circles). KB was calculated by solving simultaneous equilibria at the inflection point of each curve. Experiments were conducted in buffer (50 mM NaCl, 10 mM NaPi, pH 7.1) using 1 μM DNA and 1 μM rac-[Rh(bpy)2chrysi]3+. Uncertainties are estimated to be 10%.
(d). Cellular uptake and subcellular localization
We next explored the accumulation of rhodium in whole-cell, nuclear and mitochondrial extracts using ICP-MS. Could the difference in biological function be explained through a difference in cellular uptake?
To determine whole-cell uptake, HCT116O cells were incubated in medium containing 10 μM [Rh(DPAE)2chrysi]3+ or [Rh(PrDPA)2chrysi]3+ for 24 h. Cells were rinsed with PBS (pH 7.2) and lysed in a 1% SDS solution. Rhodium content was quantified using ICP-MS and normalized to cellular protein content as determined by a BCA assay [23].
As is evident in figure 5, it is apparent that it is [Rh(PrDPA)2chrysi]3+ that is more efficiently taken up inside cells. [Rh(PrDPA)2chrysi]3+ exhibits significantly more cellular rhodium accumulation than [Rh(DPAE)2chrysi]3+. The whole-cell uptake of [Rh(PrDPA)2chrysi]3+ was measured to be 705±140 ng Rh mg−1 cellular protein, whereas accumulation of [Rh(DPAE)2chrysi]3+ was determined to be 165±65 ng Rh mg−1 cellular protein. HCT116N cells were treated similarly, and the same trends in uptake and localization were observed for both cell lines. The increased lipophilicity of [Rh(PrDPA)2chrysi]3+ afforded by the alkyl moieties probably contributes to this enhanced cellular accumulation. Based upon cellular accumulation, then, [Rh(PrDPA)2chrysi]3+ might be expected to show greater biological efficacy, contrasting what we observe.
Figure 5.
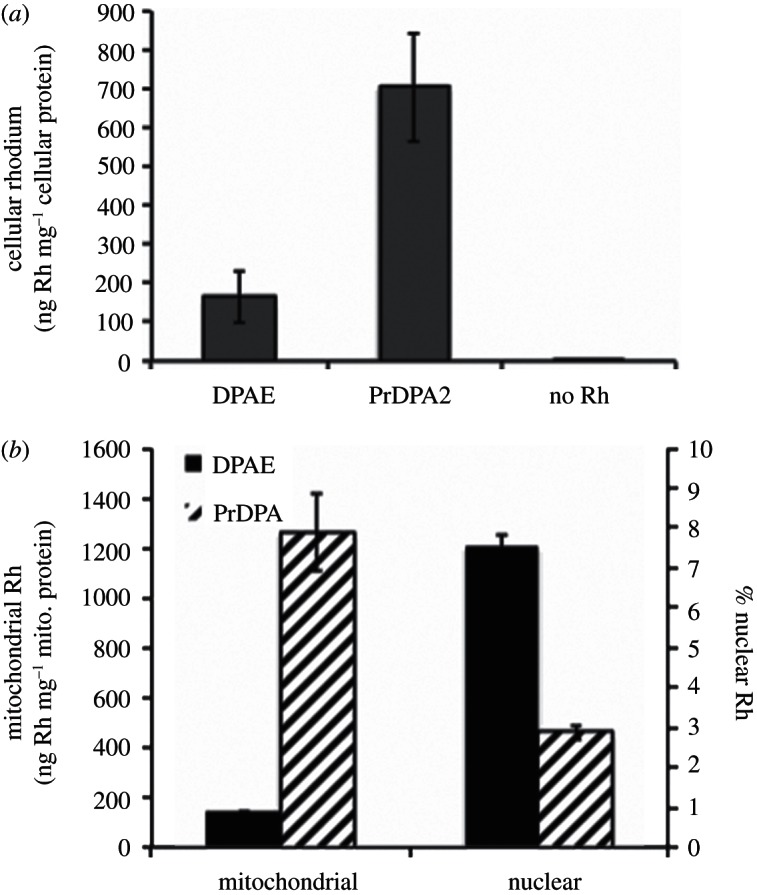
ICP-MS assay for rhodium uptake in (a) whole-cell extracts and (b) nuclear and mitochondrial fractions. HCT116O cells were incubated in medium containing 10 μM of either [Rh(DPAE)2chrysi]3+ (black) or [Rh(PrDPA)2chrysi]3+ (hashed) for 24 h. Rhodium content was quantified by ICP-MS and normalized to (a) cellular protein content or (b) mitochondrial protein content.
