

Hereditary hearing loss is one of the most common neurosensory defects in humans. Approximately 70% of cases are nonsyndromic and could be inherited in autosomal dominant, autosomal recessive, mitochondrial, X-linked, and Y-linked manners (Wang et al., 2004; Alford, 2011). The autosomal dominant type, comprising 15%–20% of nonsyndromic hearing loss, is monogenic and genetically heterogeneous. Since the first dominant deafness locus (DFNA1) was identified in 1992, a total of 64 DFNA loci have been mapped (DFNA1–DFNA64), and 27 corresponding genes have been identified (http://hereditaryhearingloss.org). Previous studies have revealed that one deafness locus can be linked to more than one gene (Bayazit and Yilmaz, 2006), and the question “one locus, how many genes?” was first raised about a decade ago (Van-Hauwe et al., 1999). So far, several loci, including DFNA2 and DFNA3, have been shown to be related to one or more genes, showing high genetic heterogeneity in hereditary hearing loss (Grifa et al., 1999; Goldstein and Lalwani, 2002; Yan et al., 2011).
The DFNA4 locus was mapped to chromosome 19q13 with progressive sensorineural hearing loss (Chen et al., 1995; Mirghomizadeh et al., 2002; Pusch et al., 2004). However, it took almost 10 years to uncover the corresponding gene; finally, the nonmuscle myosin heavy-chain gene MYH14 was identified as a causal gene of DFNA4 deafness (Bearer et al., 2000; Donaudy et al., 2004; Mhatre et al., 2004). However, causal mutations of the MYH14 gene have been revealed only in families from Western European countries (Fig. S1). Genetic analysis in the American family in which this locus was originally defined excluded MYH14 as the possible causal gene. The DFNA4 locus has been proposed to be associated with other responsible genes including the CEACAM16 gene (Zheng et al., 2011). In this study, we ascertained and located a large Chinese family with autosomal dominant nonsyndromic hearing loss. Linkage analysis and mutation screening identified the linkage to the DFNA4 locus rather than the known DFNA4 genes, MYH14 and CEACAM16. Our results support the hypothesis that there is another DFNA gene upstream of the MYH14 gene and it may be linked to the DFNA4 locus.
We first collected and evaluated a six-generation Chinese family (family Z002) with autosomal dominant sensorineural hearing loss living in a county of South China. Phenotype of this family was similar but different to that of the reported DFNA4 families. Affected family members showed postlingual, symmetrical, and bilateral nonsyndromic sensorineural hearing loss without vestibular symptoms (Fig. 1A). Hearing loss occurred in the second to third decades of life. Ages of the affected members in family Z002 ranged from 22 to 61 years at the time of our investigation. Hearing loss varied between the fourth and fifth generations. This audiological variation between generations indicated progressive hearing loss with increasing age, which began in the high frequencies and gradually developed to all frequencies (Table S1). The level of hearing loss became severe at later ages. Speech discrimination scores were low in all patients. Vestibular function was normal in all affected family members, and high-resolution computed tomography (HRCT) of the temporal bone in the proband (IV:1) showed normal inner and middle ear structures, including the vestibular aqueduct and internal auditory canal. None of the affected members had a history of exposure to aminoglycosides, noise, or other causes that may account for the hearing impairment. Electrocardiography studies and color Doppler cardiogram indicated normal cardiovascular functions in affected members.
Fig. 1. Gene fine mapping and screening in family Z002 and the other DFNA4 families.
