

Abstract
Latent HIV persists in CD4+ T cells in infected patients under antiretroviral therapy (ART). Latency is associated with transcriptional silencing of the integrated provirus and driven, at least in part, by histone deacetylases (HDACs), a family of chromatin associated proteins that regulate histone acetylation and the accessibility of DNA to transcription factors. Remarkably, inhibition of HDACs is sufficient to reactivate a fraction of latent HIV in a variety of experimental systems. This basic observation led to the shock and kill idea that forcing the transcriptional activation of HIV might lead to virus expression, to virus-or host-induced cell death of the reactivated cells, and to the eradication of the pool of latently infected cells. Such intervention might possibly lead to a cure for HIV infected patients. Here, we review the basic biology of HDACs and their inhibitors, the role of HDACs in HIV latency and recent efforts to use HDAC inhibitors to reactivate latent HIV in vitro and in vivo.
Keywords: HIV, Latency, HDAC, HDAC inhibitors
Introduction: HIV persistence
With the introduction of combination antiretroviral therapy (ART) in the mid-1990s, HIV-associated mortality and morbidity dramatically decreased, and gave rise to the hope for viral eradication in infected individuals. ART is comprised of several drugs that each target a specific stage of the viral life cycle, and is effective at combating active viral replication of newly infected cells and reducing the viral load down to undetectable levels [1]. However, despite the ability of ART to stop new rounds of infection, HIV persists within the body of infected individuals undergoing therapy and cessation of ART leads to a viral rebound within 3–4 weeks [2]. The observation of viral rebound has given rise to questions surrounding the mechanisms behind this viral persistence, and subsequent studies have led to the discovery of several potential viral reservoirs within the body that may contribute to persistence. These include long-lived cells such as resting CD4+ T lymphocytes, follicular dendritic cells, and hematopoietic stem cells. In addition, the virus may persist in anatomical sanctuaries that are not reached by the ART drugs, such as the male urogenital tract, gut-associated lymphatic tissues and central nervous system (reviewed in [3]).
It is not yet fully clear whether HIV persists in the presence of ART because of ongoing low level replication or because of the episodic reactivation of a stable reservoir. A recent study by Shen and Siliciano used a detailed analysis of residual viremia and a new pharmacodynamic analysis of the effectiveness of ART to show that current therapy is fully capable of suppressing ongoing viral replication [4]. However, the possibility of residual ongoing viral replication cannot be fully excluded [5], and intensification of ART using new agents such as integrase inhibitor, fusion inhibitor, and chemokine antagonists are under clinical trials. If, as we suspect, the majority of residual viremia is the result of viral production from stable reservoirs, novel approaches will be required to target these stable latent reservoir for the eradication of HIV.
Latent infection of CD4+ T cells
Following binding to its main target cell, the CD4+ T lymphocyte, HIV fuses with the cell membrane and releases its contents into the cytoplasm. The virus reverse-transcribes its genomic RNA into double-stranded DNA that then enters into the nucleus using both host and viral factors. At this point, the double-stranded viral DNA, or provirus, integrates into the host genome, where it comes under the control of host transcriptional activation and repression mechanisms. In activated CD4+ T cells, cellular transcription factors such s NF-κB and Sp1 drive the initial transcription of viral regulatory proteins, including the viral transactivator of transcription, Tat. Once Tat production reaches sufficient levels, viral transcription is increased by greater than 100-fold, which results in virus production and death of the infected cell usually within 24 hours post infection [6].
However, in most patients, one can detect rare CD4+ T cells (frequency of 1 per 106 cells) that contain an integrated and transcriptionally silent HIV provirus [2, 7]. These proviruses can be reactivated and are replication competent. It is not clear how latency is established, particularly whether latency is established by infection of resting T cells or by infection of activated CD4+ T cells that escape the cytotoxic effects of circulating CD8+ T cells and revert back to a quiescent state. Latent HIV is primarily found in long-lived central memory and transitional memory CD4+ T cells [8]. The absence of viral evolution in latently infected cells supports the model that this reservoir is seeded early in infection and likely maintained by T cell survival and low levels of antigen-driven homeostatic proliferation.
Because of the failure of ART to eradicate HIV from infected individuals, new strategies are being developed with the aim of curing HIV. One such strategy is to purge, or reactivate, the pool of latently infected cells in the presence of ART. To reach this goal, a comprehensive understanding of the mechanisms that govern HIV latency is required.
Several mechanisms have been identified that play a role in the establishment and maintenance of HIV latency. These include a variety of cis- and trans-acting mechanisms. Cis-acting mechanisms include the site of integration of the provirus in the genome and the possible role of the local chromatin environment [9, 10], or possible transcriptional interference mediated by an adjacent gene [11]. Trans-acting mechanisms reflect the state of activation of the CD4+ T lymphocyte and include the level of transcriptional activators such as NF-κB, and Tat cofactors [12] or the presence of transcriptional repressors [13, 14].
