

Abstract
The revival of interest in cancer cell metabolism in recent years has prompted the need for quantitative analytical platforms for studying metabolites from in vivo sources. We implemented a quantitative polar metabolomics profiling platform using selected reaction monitoring with a 5500 QTRAP hybrid triple quadrupole mass spectrometer that covers all major metabolic pathways. The platform uses hydrophilic interaction liquid chromatography with positive/negative ion switching to analyze 258 metabolites (289 Q1/Q3 transitions) from a single 15-min liquid chromatography–mass spectrometry acquisition with a 3-ms dwell time and a 1.55-s duty cycle time. Previous platforms use more than one experiment to profile this number of metabolites from different ionization modes. The platform is compatible with polar metabolites from any biological source, including fresh tissues, cancer cells, bodily fluids and formalin-fixed paraffin-embedded tumor tissue. Relative quantification can be achieved without using internal standards, and integrated peak areas based on total ion current can be used for statistical analyses and pathway analyses across biological sample conditions. The procedure takes ~12 h from metabolite extraction to peak integration for a data set containing 15 total samples (~6 h for a single sample).
INTRODUCTION
This protocol addresses the need to quantitatively profile metabolites extracted from cell lines, bodily fluids, tumors and formalin-fixed tissue1. Renewed interest in cancer cell metabolism over the last few years has prompted requests for technology capable of profiling both intra- and extracellular metabolites from cultured cells and in vivo sources in order to assess altered central metabolic networks that contribute to cell proliferation, growth and survival2,3. These analyses are useful for determining which metabolic pathways are affected by, for example, small molecules or drugs, genetic alterations, or environmental stimuli. Metabolomics analyses are routinely performed using several different platforms, including nuclear magnetic resonance, gas chromatography–mass spectrometry and liquid chromatography–mass spectrometry (LC-MS), each with its advantages and disadvantages4-6.
This protocol uses a hybrid dual quadrupole linear ion trap mass spectrometer for steady-state profiling of endogenous polar metabolites from organic (methanol or acetonitrile) extractions from biological samples. It requires no sample manipulation other than metabolite extraction. The targeted metabolites cover all major metabolic pathways, including glycolysis, the tricarboxylic acid cycle, the pentose-phosphate pathway, and metabolism of amino acids, nucleotides and so on. Although other mass spectrometers can be used, triple quadrupole mass spectrometers are excellent for quantification because they isolate and target biomolecules of interest, exclude signal from the background matrix, and are sensitive, fast-scanning and reproducible with high dynamic range. For that reason, triple quadrupoles are routinely used for developing mass spectrometry–based quantitative assays for metabolites and peptides via multiple reaction monitoring (MRM) and/or selected reaction monitoring (SRM)7-9. Recent developments in mass spectrometry instrumentation such as fast–scanning, high-resolution orbitrap mass spectrometers and hybrid quadrupole–time of flight mass spectrometers have made it possible to profile metabolites in MS mode with identification in MS/MS mode with high resolution10-12.
We use relative quantification across biological conditions wherein one or more of the profiled biological samples serve as a reference control. Relative quantification relies on a reproducible platform and it is recommended that samples be run in triplicate so that statistics such as t tests can be applied for quality control and three samples per biological condition are required for some informatics software packages such as MetaboAnalyst (http://www.metaboanalyst.ca, free online software)13. Biological triplicates rather than technical triplicates are suggested, as the platform routinely shows R2 values ≥0.97 for replicate analyses. If one chooses to use a simple ratio or fold change analysis for quantifying between samples, it is best to first normalize all samples, including replicates and/or triplicates, according to the median peak area in each sample; this should be followed by taking an average of the triplicates per biological sample. One can then calculate fold changes between biological sample conditions. Alternatively, we recommend that MetaboAnalyst be used for statistically analyzing the data as well as for creating heat maps and clustergrams, and for performing principal component analysis (PCA) and pathway analysis13,14. Absolute quantification (not covered here) can be achieved using a concentration curve for each metabolite of interest or by spiking the samples using an internal standard15.
The important features of our platform are that (i) this is a single normal-phase hydrophilic interaction liquid chromatographic (HILIC) run, (ii) the MS acquisition time per sample is short (~15 min) and (iii) more than 250 compounds are targeted without chromatographically scheduled SRMs.