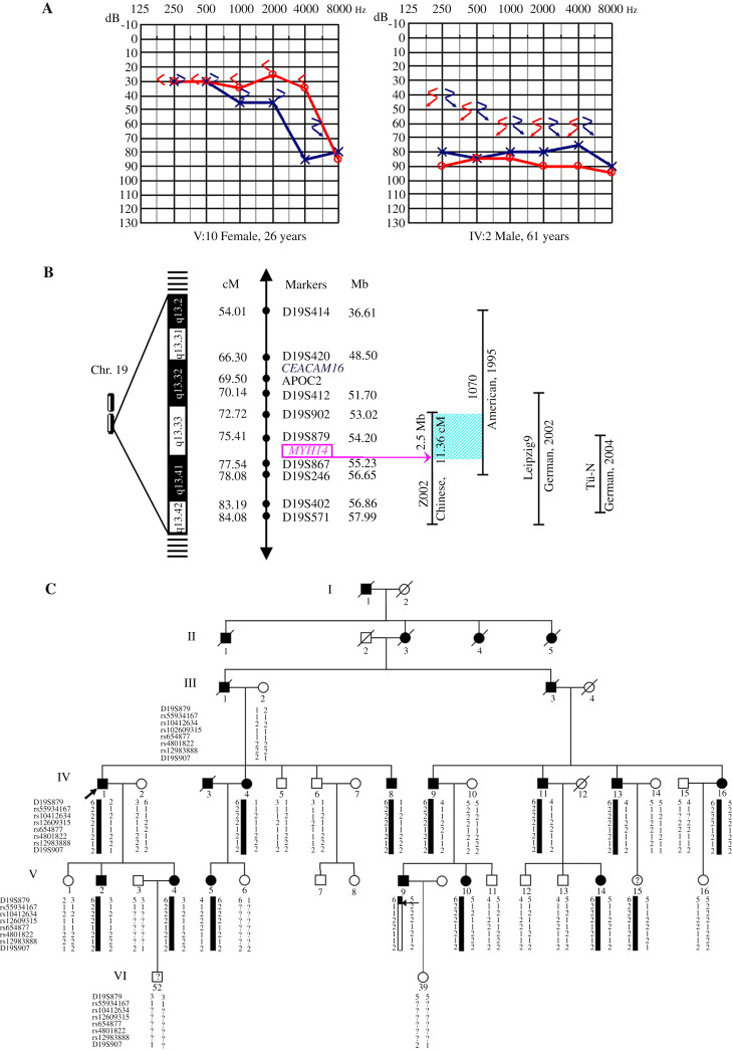
A: typical pure-tone audiograms of both ears from two affected subjects (V:10 and IV:2) in family Z002. ‘○’ connected by lines, right-ear air conduction; ‘×’ connected by lines, left-ear air conduction; ‘<’, right-ear bone conduction; ‘>’, left-ear bone conduction; ‘↙’ and ‘↘’, no response at maximum bone conduction output. B: schematic genetic and physical map of the DFNA4 locus on chromosome 19q13.2-q13.4 in families Z002, 1070, Leipzig9, and Tü-N. The shadowed area represents a 2.5-Mb region from marker D19S902 to the MYH14 gene shared by families Z002 and 1070. The genetic distances of microsatellite markers were obtained from the Marshfield genetic maps, and the physical distances of the markers were obtained from the NCBI database. cM, centimorgan; Mb, megabase. C: SNP haplotype analysis in family Z002. The analysis indicates a recombination (denoted by the horizontal arrow) between D19S879 and rs55934167 in affected subject V:9. The recombination allows exclusion of MYH14 from the candidate region of family Z002 and refines the region to ~2.5 Mb.
To identify the causal gene, we performed the genome-wide linkage scan, fine mapping, and haplotype analysis in family Z002. The linkage study located the deafness gene on the long arm of chromosome 19 (19q13.33-q13.4) and excluded the remainder of the genome. Positive two-point LOD scores of 3.54 and 3.03 (θ = 0) were observed at markers D19S902 and D19S420, respectively. Fine mapping of the region using additional microsatellite markers confirmed the linkage. A maximum two-point LOD score of 5.30 (θ = 0) was obtained for marker D19S879 (Table S2). Haplotype analysis disclosed a key recombination event between markers D19S902 and D19S879 in subject V:9, defining the centromeric limit of the critical interval (Fig. S2). The same recombination was observed in subject V:15. However, she was 10 years old, below the age of onset of the deafness in the family, and therefore her diagnosis was not clear. Subjects IV:4, IV:6, IV:8, IV:9, IV:13, and IV:16 provided the telomeric boundary of the linked interval at marker D19S571. These recombinations refined the disease-linked region to 11.36 cM (4.97 Mb), flanked by markers D19S902 (centromeric) and D19S571 (telomeric). This candidate region showed partial overlap with the critical intervals identified in the previously studied American and German DFNA4 large families, and included the reported deafness-causal gene MYH14 (Fig. 1B).