Mitochondrial and nuclear localization studies, however, offered further insight into the behaviour of rhodium metalloinsertors in cellulo. HCT116O cells were incubated in medium containing 10 μM [Rh(DPAE)2chrysi]3+ or [Rh(PrDPA)2chrysi]3+ for 24 h and harvested by trypsinization. After washing with PBS, nuclei and mitochondria were isolated via differential centrifugation, and rhodium content was quantified by ICP-MS. Mitochondrial rhodium accumulation is normalized to mitochondrial protein content, and nuclear rhodium accumulation is expressed as a percentage of the total whole-cell rhodium. Here, too, HCT116N cells were treated similarly, and the same trends in uptake and localization were observed for the two cell lines.
As illustrated in figure 5b, mitochondrial rhodium content in cells incubated with [Rh(PrDPA)2chrysi]3+ exceeds that of cells grown in the presence of [Rh(DPAE)2chrysi]3+ by nearly 10-fold. As with whole-cell uptake, the mitochondrial accumulation of [Rh(PrDPA)2 chrysi]3+ can probably be attributed to the lipophilic ancillary ligands, facilitating uptake of the lipophilic cation in response to mitochondrial membrane potential. It is understandable that this greater accumulation of [Rh(PrDPA)2chrysi]3+ in mitochondria is likely to account for the MTT results.
For the DPAE complex, in contrast, a larger percentage of Rh appears in the nucleus. Clearly, it is this nuclear trafficking that is responsible for the biological efficacy of [Rh(DPAE)2chrysi]3+. Interestingly, the nuclear rhodium concentrations are actually quite similar for both complexes. However, the significantly increased cellular accumulation of [Rh(PrDPA)2chrysi]3+ results in a higher proportion of rhodium in the cytosol and mitochondria, and it is here where cytotoxic effects that are not cell selective must be triggered. By contrast, there is a smaller amount of extranuclear [Rh(DPAE)2chrysi]3+, which by extension results in a lower mitochondrial concentration. For the DPAE complex, MMR-selective effects of the complex prevail over any non-specific consequences of mitochondrial accumulation.
Perhaps most significantly, these data identify quite simply that metalloinsertors target mismatch lesions in genomic DNA rather than those in mitochondrial DNA. It is this nuclear mismatch targeting that is responsible for the differential biological activity in MMR-deficient cells that we observe.
4. Implications and conclusions
We have characterized two metalloinsertors, similar in structure and in their binding to DNA mismatches, but we have found only of one of the complexes, [Rh(DPAE)2chrysi]3+, with a highly selective biological response in MMR-deficient cells, whereas the other, [Rh(PrDPA)2chrysi]3+, shows no cell selectivity. It is remarkable that this biological effect depends so sensitively on the chemical structure of the ancillary ligands. Substitution of the terminal alcohols on the DPA ligands for methyl groups is sufficient to extinguish the differential inhibition of cellular proliferation. It is, moreover, neither mismatch binding nor whole-cell uptake that is responsible for this effect; the complexes show quite similar DNA binding affinities and, indeed, there is superior whole-cell uptake of [Rh(PrDPA)2chrysi]3+.