For HILIC-based chromatography, we use amide-capped ethylene-bridged hybrid (BEH) particles (XBridge; http://www.waters.com/webassets/cms/library/docs/720003232en.pdf) and amino-capped silica particles (Luna; http://www.phenomenex.com/Products/HPLCDetail/Luna/NH2?returnURL=/Products/Search/HPLC) Other modes of chromatography such as reversed-phase (C18) can be used effectively, but HILIC chromatography at high pH (~9.0) captures the highest number of metabolites from a single analysis, as demonstrated in several of our publications relating to cancer metabolism16-23.
Mass spectrometry with positive/negative polarity switching allows for the acquisition of Q1/Q3 MRM transition mass spectra in both ionization modes from a single LC-MS/MS analysis24-26. The 5500 QTRAP hybrid dual quadrupole linear ion trap mass spectrometer has the ability to switch polarities in <50-ms, allowing one to use positive/negative switching without substantial delay time in the duty cycle.
Similar platforms have been implemented27,28, whereas previous SRM-based metabolomics platforms typically use multiple LC-MS/MS runs and chromatography platforms per sample. We also show the highest number of polar metabolites from a single targeted LC-MS/MS experiment to date. It is important to note that the current number of targeted compounds (258) can be increased without adding substantial cycle time, because as low as a 2-ms dwell time can be used on the 5500 QTRAP without marked losses in sensitivity. Although this protocol focuses on formalin-fixed paraffin-embedded (FFPE) tissue, fresh and/or frozen bodily tissue, bodily fluids and cultured cells, the platform could be used to analyze polar metabolites from any biological source regardless of organism or tissue. However, we recommend that more than two to three times the amount (weight) of recommended tissue be used when analyzing adipose tissue because of the very high lipid content. Also, we have not worked out metabolite extraction methods from bone or related skeletal material.
MATERIALS
REAGENTS
LC/MS-grade water (Fisher Scientific, cat. no. MWX00011)
LC/MS-grade acetonitrile (Fisher Scientific, cat. no. AC61514-0025)
HPLC-grade methanol (Fisher Scientific, cat. no. AC61009-0040)
Ammonium hydroxide (25% (wt/vol) solution, puriss grade; Fisher Scientific, cat. no. 1336-21-6)
Ammonium acetate crystals (puriss grade; Fisher Scientific, 09688)
EQUIPMENT
Amide XBridge HPLC column (3.5 μm; 4.6 mm inner diameter (i.d.) × 100 mm length; Waters, cat. no. 186004868)
Luna NH2 HPLC column (5.0 μm; 4.6 mm i.d. × 50 mm length; Phenomenex, cat. no. 00B-4378-E0) ▲ CRITICAL The Waters Amide Xbridge columns are robust, reproducible and have a long life for metabolomics analyses (≥1,000 analyses with high reproducibility). Alternatively, the Phenomenex Luna NH2 columns can be used as they produce slightly higher metabolite peak separation than the amide column in our hands; however; the NH2 columns have a considerably shorter life at pH = 9.0 with typically approximately 500–700 highly reproducible analyses.
Security Guard universal HPLC guard cartridge (Phenomenex, cat. no. AJ0-4301)
5500 QTRAP hybrid dual quadrupole ion trap mass spectrometer (AB/SCIEX) capable of polarity switching times ≤50 ms at 3- to 4-ms dwell time or other triple quadrupole mass spectrometer with similar specifications (http://www.absciex.com/products/mass-spectrometers/qtrap-systems/ab-sciex-qtrap-5500-lcmsms-system)
Prominence HPLC (Shimadzu) or generic HPLC instrument capable of high run-to-run reproducibility and capable of flow rates in the several hundred microliters per minute range (up to ~1 ml min−1). The HPLC must have a chilled (4–8 °C) autosampler attached to the unit capable of holding all of the metabolite samples in your sample set (approximately 24–96). Note that equipment from many HPLC vendors can be used for this purpose
SpeedVac concentrator or lyophilizer (for drying metabolite supernatants to a pellet; Thermo Electron or other vendor)
Polypropylene microcentrifuge tubes (1.5 ml; Fisher Scientific, cat. no. 05-408–129)
Polypropylene microcentrifuge tubes (2.0 ml; Fisher Scientific, cat. no. 02-682–558)
Polypropylene autosampler vials with caps (12 × 32 mm2; National Scientific, cat. no. C4000-87)
Small pestle or tissue grinder for use in 1.5–2.0 ml microcentrifuge tubes
MATLAB R2010a (MathWorks)
Cell scraper
REAGENT SETUP
Tissue
For soft bodily tissue samples including tumor tissue, start with approximately 10–15 mg of solid tissue or the equivalent of at least two million cells, place in a 1.5-ml microcentrifuge tube and snap-freeze the tissue sample in liquid nitrogen (− 196 °C).