To test whether MYH14 is responsible for the hearing loss phenotype in family Z002, we did direct sequencing of the gene and revealed no disease-causing mutations. However, 20 nucleotide variants were detected in both affected and unaffected family members (Fig. S1). Among these variants, two synonymous substitutions, 657G > A (rs4801822) in exon 5 and 2127A > G (rs1651553) in exon 17, were detected. The other 18 intronic nucleotide variants identified in the family were all known SNPs. These SNPs did not co-segregate with the deafness phenotype of family Z002.
To further define the candidate region of this family, we performed SNP haplotype analysis. The SNP analysis revealed an important recombination between D19S879 and rs55934167 (Fig. 1C, horizontal arrow). In the SNP haplotype of subject V:9, an important recombination was present between marker D19S879 and the MYH14 gene, mapping the disease gene telomeric to MYH14. The patient was clearly diagnosed with hereditary deafness, but had a different SNP haplotype (112211) compared with the other affected family members. This haplotype had been transmitted from his unaffected mother (IV:9). This recombination excluded the MYH14 gene from the candidate region of family Z002 and placed the region between the proximal boundary marker D19S902 and the distal boundary MYH14 gene, defining a critical interval of 2.5 Mb.
The refined critical region of family Z002 included 121 known genes according to the MapViewer database. However, CEACAM16 was excluded from the area, and coding sequence screening of this gene also found no causative mutations. No other nonsyndromic deafness genes have been mapped to this critical region, with the exception of MYH14. Among these, we selected three potassium channel genes, KCNA7, KCNJ14, and KCNC3, as candidates for mutation screening in family Z002. Genes located around the refined region, such as KPTN, ERCC1, and KCNN4, were also screened. No deafness-related mutations were detected in the coding regions or splice sites of these six genes.
Autosomal dominant nonsyndromic hearing loss has very complicated genotype–phenotype correlations because of its extreme heterogeneity (Lenz and Avraham, 2011). One locus harboring more than one gene is not uncommon. The DFNA2 locus, which contains the two identified causative genes GJB3 and KCNQ4, is a good example (Xia et al., 1998; Coucke et al., 1999). Previous studies indicate that DFNA4 might be one such complex locus containing multiple deafness genes (Fig. 1B). Our genetic analysis showed that Z002 is the sixth reported family that maps to the DFNA4 locus. This locus has a distinct audiologic phenotype including later onset, slower progression, and wider affected frequency range (from high to all frequencies) of sensorineural hearing loss with reduced speech discrimination compared with the other DFNA4 families. These audiologic characteristics, when correlated with subsequent genetic findings, could provide further evidence for heterogeneity at the DFNA4 locus. Four of the previously reported families from Western European countries presented with causal mutations in the known DFNA4 gene, MYH14 (Donaudy et al., 2004; Yang et al., 2005). Further study excluded MYH14 and identified CEACAM16 as a causal gene of the American DFNA4 family (Yang et al., 2005; Zheng et al., 2011). We screened the coding and noncoding exons and 5′-upstream regulatory region of MYH14, as well as the CEACAM16 gene, in family Z002. However, no causal mutations were detected. Haplotype transmission analysis of intragenic SNPs has refined the critical interval to 2.5 Mb and excluded the two known DFNA4 genes as candidates for family Z002, implying the existence of a new deafness gene at the DFNA4 locus.