Both cis- and trans-acting mechanisms affect the chromatin environment at the HIV promoter and the transcriptionally active and inactive HIV promoters show significant differences in terms of their chromatin organization. For example, the nuc-1 nucleosome is present immediately downstream of the HIV promoter under latency conditions and is disrupted in the activated provirus [15]. Supporting a role for chromatin in the establishment and maintenance of HIV latency, treatment of HIV latently infected cells with small molecule inhibitors of histone deacetylases (HDACs), a family of histone modifying enzymes, is sufficient to reactivate latent HIV [16]. These early observations led to current studies of the role of HDAC inhibitors in patients with the aim of eradicating latently infected cells.
Here, we review the role of HDACs and their inhibitors in HIV transcription, focusing specifically on transcriptional repression leading to latency.
Histone acetylation and characteristics of HDACs
Histone acetylation consists in the addition of an acetyl group to the ε-amino group of lysine residues. Histone lysine acetylation, on lysine 9 or 14 of histone H3 (H3K9, H3K14) or on lysine 16 of histone H4 (H4K16), is generally associated with active gene transcription. The positively charged ε-amino group of lysine residues on the histone tails are thought to regulate the degree of compaction of chromatin and thereby its accessibility to transcriptional factors [17]. In addition, a protein domain known as the bromodomain specifically binds to acetylated lysines in proteins and thereby enables the docking of transcriptional co-activators, such as ATP-dependent chromatin-remodeling complexes, to acetylated chromatin [18].
Histones and other proteins are reversibly acetylated by lysine acetyltransferases (KATs) and deacetylated by lysine deacetylases (KDACs) [19]. Because these activities were initially identified and studied in the context of histone proteins in chromatin, these enzymes are commonly referred to as histone acetyltransferases (HATs) and HDACs, a practice that we will follow here. HDACs are grouped into four classes based on their homology to the yeast deacetylases RPD3 (class I, IV), HDAI (class II) and Sir2 (class III) [20] (Figure 1). Class I includes HDAC1, 2, 3 and 8 and represents a subset of mostly nuclear and ubiquitous enzymes [21]. Class II HDACs are divided in two subclasses: class IIa includes HDAC4, 5, 7, 9, while class IIb contains HDAC6 and 10. Class II HDACs are found both in the nuclei and cytoplasm and are expressed in a more tissue-specific manner and regulate tissue differentiation in a variety of organs [22]. Class IIa HDACs contain a large regulatory N-terminal domain while class IIb HDACs have two tandemly arranged deacetylase domains (Figure 1). Class IV contains only HDAC11 and is related to both RPD3 and HDA1. HDACs from classes I, II, and IV HDACs all share some sequence homology and are Zn2+-dependent enzymes. They harbor a catalytic pocket with a Zn2+ ion at its base that can be inhibited by Zn2+-chelating compounds. In contrast, the class III HDACs, which are also called sirtuins, are nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases. The seven mammalian sirtuins (SIRT1-7) are important and widely expressed enzymes that target histone and non-histone proteins for deacetylation [23].
Figure 1. Classification of HDAC isotypes.
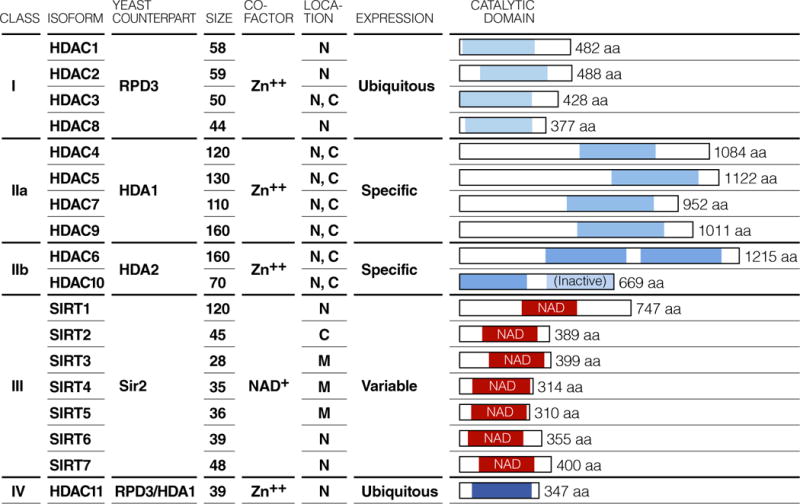
18 HDACs have been identified in mammalian cells, and classified into four groups based on sequence similarity of the catalytic domain to yeast prototypes. Class I HDACs include the yeast RPD3 homologues, HDAC1, 2, 3, 8. Class II HDACs include the yeast HDA1/2 homologues, HDAC4, 5, 6, 7, 9, 10, and are further subclassified as IIa (HDAC4, 5, 7, 9) and IIb (HDAC6, 10). Class III HDACs (also known as sirtuins) include the yeast Sir2 orthologues, SIRT1, 2, 3, 4, 5, 6, 7. Class IV HDACs include HDAC11, which has sequence similarity to both RPD3 and HDA1. N, M and C in localization indicate nuculear, mitochondrial and cytoplasmic, respectively. Sizes are in kDa.