Instead we attribute the high level of cell-selective inhibition of cellular proliferation by [Rh(DPAE)2chrysi]3+ to the subcellular localization of the complex to the nucleus rather than mitochondrion. This subtle variation in ligand structure appears to be responsible for mitochondrial versus nuclear accumulation. Interestingly, similarly small changes in subcellular localization have been demonstrated with peptide–fluorophore conjugates [28]. The enhanced cellular uptake of [Rh(PrDPA)2chrysi]3+ magnifies this difference in biological effect, resulting in a substantial amount of mitochondrial rhodium relative to nuclear rhodium with associated cytotoxicity that is not cell selective. Indeed, for [Rh(PrDPA)2chrysi]3+, the nuclear rhodium content may largely reside in the membrane. For [Rh(DPAE)2chrysi]3+, instead, a greater percentage of cellular rhodium accrues in the nucleus, available to target the DNA mismatches.
Importantly, these results are also consistent with the model that differential anti-proliferative activity is the result of mismatch recognition by metalloinsertors within genomic, rather than mitochondrial, DNA. Mismatches in the nucleus that result from MMR deficiencies are those that are targeted.
This study underscores the subtle variations in chemical structure that must be considered in the rational design of novel chemotherapeutics. Not only must macromolecular targets, in our case the DNA mismatches, be recognized with high specificity, but also whole-cell uptake and specific trafficking to the organelle of interest must be taken into account. Subcellular localization too is seen to change with very small changes in ligand functionality. These factors require consideration in the design of novel DNA binding probes.
Acknowledgements
Financial support from the NIH (GM33309) is gratefully acknowledged, and we thank the NSF for a predoctoral fellowship to A.C.K. This project benefited from the use of instrumentation made available by the Caltech Environmental Analysis Center.
References
- 1.Wildenberg J, Meselson M. 1975. Mismatch repair in heteroduplex DNA. Proc. Natl Acad. Sci. USA 72, 2202–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner R, Jr, Meselson M. 1976. Repair tracts in mismatched DNA heteroduplexes. Proc. Natl Acad. Sci. USA 73, 4135–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer RR, Pluciennik A, Burdett V, Modrich PL. 2006. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323. 10.1021/cr0404794 ( 10.1021/cr0404794) [DOI] [PubMed] [Google Scholar]
- 4.Parsons R, Li G-M, Longley M, Modrich P, Liu B, Berk T, Hamilton SR, Kinzler KW, Vogelstein B. 1995. Mismatch repair deficiency in phenotypically normal human cells. Science 268, 738–740. 10.1126/science.7632227 ( 10.1126/science.7632227) [DOI] [PubMed] [Google Scholar]
- 5.Fink D, Aebi S, Howell SB. 1998. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 4, 1–6 [PubMed] [Google Scholar]
- 6.Fram RJ, Cusick PS, Wilson JM, Marinus MG. 1985. Mismatch repair of cis-diammine-dichloroplatinum(II)-induced DNA damage. Mol. Pharmacol. 28, 51–55 [PubMed] [Google Scholar]
- 7.Zeglis BM, Pierre VC, Barton JK. 2007. Metallo-intercalators and metallo-insertors. Chem. Commun. 44, 4565–4579. 10.1039/b710949k ( 10.1039/b710949k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson BA, Barton JK. 1997. Recognition of DNA base mismatches by a rhodium intercalator. J. Am. Chem. Soc. 119, 12 986–12 987( 10.1021/ja972489a) [DOI] [Google Scholar]
- 9.Jackson BA, Alekseyev VY, Barton JK. 1999. A versatile mismatch recognition agent: specific cleavage of a plasmid DNA at a single base mispair. Biochemistry 38, 4655–4662. 10.1021/bi990255t ( 10.1021/bi990255t) [DOI] [PubMed] [Google Scholar]
- 10.Jackson BA, Barton JK. 2000. Recognition of base mismatches in DNA by 5,6-chrysenequinone diimine complexes of rhodium(III): a proposed mechanism for preferential binding in destabilized regions of the double helix. Biochemistry 39, 6176–6182. 10.1021/bi9927033 ( 10.1021/bi9927033) [DOI] [PubMed] [Google Scholar]