Fluid
For bodily fluid samples, start with 150–250 μl of serum, plasma, urine or cerebrospinal fluid (CSF) in a 1.5-ml microcentrifuge tube.
FFPE
For formalin-fixed tissue samples, start with approximately 30–40 μm FFPE slice for extraction (discard the top slice) from the tissue block. Alternatively, approximately 1–2 mm2 cores can be extracted from the block in lieu of slices after discarding a top slice layer to reduce contamination.
Cells
For cultured cells, use the equivalent of > 2–3 million cells, or one 10-cm2 plate at ~80% confluence.
Methanol (80% (vol/vol))
To prepare the 80% (vol/vol) methanol solution for metabolite extraction, add 40 ml of HPLC-grade methanol to a 50-ml polypropylene conical tube, followed by 10 ml of LC/MS-grade water solution; cap and gently shake to mix. Store in − 80 °C freezer for 4–6 h for further use. Note that methanol can be replaced or combined with acetonitrile to an 80% (vol/vol) total organic concentration for metabolite extractions, although methanol is preferred for intracellular metabolites and most applications29,30.
HPLC buffer A (pH = 9.0: 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate)
To prepare HPLC A buffer, add 1.54 g of ammonium acetate to a 1-liter HPLC bottle and add 950 μl of LC/MS grade water and gently stir by rotating the bottle. Next, add 2.8 ml of 25% (wt/vol) ammonium hydroxide solution followed by 50 ml of HPLC grade acetonitrile. Cap the bottle and mix by gently shaking. Check the pH to assure it is at 9.0. Buffer A can be stored for up to 2 weeks at room temperature (~20–25 °C).
HPLC B buffer B (100% acetonitrile)
To prepare HPLC B buffer, add 1 liter of LC/MS grade acetonitrile to a 1-liter HPLC bottle, and then cap the bottle. Buffer B (acetonitrile) can be stored for up to 6 months at room temperature.
EQUIPMENT SETUP
Mass spectrometer
Use a triple quadrupole mass spectrometer capable of a positive/negative polarity switching time of 50 ms or below and an approximately 3- to 4-ms dwell time, such as the 5500 QTRAP or other triple quadrupole style mass spectrometer with similar capabilities. This allows for up to ~350 unscheduled SRM scans resulting in a sufficient number of scans (approximately 10–12) per metabolite peak in < 2.0 s using a standard HPLC. Chromatographic scheduling of SRM is recommended if duty cycle times are close to ~2.0 s, because an insufficient number of data points will be acquired per metabolite.
HPLC
For the procedure described, a HILIC HPLC column is needed (see EQUIPMENT). Ultra-high pressure HPLC (UHPLC) is not suitable for this application, as the unscheduled SRM mode described here benefits from peak widths of at least 8–10 s at full width at half height (FWHH). UHPLC would limit the number of scans per metabolite Q1/Q3 transition.
Software
Software for SRM Q1/Q3 peak integration is needed, such as Multi-Quant 2.0 (http://www.absciex.com/products/software/multiquant-software). In addition, you will need software for computational biology calculations and statistics, such as free online software from http://www.metaboanalyst.ca/MetaboAnalyst/ that includes normalization, clustering tools, heat maps and pathway analysis.
Additional software for performing PCA clustering can be used, such as Marker-View 1.1 (http://www.absciex.com/products/software/markerview-software) in addition to customized tools for creating heat maps, such as Clustergram in MAT-LAB R2010a; R, a freeware biostatistical suite (http://www.r-project.org/); and pathway analysis from KEGG IDs in TICL (http://mips.helmholtz-muenchen.de/proj/cmp/), which is a freeware online tool.