Different HDACs have been implicated as relevant drug targets in cancer, inflammation, cardiovascular and neurological conditions [24]. Two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA)/vorinostat and romidepsin/istodax, have been approved for the treatment of cutaneous T cell lymphoma. Furthermore, a large number of other HDAC inhibitors are currently at different preclinical and clinical stages as novel therapies for various conditions [25].
HDAC inhibitors are classified into four major structural families (Figure 2): short-chain aliphatic acids (including valproic acid and butyric acid), hydroxamic acids (such as trichostatin A and vorinostat), benzamides (including entinostat), and cyclic tetrapeptides and depsipeptides (such as trapoxin B and romidepsin). Different HDAC inhibitors share the same molecular structure consisting of a cap, which targets the surface of the enzyme, a hydrophobic aliphatic chain called the linker (optimal length equals 5–6 carbons) and a head or functional group, which can be either a hydroxamic acid, a benzamide group, a phenylene diamine, a carboxylic acid group or an epoxide group. Most of these groups chelate the catalytic Zn2+ ion in different manners (hydroxamic acid or benzamide) or become covalently linked at the active site (epoxide) [26] (Figure 2).
Figure 2. Basic structure of HDAC inhibitors.
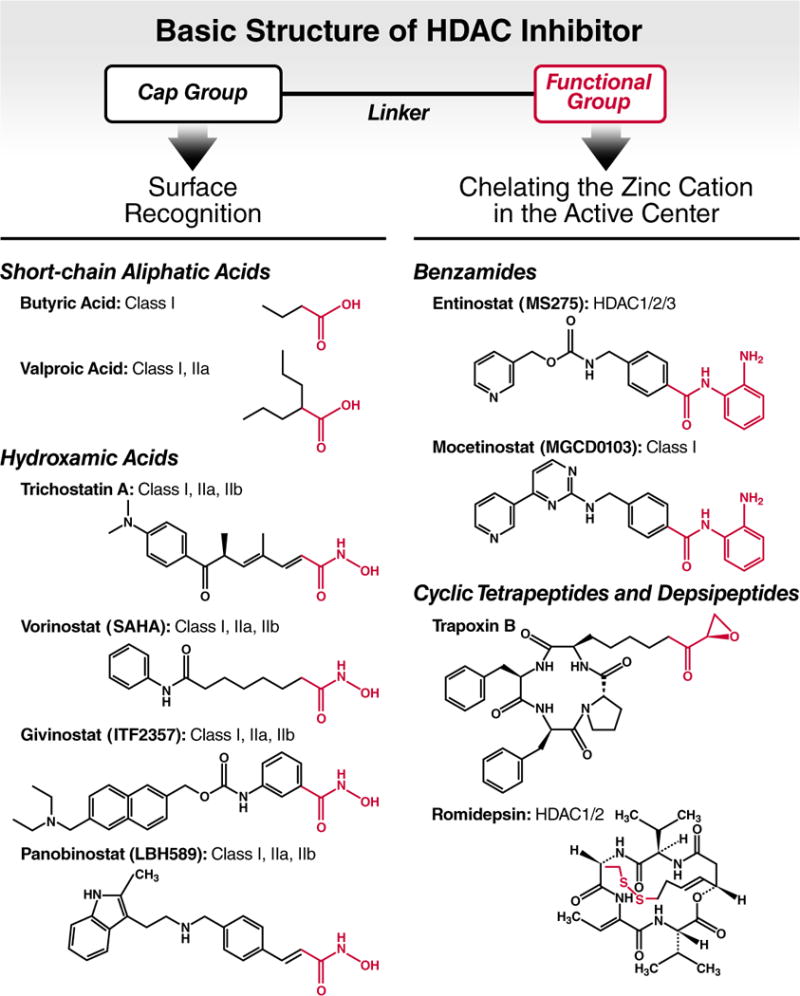
HDAC inhibitors possess a cap group for HDAC surface recognition, a linker (aliphatic chain) and a functional group (highlighted in red) that chelates the zinc cation in the active enzymatic center. The thiol group of romidepsin serves as functional group after it becomes reduced in the intracellular environment. Known HDAC targets of the HDAC inhibitors are listed after each compound.
Importantly, recent large-scale mass spectrometry efforts have shown that 10 to 15% of the mammalian cell proteome undergoes reversible acetylation [27]. In parallel, evidence has accumulated that HDAC proteins target both histone and non-histone proteins for deacetylation [28]. These recent observations suggest that inhibition of HDACs by small molecules is likely to modify not only histone but also non-histone protein acetylation and their biological functions. Understanding the true mechanism of action of HDAC inhibitors will therefore have to take this into account.
HDACs and HIV transcriptional regulation
HDACs regulate HIV latency directly by inducing histone deacetylation at HIV integrated sites (Figure 3A) and indirectly through non-histone protein modification such as NF-κB (Figure 3B). HDAC inhibitors also induce global histone acetylation resulting gene expression changes, and possibly affect HIV latency (Figure 3C). In this section, we discuss mechanisms how HDACs may contribute to the latent state of HIV and mechanism by which HDAC inhibitor treatment may lead to the reactivation of latent HIV.