- 11.Keilkopf CL, Erkkila KE, Hudson BP, Barton JK, Rees DC. 2000. Structure of a photoactive rhodium complex intercalated into DNA. Nat. Struct. Biol. 7, 117–121. 10.1038/72385 ( 10.1038/72385) [DOI] [PubMed] [Google Scholar]
- 12.Pierre VC, Kaiser JT, Barton JK. 2007. Insights into finding a mismatch through the structure of a mispaired DNA bound by a rhodium intercalator. Proc. Natl Acad. Sci. USA 104, 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeglis BM, Pierre VC, Kaiser JT, Barton JK. 2009. A bulky rhodium complex bound to an adenosine-adenosine DNA mismatch: general architecture of the metalloinsertion binding mode. Biochemistry 48, 4247–4253. 10.1021/bi900194e ( 10.1021/bi900194e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song H, Kaiser JT, Barton JK. 2012. Crystal structure of Δ-[Ru(bpy)2dppz]2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nat. Chem. 4, 615–620. 10.1038/nchem.1375 ( 10.1038/nchem.1375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. 2006. DNA mismatch-specific targeting and hypersensitivity of mismatch-repair-deficient cells to bulky rhodium(III) intercalators. Proc. Natl Acad. Sci. USA 103, 15 359–15 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst RJ, Song H, Barton JK. 2009. DNA mismatch binding and antiproliferative activity of rhodium metalloinsertors. J. Am. Chem. Soc. 131, 2359–2366. 10.1021/ja8081044 ( 10.1021/ja8081044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst RJ, Komor AC, Barton JK. 2011. Selective cytotoxicity of rhodium metalloinsertors in mismatch repair-deficient cells. Biochemistry 50, 10 919–10 928( 10.1021/bi2015822) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. 1994. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N'-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 54, 4308–4312 [PubMed] [Google Scholar]
- 19.Kirin SI, Yennawar HP, Williams ME. 2007. Synthesis and characterization of CuII complexes with amino acid substituted di(2-pyridyl)amine ligands. Eur. J. Inorg. Chem. 23, 3686–3694 [Google Scholar]
- 20.Gratzner HG. 1982. Monoclonal antibody to 5-bromo and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science 218, 474–475. 10.1126/science.7123245 ( 10.1126/science.7123245) [DOI] [PubMed] [Google Scholar]
- 21.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. 10.1016/0022-1759(83)90303-4 ( 10.1016/0022-1759(83)90303-4) [DOI] [PubMed] [Google Scholar]
- 22.Zeglis BM, Barton JK. 2007. DNA base mismatch detection with bulky rhodium intercalators: synthesis and applications. Nat. Protoc. 2, 357–371. 10.1038/nprot.2007.22 ( 10.1038/nprot.2007.22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith PK, et al. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85. 10.1016/0003-2697(85)90442-7 ( 10.1016/0003-2697(85)90442-7) [DOI] [PubMed] [Google Scholar]
- 24.Ahmad KA, Iskandar JL, Hirpara KB, Clement M-V, Pervaiz S. 2004. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 64, 7867–7878. 10.1158/0008-5472.CAN-04-0648 ( 10.1158/0008-5472.CAN-04-0648) [DOI] [PubMed] [Google Scholar]
- 25.Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, Golub TR, Mootha VK. 2008. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 26, 343–351. 10.1038/nbt1387 ( 10.1038/nbt1387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yakes FM, Van Houten B. 1997. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad Sci. USA 94, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madesh M, Bhaskar L, Balasubramanian KA. 1997. Enterocyte viability and mitochondrial function after graded intestinal ischemia and reperfusion in rats. Mol. Cell. Biochem. 167, 81–87. 10.1023/A:1006871622049 ( 10.1023/A:1006871622049) [DOI] [PubMed] [Google Scholar]
- 28.Yousif LM, Stewart KM, Horton KL, Kelley SO. 2009. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chem. Biochem. 10, 2081–2088. [DOI] [PubMed] [Google Scholar]