PROCEDURE
Sample preparation: polar metabolite extraction
-
1
Perform extraction from bodily tissues, bodily fluids or FFPE tissues by following the steps in options A, B or C, respectively. This protocol is also compatible with metabolite extractions from cultured cells. An example procedure for metabolite extraction from adherent cells is shown in option D. If you are using suspension cells, skip Step 1D(ii–vi), transfer the medium with cells into a 15-ml conical tube, spin at 750g for 5 min at room temperature, remove the medium, add 4 ml of 80% (vol/vol) methanol (− 80 °C), vortex gently to stir cells, incubate for 20 min at −80 °C and proceed with 1D(vii).
-
Extraction from bodily tissues (tumors/liver/brain/fat)
Add 500 μl of 80% (vol/vol) HPLC-grade methanol (cooled to − 80 °C) to fresh or frozen tissue piece(s) in a 1.5-ml or 2.0-ml microcentrifuge tube.
Smash/grind for 1–2 min with small pestle/tissue grinder on dry ice in the tube, vortex for 1 min at 4–8 °C and incubate for 4 h at − 80 °C.
Centrifuge at 14,000g (or the highest speed) for 10 min using a refrigerated centrifuge (4–8 °C).
Transfer the supernatant to a new 1.5-ml microcentrifuge tube; store at − 80°C until Step 1A (viii).
Add 400 μl of 80% (vol/vol) methanol (cooled to − 80 °C) to the precipitate.
Vortex for 1 min at 4–8 °C, and then incubate for 30 min at − 80 °C.
Centrifuge at 14,000g for 10 min (4–8 °C).
Transfer and combine the supernatant from both extractions.
Centrifuge again at 14,000g for 10 min (4–8 °C).
Transfer the supernatant to a new 1.5-ml microcentrifuge tube.
-
SpeedVac/lyophilize to a pellet using no heat.
■ PAUSE POINT Dried metabolite samples can be stored at − 80 °C for several weeks.
-
Extraction from bodily fluids (serum/urine/CSF)
Centrifuge at 14,000g for 10 min in a cold room (4–8 °C).
Transfer the supernatant to a new 1.5-ml microcentrifuge tube.
Add enough methanol (cooled to − 80 °C) to the supernatant to make a final 80% (vol/vol) methanol solution.
Gently shake to mix and incubate for 6–8 h at − 80 °C.
Centrifuge at 14,000g for 10 min (4–8 °C).
Transfer the supernatant to a new 1.5-ml microcentrifuge tube.
-
SpeedVac/lyophilize to a pellet using no heat.
■ PAUSE POINT Dried metabolite samples can be stored at − 80 °C for several weeks.
-
Extraction from FFPE tissue
Place curled FFPE slices or cores in a 1.5-ml microcentrifuge tube.
Prepare a water bath or heat block to 70 °C.
Add approximately 1–1.5 ml of 80% (vol/vol) methanol to the tube (enough volume to cover the slices completely) and place the tube at 70 °C for 30–45 min. Note: less volume (approximately 0.5–1.0 ml) can be used for FFPE cores because of their smaller size.
Remove from heat and put tube in ice to congeal the wax for 15 min.
Centrifuge at 14,000g for 10 min (4–8 °C).
Transfer the supernatant to a new 1.5-ml microcentrifuge tube; chill on ice for 10 min.
Centrifuge again, cold (4–8 °C), at 14,000g for 5 min.
-
Transfer to a new tube and SpeedVac/lyophilize the sample to dryness.
■ PAUSE POINT Dried metabolite samples can be stored at − 80 °C for several weeks.
-
Extraction from adherent cell lines
Change the medium of the cell plate(s) 2 h before metabolite extraction.
Aspirate the medium completely.
Put the plates on dry ice and add 4 ml of 80% (vol/vol) methanol (cooled to − 80 °C).
Incubate the plates at − 80 °C for 20 min.
Scrape the plates on dry ice with cell scraper.
Transfer the cell lysate/methanol mixture to a 15-ml conical tube on dry ice.
Centrifuge the tube at 14,000g for 5 min at 4–8 °C to pellet the cell debris.