According to these results, we deduced that the causal gene in family Z002 is proximal to MYH14. This refined candidate region contains more than 100 functional genes. We have screened several genes as candidates according to their functions or specific expression in the inner ear of mice within and even around the refined region. However, no pathologic mutations were detected, with the exception of reported SNPs (Table S3), in these genes, suggesting that they are not involved in DFNA4 deafness. In addition, there are six genes (RPL13A, RPS11, HRC, NUP62, FTL, and IL4I1) within the interval included in the expression database for the human cochlea (http://www.brighamandwomens.org/bwh_hearing/default.aspx) that are thought to be good candidates. SLC17A7, which encodes the vesicular glutamate transporter VGLUT1, is a paralog for SLC17A8, which encodes the vesicular glutamate transporter-3 (VGLUT3) and is responsible for human deafness DFNA25 (Ruel et al., 2008). SLC17A7 is specifically expressed in the neuron-rich regions of the brain and mediates the uptake of glutamate into synaptic vesicles at presynaptic nerve terminals of excitatory neural cells. Thus, it may be involved in glutamate metabolism in the inner ear (Schenck et al., 2009). Another possibility is GRIN2D, which belongs to the ionotropic glutamate receptor, N-methyl-D-aspartate (NMDA) receptor-channel gene family. Studies have shown that the glutamate receptor plays an essential role in high-frequency hearing and ionic homeostasis in the basal cochlea, and glutamate receptor antagonists can have a protective effect on the inner ear (Gao et al., 2007). Screening of these genes is currently being performed. With the development of next generation sequencing technology in recent years (Zhang et al., 2011), target next-generation sequencing of the critical region would be an appropriate choice for us to identify the causal gene of family Z002.
In conclusion, our results provide further clues to the identification of the new DFNA4 gene. Because of the diversity of functional genes and the proteins encoded that are involved in the hearing system, identification of this novel DFNA4 gene may provide new insight into the molecular mechanism of hearing loss.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients and their families for their cooperation during this work. This work was supported by the grants from the National Natural Science Foundation of China, key project (No. 30830104) and major project (No. 81120108009).
Footnotes
SUPPLEMENTARY DATA
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.jgg. 2012.11.002.
REFERENCES
- Alford RL. Nonsyndromic hereditary hearing loss. Adv. Otorhinolaryngol. 2011;70:37–42. doi: 10.1159/000322867. [DOI] [PubMed] [Google Scholar]
- Bayazit YA, Yilmaz M. An overview of hereditary hearing loss. ORL. J. Otorhinolaryngol. Relat. Spec. 2006;68:57–63. doi: 10.1159/000091090. [DOI] [PubMed] [Google Scholar]
- Bearer EL, Chen AF, Chen AH, Li Z, Mark HF, Smith RJ, Jackson CL. 2E4/Kaptin (KPTN) – a candidate gene for the hearing loss locus, DFNA4. Ann. Hum. Genet. 2000;64:189–196. doi: 10.1046/j.1469-1809.2000.6430189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AH, Ni L, Fukushima K, Marietta J, O’Neill M, Coucke P, Willems P, Smith RJ. Linkage of a gene for dominant non-syndromic deafness to chromosome 19. Hum. Mol. Genet. 1995;4:1073–1076. doi: 10.1093/hmg/4.6.1073. [DOI] [PubMed] [Google Scholar]
- Coucke PJ, Van-Hauwe P, Kelley PM, Kunst H, Schatteman I, Van- Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJ, Cremers CW, Vande- Heyning P, Willems PJ, Smith SD, Van-Camp G. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Mol. Genet. 1999;8:1321–1328. doi: 10.1093/hmg/8.7.1321. [DOI] [PubMed] [Google Scholar]
- Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di-Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, Pusch CM, Nürnberg P, Melchionda S, Zelante L, Ballana E, Estivill X, Van- Camp G, Gasparini P, Savoia A. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4) . Am. J. Hum. Genet. 2004;74:770–776. doi: 10.1086/383285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Maison SF, Wu X, Hirose K, Jones SM, Bayazitov I, Tian Y, Mittleman G, Matthews DB, Zakharenko SS, Liberman MC, Zuo J. Orphan glutamate receptor δ1 subunit required for high-frequency hearing. Mol. Cell. Biol. 2007;27:4500–4512. doi: 10.1128/MCB.02051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JA, Lalwani AK. Further evidence for a third deafness gene within the DFNA2 locus. Am. J. Med. Genet. 2002;108:304–309. [PubMed] [Google Scholar]
- Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause non-syndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 1999;23:16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- Lenz DR, Avraham KB. Hereditary hearing loss: from human mutation to mechanism. Hear. Res. 2011;281:3–10. doi: 10.1016/j.heares.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Mhatre AN, Li J, Chen AF, Yost CS, Smith RJ, Kindler CH, Lalwani AK. Genomic structure, cochlear expression, and mutation screening of KCNK6, a candidate gene for DFNA4. J. Neurosci. Res. 2004;75:25–31. doi: 10.1002/jnr.10839. [DOI] [PubMed] [Google Scholar]
- Mirghomizadeh F, Bardtke B, Devoto M, Pfister M, Oeken J, König E, Vitale E, Riccio A, De-Rienzo A, Zenner HP, Blin N. Second family with hearing impairment linked to 19q13 and refined DFNA4 localisation. Eur. J. Hum. Genet. 2002;10:95–99. doi: 10.1038/sj.ejhg.5200769. [DOI] [PubMed] [Google Scholar]
- Pusch CM, Meyer B, Kupka S, Smith RJ, Lalwani AK, Zenner HP, Blin N, Nürnberg P, Pfister M. Refinement of the DFNA4 locus to a 144 Mb region in 19q13.33. J. Mol. Med. 2004;82:398–402. doi: 10.1007/s00109-004-0538-z. [DOI] [PubMed] [Google Scholar]
- Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van-Rybroek JM, Rebillard G, Lenoir M, Eybalin M, Delprat B, Sivakumaran TA, Giros B, El-Mestikawy S, Moser T, Smith RJ, Lesperance MM, Puel JL. Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am. J. Hum. Genet. 2008;83:278–292. doi: 10.1016/j.ajhg.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck S, Wojcik SM, Brose N, Takamori S. A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat. Neurosci. 2009;12:156–162. doi: 10.1038/nn.2248. [DOI] [PubMed] [Google Scholar]
- Van-Hauwe P, Coucke PJ, Declau F, Kunst H, Ensink RJ, Marres HA, Cremers CW, Djelantik B, Smith SD, Kelley P, Van-de-Heyning PH, Van-Camp G. Deafness linked to DFNA2: one locus but how many genes? Nat. Genet. 1999;21:263. doi: 10.1038/6778. [DOI] [PubMed] [Google Scholar]
- Wang Q, Lu C, Li N, Rao S, Shi Y, Han D, Li X, Cao J, Yu L, Li Q, Guan M, Yang W, Shen Y. Y-linked inheritance of non-syndromic hearing impairment in a large Chinese family. J. Med. Genet. 2004;41:e80. doi: 10.1136/jmg.2003.012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein β-3 associated with autosomal dominant hearing impairment. Nat. Genet. 1998;20:370–373. doi: 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- Yan XK, Zhang TY, Wang ZM, Jiang Y, Chen Y, Wang HY, Ma D, Wang L, Li HW. A novel mutation in the MITF may be digenic with GJB2 mutations in a large Chinese family of Waardenburg syndrome type II. J. Genet. Genomics. 2011;38:585–591. doi: 10.1016/j.jgg.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Yang T, Pfister M, Blin N, Zenner HP, Pusch CM, Smith RJ. Genetic heterogeneity of deafness phenotypes linked to DFNA4. Am. J. Med. Genet. 2005;139:9–12. doi: 10.1002/ajmg.a.30989. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chiodini R, Badr A, Zhang GF. The impact of next-generation sequencing on genomics. J. Genet. Genomics. 2011;38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Miller KK, Yang T, Hildebrand MS, Shearer AE, DeLuca AP, Scheetz TE, Drummond J, Scherer SE, Legan PK, Goodyear RJ, Richardson GP, Cheatham MA, Smith RJ, Dallos P. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with α-tectorin and is mutated in autosomal dominant hearing loss (DFNA4) Proc. Natl. Acad. Sci. USA. 2011;108:4218–4223. doi: 10.1073/pnas.1005842108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.