Figure 3. Mechanisms for reactivation of latent HIV by HDAC inhibitors.
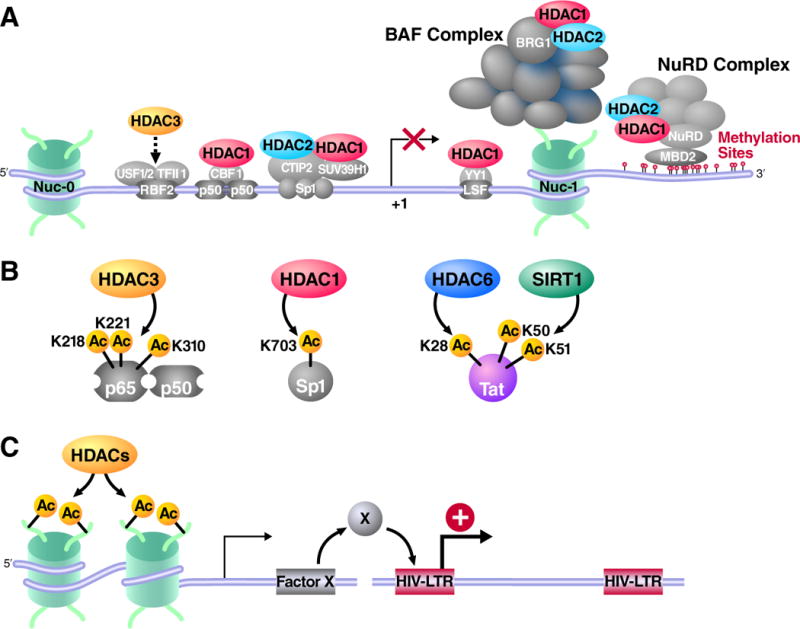
(A) Transcription factors such as RBF2, NF-κB p50, CBF1, Sp1 and YY1 recruit HDACs to the HIV LTR. The BAF complex, which is necessary for the position of the repressor nucleosome nuc-1 at the HIV transcriptional start site, also contains HDAC1 and 2. MBD2 recognizes methylated DNA downstream of nuc-1 and recruits the NuRD complex, which also contains HDAC1 and HDAC2. HDAC inhibitors block these HDACs and directly induce histone acetylation, which leads to the reactivation of the HIV LTR promoter. (B) Full activation of NF-κB (p65–p50) is negatively regulated by HDAC3 through deacetylation. HDAC3 inhibition induces the hyperactivation of the NF-κB factor and to latent HIV reactivation. Sp1 is negatively regulated by HDAC1. HIV Tat is also regulated by acetylation. (C) HDAC inhibitors may also activate the expression of a gene whose product positively regulates HIV transcription.
Recruitment of HDACs to the HIV promoter via transcription factors
The HIV promoter is packaged in chromatin, and nucleosomes are deposited at defined positions within the HIV long terminal repeat [29]. Importantly, a single nucleosome positioned downstream of the HIV transcription start site, called nuc-1, is present on the latent proviral genome and displaced when it is transcriptionally activated. Nuc-1 remodeling is an early event in the reactivation of latent HIV and modification of histone acetylation alone by treatment with an HDAC inhibitor is sufficient to activate latent HIV, indicating that chromatin plays a dominant role in the control of HIV latency [15, 16]. Several transcription factors can recruit HDACs to the HIV long terminal repeat (LTR) promotor under basal conditions (Figure 3A). These include Ying-Yang 1 (YY1) [30], late SV40 factor (LSF) [31], COUP-TF interacting protein (CTIP2) [32], c-promotor-binding factor (CBF-1) [33], NF-κB p50 homodimer [34], and c-myc and Sp1 [35]. These transcription factors recruit HDAC1 to the HIV promoter, locally deacetylate histone proteins locally and lead to transcriptional silencing. HDAC2 is also recruited to the HIV promoter via CTIP2 and Sp1 [32]. HDAC3 also associates with the HIV promoter [36] (Figure 3B).
Importantly, the relative contribution of each HDAC to HIV transcriptional silencing during latency has not been fully elucidated, particularly in primary lymphocytes models of latency or in patients’ cells. It is also not fully clear which of the transcription factors described above is critical for HDAC recruitment to the HIV promoter. In recent experiments, Margolis and colleagues showed that short hairpin RNA (shRNA)-mediated knockdown of YY1 caused an increase in HIV expression in latent cell lines [37]. However, knockdown of YY1 alone or in combination with c-myc showed no change in HDAC occupancy on the HIV LTR indicating that other factors than HDACs contribute to the suppression of HIV expression mediated by YY1 [37].