Transfer the metabolite-containing supernatant to a new 15-ml conical tube on dry ice.
Add 500 μl 80% (vol/vol) methanol (− 80 °C) to the pellet in a 15-ml tube and vortex for 1 min at 4–8 °C.
Spin the tubes at 14,000g for 5 min at 4–8 °C.
Transfer the supernatant to a 50-ml conical tube on dry ice (from Step 1D(viii)).
Divide and transfer 4.5 ml of total extraction buffer into three 1.5-ml microcentrifuge tubes (1.5 ml in each tube).
-
SpeedVac/lyophilize to a pellet using no heat.
■ PAUSE POINT Dried metabolite samples can be stored at − 80 °C for several weeks.
-
Preparing the instrument for acquisition
-
2
Create an instrument method in SRM mode (also called MRM when more fragments are incorporated) according to the manufacturer’s acquisition software. Enter the Q1 (precursor ion) and Q3 (fragment ion) transitions, the metabolite name, the dwell time and the appropriate collision energies (CEs) for both positive and negative ion modes. It is not necessary to use scheduled SRM mode for this method, as the instruments cited are capable of scanning the 289 SRM transition list in 1.55 s using a 3-ms dwell time, producing approximately 10–14 scans per metabolite peak.
-
3
Always select Q1 and Q3 transitions to UNIT resolution for optimal metabolite ion isolation and selectivity. In addition, ensure that the polarity switching (settling) time is set to the lowest setting possible (50 ms for the 5500 QTRAP).
-
4Check that the source conditions are suited to high flow rates from the HPLC. For the 5500 QTRAP only, the source and compound settings should be as follows for both positive and negative ion modes:
Temperature 475 °C Curtain gas 20–25 (nitrogen) Collision gas High (nitrogen) Ion source gas 1 33 Ion source gas 2 33 Declustering potential + 93 in positive ion mode/ − 93 in negative ion mode Entrance potential + 10 in positive ion mode/ − 10 in negative ion mode Collision cell exit potential + 10 in positive ion mode/ − 10 in negative ion mode -
5Set up the generic HPLC gradient as follows:
Flow rate Approximately 350–400 μl min−1 (back pressure should not exceed ~3,000 p.s.i. at 2% (vol/vol) B) 85% (vol/vol) B 0.0 min 85% B to 30% B 3.0 min 30% B to 2% B 12.0 min 2% B 15.0 min 2% B to 85% B 16.0 min 85% 23.0 min
Metabolite sample preparation
-
6
For each metabolite of interest, prepare and run a standard compound to ensure the proper chromatographic elution time on your particular system. It should be prepared in LC/MS-grade water at a concentration of ~1 μM. Before running a real biological sample, one should also prepare a ‘standard’ sample to be run as the first sample preceding a biological sample set to ensure that the LC-MS system is operating efficiently (sufficient metabolite separation on the HPLC column, well-calibrated MS with proper sensitivity). The sample could be a well-characterized metabolite extract from a cell line or tissue source. One should achieve at least 180–200 unique quantifiable metabolites from this test sample. As the -coA derivatives tend to be some of the weakest metabolite responses by SRM in our platform, one should be able to quantify acetyl-coA in positive ion mode (peak area in the 104 range or greater) if samples were prepared properly and sufficient metabolite material is available. In addition, hexose phosphate, a metabolite with a good response in our platform, should be present with peak areas in the lower 106 range or greater.
-
7
Add ~20 μl of LC/MS grade water to resuspend each standard sample and each biological sample just before LC-MS/MS analysis.
-
8
Inject approximately 5–10 μl of sample onto the LC-MS/MS system using an autosampler (do not inject more than half of the sample in case of a system failure, and inject no more than ~5 μl if technical triplicate runs are desired).
Peak integration
-
9
Once the SRM data are acquired, peaks must be integrated in order to generate chromatographic peak areas used for quantification across the sample set. Use a suitable software platform for peak area integration, such as MultiQuant 2.0 from AB/SCIEX (http://www.absciex.com/products/software/multiquant-software). Ideally, standard compounds from the metabolites of interest should be tested on the platform to assure proper chromatographic elution times; however, the largest SRM peak in the chromatogram is typically correct for approximately 80–90% of the targeted metabolites for carefully selected Q1/Q3 transitions, typically between ~4 and 11 min (the majority of peaks appear between 6.0 and 8.0 min) in elution time.