DNA methylation and histone methylation
DNA methylation of two CpG islands in the HIV promoter has been observed in latently infected cell lines and in patients samples and can mediate the recruitment of the NuRD complex via the methyl binding protein MBD2 [38, 39] (Figure 3A). Importantly, the NuRD complex exists in vertebrates in different compositions in a cell-type-dependent manner and contains HDAC1 and HDAC2 [40]. Histone methylation is linked to DNA methylation. In the HIV latent cell line, 2D12, which has a hypermethylated HIV promoter, histone H3 lysines 9 (H3K9) and 27 (H3K27) are methylated. H3K9 methylation is mediated by Suv39H1 (di- and tri-methylation) and G9a (mono- and di-methylation) while H3K27 methylation is partly mediated by EZH2 (di- and tri-methylation) [41]. These histone methyltransferases are recruited to latent HIV and have been both implicated in HIV latency [42–44]. Recent reports have questioned whether latent HIV is methylated in patients [45]. However, since defective proviruses outnumber latent proviruses by a factor of 100:1 [7], and since latent cells can only be identified a posteriori, methylation analysis of total patient DNA is likely to reflect the defective HIV pool rather than the latent pool. The exact role of DNA methylation in patients therefore remains undefined and a challenge for future investigations.
Remodeling of nuc-1 in the HIV promoter by the BAF/PBAF complexes and protein acetylation
Remodeling of nuc-1 occurs in response to a variety of agents that activate latent HIV, including tumor necrosis factor-α (TNF-α), phorbol esters and the HIV transactivator Tat [15, 46]. The chromatin region encompassing the nuc-1 region becomes acetylated in a transcription factor and HAT-dependent manner [47, 48]. Remarkably, treatment of latently infected cells with HDAC inhibitors is sufficient to induce the chromatin remodeling of nuc-1 [16]. A SWI/SNF chromatin remodeling complex called BAF is associated with the latent HIV promoter and may help to stabilize nuc-1 downstream of the transcriptional start site, on a region of DNA that is thermodynamically unfavored for nucleosome incorporation (Figure 3A) [14]. Interestingly, the BAF complex and HDAC1 and 2 interact [49].
Regulation of trans-acting factors by acetylation: NF-κB, Sp1 and HIV Tat
Transcription factors are also regulated by reversible lysine acetylation. Lysine acetyltransferase p300/CBP acetylates the RelA subunit of NF-κB and enhances its transcriptional activity [50]. Acetylation of lysine residues 210 and 218 of RelA controls its interaction with its endogenous inhibitor 1κB [51], and acetylated RelA persists longer in the nucleus. Acetylation of lysine 310 is required for full activation of its transcriptional activity [52]. HDAC3 deacetylates RelA and negatively regulates its activity (Figure 3B) [51]. Sp1 is also acetylated at lysine 703 in its DNA binding domain leading to increase DNA affinity (Figure 3B) [53]. Inhibition of HDACs causes hyperacetylation of these factors, and likely enhances their ability to activate transcription and cause reversal of repression on the HIV LTR.
HIV-1 Tat is also regulated by posttranslational modifications including reversible acetylation (Figure 3B) [54]. Lysine 28 in the cysteine-rich, transactivation domain of Tat is acetylated by the acetyltransferase PCAF, and this modification is critical for high affinity binding to TAR and P-TEFb. HDAC6 deacetylates lysine 28 of Tat in the nucleus and inhibits Tat transcriptional activity by triggering its export to the cytoplasm [55]. Lysines 50 and 51 in the arginine-rich RNA binding domain, which is required for TAR binding, are acetylated by acetyltransferases p300/CBP and GCN5/PCAF. Acetylation of lysines 50 and 51 is recognized by the bromodomain of the PCAF or SWI/SNF chromatin-remodeling complex, and further facilitate HIV transcription [56]. The class III HDAC, SIRT1 deacetylates Tat [57] and inhibits Tat transactivation in the latently infected cell model [58].
Indirect effects of HDAC inhibitors on latent HIV
HDAC inhibitors induce histone acetylation and affect gene expression in a small proportion (2%–20%) of genes [59, 60]. Genes induced by HDAC inhibitors may regulate the state of HIV latency (Figure 3C).
HDAC inhibitors in basic and clinical research
Various HDAC inhibitors have been shown to reactivate latent HIV in a variety of experimental systems and in cells from HIV-infected individuals. These studies are described next. Current ongoing and completed clinical trials of HDAC inhibitors in HIV infected patients are summarized in Table 1.
Table 1.
Completed and ongoing clinical trials of HDAC inhibitors in HIV-infected patientsa
| Agent | Regimen | Intensification | Phase | No. | Results | Refs |
|---|---|---|---|---|---|---|
| Valproic acid | 500–750 mg bid 4–6 weeks | Enfuvirtide (90 μg) | I/II | 4 | Mean 75% reduction in IUPB | [61] |
|
|
||||||
| 1000 mg od 16 weeks | None | II | 12 | More than 50% reduction in IUPB of 4 patients out of 12 | [62] | |
|
|
||||||
| Plasma valproic acid, mean 58 mg/L | None | II | 11 (13)b |
No significant reduction in HIV DNA copy per million cells | [63] | |
|
|
||||||
| 500–750 mg bid 96 weeks | Raltegravir (400 mg bid) | II | 18 | No significant reduction in IUPB | [64] | |
|
|
||||||
| 500 mg bid 16 or 32 weeks | None | II | 56 | No significant reduction in IUPB | [65] | |
|
| ||||||
| Vorinostat | 400 mg given 3 times a week 8 weeks | None | I/II | 8 | Mean 4.8 fold increase in HIV RNA in resting CD4 T cells | [68] |
Abbreviations: bid, twice a day; od, once daily; IUPB, infectious unit per billion cells.
Control.
Valproic acid
Valproic acid is an FDA-approved anti-epileptic agent that inhibits class I and II HDACs at millimolar concentrations. In a pilot clinical study, valproic acid was administered to four patients on ART [61]. These patients were also given the fusion inhibitor drug enfuvirtide to intensify ART. The frequency of latent infection was measured and significantly declined in three of four patients [61]. However, several follow up studies of valproic acid have failed to detect reduction in latent reservoir size (Table 1) [62–65].
SAHA/vorinostat
The most studied HDAC inhibitor in the context of HIV latency is SAHA, an FDA-approved HDAC inhibitor named vorinostat currently approved for the treatment of cutaneous T cell lymphoma [66]. It reactivates latent HIV in a variety of in vitro experimental models including J89, ACH-2, U1 and J-LAT [67]. Vorinostat also reactivates latent HIV in primary CD4+ T cell models of HIV latency and in CD4 T+ cells isolated from patients on ART [68, 69]. Vorinostat activates the HIV promoter, induces hyperacetylation of the HIV promoter nucleosomes and suppresses HDAC binding to the HIV promoter [70]. In an important pilot experiment (Table 1), a group of 11 ART-treated HIV infected patients were selected based on the absence of measurable viral plasma RNA and increased HIV RNA production ex vivo in response to vorinostat. Treatment of this select group of patients with a single dose of vorinostat triggered a median 4.8-fold increase of HIV mRNA expression in resting CD4+ T cells (range: 1.5 to 10.0) [68, 69]. Among these patients, vorinostat was well tolerated with no significant adverse event. However, it should be noted that the patients were selected based on responsiveness to vorinostat, that we do not know whether this treatment resulted in a decrease in reservoir size, and that we do not know what fraction of latent HIV responds to HDAC inhibitor.
Other hydroxamic acid HDAC inhibitors: givinostat (ITF2357) and panobinostat (LBH-589)
Two other promising HDAC inhibitors are givinostat and panobinostat. Givinostat is currently in phase II clinical trials for relapsed hematological malignancies [71]. Givinostat also induces HIV transcription in latently infected T cells and monocytic cells [72]. Although givinostat needs to be further evaluated for efficacy in aviremic patients on ART, experiments in ACH2 cells and U1 latent cells showed that this inhibitor effectively induces HIV transcription at therapeutically relevant concentrations. Importantly, givinostat decreases the expression of the HIV co-receptors CXCR4 and CCR5 on the cell surface of primary CD4+ T cells [72]. Another hydroxamic acid HDAC inhibitor, panobinostat is a pan-HDAC inhibitor in phase I/II clinical trials for relapsed hematological malignancies and solid tumors. Panobinostat targets class I, II and IV HDACs at low nanomolar range except for HDAC4, 7 and 8 [73]. Panobinostat is at least 10 times more potent than vorinostat and is a good candidate to test in reactivation of latent HIV.
Benzamide HDAC inhibitors: entinostat (MS275) and mocetinostat (MGCD0103)
Entinostat induces HIV expression in latently infected cell lines and in primary cell models of latency [67]. Entinostat preferentially targets class I HDACs (HDAC1, 2 and 3) [69], and shows little toxicity in the models tested [67]. Since entinostat is selective for only HDAC1, 2 and 3, which are most relevant to HIV regulation [74], entinostat might be more efficacious and less toxic. Of note, mocentinostat is another benzamide HDAC inhibitor that also targets class I HDACs and is undergoing clinical trials on Hodgkin lymphoma and other cancers [75].
Romidepsin (FK228)
Romidepsin was originally discovered as an antibiotic produced by Chromobacterium violaceum and is another FDA-approved HDAC inhibitor for the treatment of recurrent cutaneous T cell lymphoma. Romidepsin targets class I HDACs more potently than vorinostat with IC50 of 1 nM in in vitro assays [76]. Unlike vorinostat, romidepsin is Ames test negative. Both basic and clinical studies are required to test this HDAC inhibitor in the context of HIV reactivation.
Novel class I selective HDAC inhibitors
Recent efforts in HDAC inhibitor development have focused increased specificity against unique HDACs. Several of these compounds that selectively target class I HDACS induce HIV transcription in resting T cells from patients receiving ART [77]. This observation suggests that class I HDACs represent promising targets for anti-HIV-latency therapies.