-
10
Use the MultiQuant v2.0 software to integrate the peak areas from the Q3 TIC values across the chromatographic elution. If sufficient sample is present, a single dominant peak will typically be present for most detectable compounds.
For MultiQuant peak integration settings, use the following:Algorithm MQ4 Smoothing points 2.0 Retention time (RT) half window 15–20 s (varies depending on chromatographic reproducibility) Update expected RT No Do not check ‘report largest peak’ box Min peak width 8 points Min peak height 750 (varies depending on sensitivity) Noise (%) 40 Baseline subwindow 1.7 min Peak splitting 3 points -
11
Select a representative control sample and manually scroll through each metabolite peak to ensure that the proper peaks are selected for integration over the entire data set. If there is an error with peak selection, one can manually select the peak of interest or change the integration parameters for a particular Q1/Q3 transition.
▲ CRITICAL STEP It is important to run a standard compound to ensure the proper chromatographic elution time on your particular system, especially if multiple peaks are present per Q1/Q3 transition. This is essential when incorporating new compounds into your transition list. Previous knowledge of the compound of interest and the chromatography can also be used to identify the correct peak for integration. SRM transitions can be obtained from public mass spectral databases such as MassBank (http://www.massbank.jp/) or the Human Metabolome Database (HMDB; http://www.hmdb.ca/).
? TROUBLESHOOTING
? TROUBLESHOOTING
The dried metabolite pellet appears to be very large and fluffy
This may indicate contamination with large molecules (protein, DNA, lipids and so on). Ensure that the appropriate amount of methanol and/or acetonitrile was used in extraction (at least 80% (vol/vol)), incubation time was sufficient at − 80 °C, and that the sample was spun at the highest speed before removing the supernatant (polar metabolites). The pellet should be less than 15 μl liquid equivalent in volume in a 1.5-ml microcentrifuge tube.
The mass spectrometer is not showing any Q1/Q3 transition peaks above the baseline
This would indicate that not enough metabolite content is on the HPLC column during the acquisition. Ensure that HPLC buffers and collision and source gases are sufficient, and that the column or tubing is not leaking. Is the instrument well calibrated in both positive and negative ion mode? How many cells or how much tissue was used? It is possible that more sample quantity is needed (equivalent of > 2 million cells). In addition, the ion source area has a tendency to get very dirty using the conditions described in this protocol, so routine source cleaning is necessary.
The metabolite peaks are either not being retained or eluting very late on the HPLC column
Are your HPLC buffer concentrations and pH correct? Are the HPLC A and B pumps properly purged to remove air in pumps? If so, this may indicate that either your pre-column filter is clogged or that you need to change or clean your HPLC column due to passivation or contamination. First, try changing the pre-column filter followed by an analytical column change.
Some metabolites are never detected no matter what I change or what concentrations I inject
It is possible that the metabolite Q1/Q3 values are not optimal or incorrect or that the CE selected is not ideal for the selected fragmentation event. Also, the particular metabolite of interest may exist in very low concentrations in your biological matrix or it may degrade quickly. For example, the -coA derivatives degrade quickly and are more difficult to detect than other metabolites. Other possibilities are that methanol or acetonitrile are not ideal for the metabolite extraction or that the chromatography used is not suitable for the compound of interest.