Other compounds to reactivate HIV latency
Chemical compounds that target unique HIV transcription factors and co-factors have been identified for their ability to reactivate latent HIV. Prostratin is a PKC activator from mamala tree bark that induces nuclear accumulation of the NF-κB p65–p50 heterodimer [78]. Synergy is observed when it is used with an HDAC inhibitor [69]. Recently, the acetaldehyde dehydrogenase inhibitor disulfiram was identified in a screen for small molecules that reactivate latent HIV [79]. Disulfiram activates the Akt signaling pathway through downregulation of a phosphatase called PTEN, resulting in latent HIV reactivation [80]. Interestingly, it has been proposed that vorinostat may also activate latent HIV via phosphatidylinositol 3-kinase/Akt pathway activation [81]. Furthermore, other epigenetic modifiers such as histone methyltransferases and DNA methyltransferases are also involved in HIV transcriptional repression. The DNA cytosine methylation inhibitor 5-aza-2′deoxycytidine synergizes with either prostratin or TNF-α to reactivate latent HIV in J-LAT cell lines [38, 39]. Histone methyltransferase inhibitors such as the Suv39H1 inhibitor chaetocin [82], G9a inhibitor BIX01294 [43], and EZH2 inhibitor DZNep [44] are also reported to reactivate latent HIV in a latent cell line model or primary CD4+ T cell model. Combining these small molecules with HDAC inhibitors may prove to be an even more effective inducer of latent HIV.
Concluding remarks: shock and kill approach and future directions
The shock and kill approach combines the induction of latent HIV, by HDAC inhibitors and other drugs, in combination with ART therapy. It is hoped that reactivation of latent HIV will lead to the killing of the productively infected cells by the virus itself, or by the host immune system, and that the ART will block novel infection from the released virus. If effective, this approach might lead to the elimination of the latent viral reservoir and represent a first step in our search for an HIV cure. The recently reported vorinostat study is the first promising example of this shock and kill approach [68], but it raises many new questions. First, as a proof of concept, further clinical studies are required to test whether the addition of vorinostat increased virus production, whether it induced cell death in the latent reservoir and whether it is sufficient to reduce the size of the latent reservoir. Second, additional clinical studies are required to test other drugs and other combinations of drugs and measure their effect on the latent reservoir. Since vorinostat is a toxic anticancer drug and is mutagenic (Ames test-positive), safety issues must also be clarified. In particular, there is concern that HDAC inhibitors might lead to the reactivation of endogenous viruses in the host genome that are under the control of HDACs as well [83]. Finally, a recent report from the Siliciano lab, using an in vitro model of HIV latency in primary lymphocyte, showed that reactivated latently infected resting T cells survive viral cytopathic effects [84]. They further showed that antigen-specific stimulation of cytotoxic T cells from patients on ART was required to efficiently kill latently infected cells that contained reactivated provirus. A combination of virus inductive therapy, such as HDAC inhibitors or prostratin, along with immune stimulators might therefore be necessary for this type of therapeutic approach to be successful.
Acknowledgments
We thank Gary Howard for editorial assistance, John Carroll and Teresa Roberts for graphics and Veronica Fonseca for administrative assistance. Kotaro Shirakawa is funded by a fellowship from CFAR Basic Science Award Program in HIV/AIDS, P30AI027763. Eric Verdin is supported by NIDA Avant-Garde-1 DP1 DA031126, 1R01 DA030216-01, and is a member of the CARE collaborator (UNC/NIH–Federal-5-31532). We apologize to colleagues whose work we could not cite owing to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 2.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary SK, Margolis DM. Curing HIV: Pharmacologic approaches to target HIV-1 latency. Annu Rev Pharmacol Toxicol. 2011;51:397–418. doi: 10.1146/annurev-pharmtox-010510-100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. The Journal of allergy and clinical immunology. 2008;122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tobin NH, et al. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. Journal of virology. 2005;79:9625–9634. doi: 10.1128/JVI.79.15.9625-9634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muesing MA, et al. Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell. 1987;48:691–701. doi: 10.1016/0092-8674(87)90247-9. [DOI] [PubMed] [Google Scholar]
- 7.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 8.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature medicine. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jordan A, et al. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan A, et al. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. Embo J. 2001;20:1726–1738. doi: 10.1093/emboj/20.7.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenasi T, et al. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell host & microbe. 2008;4:123–133. doi: 10.1016/j.chom.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams M, et al. Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3862–3866. doi: 10.1073/pnas.91.9.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Molecular and cellular biology. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafati H, et al. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS biology. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdin E, et al. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Lint C, et al. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 17.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 18.Mujtaba S, et al. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 19.Allis CD, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 20.Gregoretti IV, et al. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verdin E, et al. Class II histone deacetylases: versatile regulators. Trends in Genetics. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 23.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ververis K, Karagiannis TC. Potential non-oncological applications of histone deacetylase inhibitors. Am J Transl Res. 2011;3:454–467. [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 26.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 27.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 28.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdin E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. Journal of virology. 1991;65:6790–6799. doi: 10.1128/jvi.65.12.6790-6799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margolis DM, et al. Human transcription factor YY1 represses human immunodeficiency virus type 1 transcription and virion production. Journal of virology. 1994;68:905–910. doi: 10.1128/jvi.68.2.905-910.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romerio F, et al. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. Journal of virology. 1997;71:9375–9382. doi: 10.1128/jvi.71.12.9375-9382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marban C, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams SA, Greene WC. Regulation of HIV-1 latency by T-cell activation. Cytokine. 2007;39:63–74. doi: 10.1016/j.cyto.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang G, et al. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. Journal of virology. 2007;81:10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malcolm T, et al. Induction of chromosomally integrated HIV-1 LTR requires RBF-2 (USF/TFII-I) and Ras/MAPK signaling. Virus genes. 2007;35:215–223. doi: 10.1007/s11262-007-0109-9. [DOI] [PubMed] [Google Scholar]
- 37.Barton K, Margolis D. Selective targeting of the repressive transcription factors YY1 and cMyc to disrupt quiescent human immunodeficiency viruses. AIDS research and human retroviruses. 2012;29:289–98s. doi: 10.1089/aid.2012.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kauder SE, et al. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS pathogens. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blazkova J, et al. CpG methylation controls reactivation of HIV from latency. PLoS pathogens. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahringer J. NuRD and SIN3 histone deacetylase complexes in development. Trends Genet. 2000;16:351–356. doi: 10.1016/s0168-9525(00)02066-7. [DOI] [PubMed] [Google Scholar]
- 41.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews Genetics. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.du Chene I, et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imai K, et al. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedman J, et al. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. Journal of virology. 2011;85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blazkova J, et al. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. Journal of virology. 2012;86:5390–5392. doi: 10.1128/JVI.00040-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henderson A, et al. Recruitment of SWI/SNF to the human immunodeficiency virus type 1 promoter. Molecular and cellular biology. 2004;24:389–397. doi: 10.1128/MCB.24.1.389-397.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gatignol A. Transcription of HIV: Tat and cellular chromatin. Adv Pharmacol. 2007;55:137–159. doi: 10.1016/S1054-3589(07)55004-0. [DOI] [PubMed] [Google Scholar]
- 48.Ott M, et al. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Current biology: CB. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- 49.Hang CT, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, et al. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 52.Chen LF, et al. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryu H, et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ott M, et al. The control of HIV transcription: keeping RNA polymerase II on track. Cell host & microbe. 2011;10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huo L, et al. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J Biol Chem. 2011;286:9280–9286. doi: 10.1074/jbc.M110.208884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahmoudi T, et al. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Biol Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- 57.Pagans S, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS biology. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinberger LS, et al. Transient-mediated fate determination in a transcriptional circuit of HIV. Nature genetics. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- 59.Van Lint C, et al. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene expression. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 60.Peart MJ, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lehrman G, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Archin NM, et al. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS. 2008;22:1131–1135. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sagot-Lerolle N, et al. Prolonged valproic acid treatment does not reduce the size of latent HIV reservoir. AIDS. 2008;22:1125–1129. doi: 10.1097/QAD.0b013e3282fd6ddc. [DOI] [PubMed] [Google Scholar]
- 64.Archin NM, et al. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PloS one. 2010;5:e9390. doi: 10.1371/journal.pone.0009390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Routy JP, et al. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study. HIV medicine. 2012;13:291–296. doi: 10.1111/j.1468-1293.2011.00975.x. [DOI] [PubMed] [Google Scholar]
- 66.Richon VM, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wightman F, et al. HDAC inhibitors in HIV. Immunol Cell Biol. 2012;90:47–54. doi: 10.1038/icb.2011.95. [DOI] [PubMed] [Google Scholar]
- 68.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reuse S, et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PloS one. 2009;4:e6093. doi: 10.1371/journal.pone.0006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Archin NM, et al. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS research and human retroviruses. 2009;25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan J, et al. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. Journal of hematology & oncology. 2010;3:5. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matalon S, et al. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. Journal of acquired immune deficiency syndromes. 2010;54:1–9. doi: 10.1097/QAI.0b013e3181d3dca3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer letters. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 74.Keedy KS, et al. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. Journal of virology. 2009;83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boumber Y, et al. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert opinion on investigational drugs. 2011;20:823–829. doi: 10.1517/13543784.2011.577737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furumai R, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer research. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 77.Archin NM, et al. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 2009;23:1799–1806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams SA, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279:42008–42017. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 79.Doyon G, et al. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS. 2013;27:F7–F11. doi: 10.1097/QAD.0b013e3283570620. [DOI] [PubMed] [Google Scholar]
- 80.Xing S, et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. Journal of virology. 2011;85:6060–6064. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Contreras X, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem. 2009;284:6782–6789. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bouchat S, et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. AIDS. 2012;26:1473–1482. doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- 83.Montoya-Durango DE, et al. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutation research. 2009;665:20–28. doi: 10.1016/j.mrfmmm.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shan L, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]