● TIMING
From tissue/cells/fluid/FFPE collection to sample injection of metabolite extracts: ~4–6 h, mainly depending upon the drying speed of the SpeedVac/lyophilizer
Total LC-MS/MS run time: 23 min (15 min MS acquisition) from injection to injection
Peak integration and verification of raw data: ~1 h per data set
Data analysis using various bioinformatics software tools: variable, depending on the degree of interpretation
ANTICIPATED RESULTS
Figure 1 shows an overview of the metabolomics platform described in this protocol. Extracted metabolites are run using a 5500 QTRAP LC-MS/MS system via SRM followed by Q3 peak area integration using MultiQuant 2.0 software. This peak area output is considered to be the raw data from the metabolomics profiling experiment for further informatics processing. Be aware that some metabolites from some biological conditions will show no data (‘N/A’ in the table). This is due to either low metabolite concentration below the instrument threshold for detection, metabolite degradation, poor fragmentation or poor ionization efficiency for a specific metabolite. From 258 unique metabolites (289 transitions), we typically acquire robust data for ~200 unique metabolites when sufficient sample quantity is present. It is important to note that the data do not represent absolute concentrations of metabolites so peak area values can only be used relative to a control (reference) sample. However, one can perform a standard curve analysis using known concentrations of a standard compound for absolute quantification if needed, or a stable isotope-labeled metabolite can be spiked into a sample for quantitative reference31,32. Supplementary Table 1 contains 289 Q1/Q3 transition lists representing 258 unique polar metabolites using both negative and positive ion modes along with CEs, chemical formulas, dwell times and database identifiers (KEGG, HMDB or PubChem) for the targeted metabolites. Some metabolites are targeted in both positive and negative ion modes as they show response in both ionization modes; however, the chromatography mode used and the pH of HPLC buffers can influence the response in a particular ionization mode.
Figure 1.
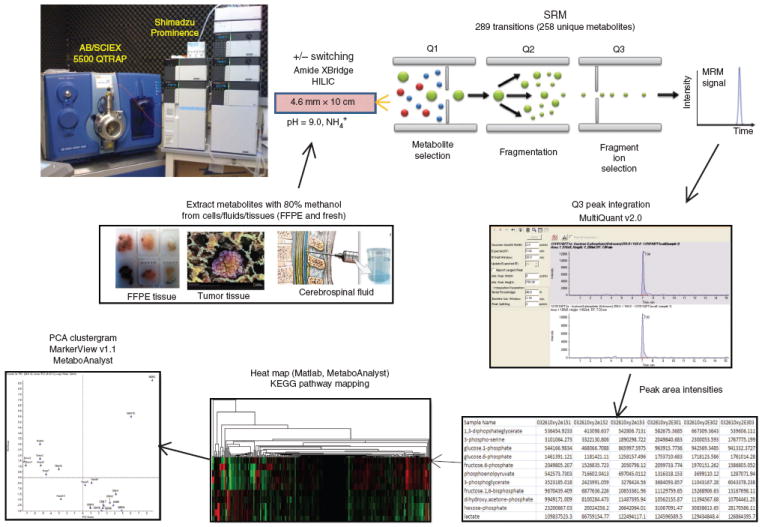
Metabolomics platform schematic. Overview of the targeted LC-MS/MS experiment for polar metabolite profiling via SRM, using positive/negative switching from a single 15-min HILIC column run while targeting more than 250 compounds.
Figure 2a shows a typical example of the reproducibility of two biological replicate analyses by Pearson correlation with ~4 orders of magnitude of SRM dynamic range. Notice the highly reproducible R2 value for both 293T and H929 cell extracts. Average replicate reproducibility is ~0.986 (R2) for most data sets and primarily represents biological sample variability, and, to a lesser extent, technical variability. Figure 2b shows an unsupervised hierarchical heat map of more than 200 unique metabolites from 293T embryonic kidney–derived cells and H929 multiple myeloma plasma cells derived from bone marrow that were treated with epidermal growth factor (EGF) and insulin at 15 min and 30 min of stimulation after overnight serum starvation. This heat map was created using MetaboAnalyst software (distance was calculated using Pearson and clustering using Ward). The data show that the metabolites mostly cluster according to cell type rather than growth factor stimulation. 293T cells were more responsive to different growth factor stimulation times. Figure 2c shows the PCA (PC1 versus PC2) clustergram created using MarkerView software from the same 293T and H929 cells treated with EGF or insulin for 15 min and 30 min as in Figure 2a. The data show three different clusters on the basis of the polar metabolic profile. Although H929 cells are less responsive to growth factor stimulation, 293T cells are responsive and form two distinct clusters, separated mainly by the time of stimulation rather than the specific growth factor used. This may be explained by noting that 293T cells have very high activation levels of both AKT and extracellular signal-regulated kinase (ERK) compared with H929 cells. Figure 2d shows the fold change of glycolytic intermediates for 20 min of acute serum stimulation with 10% (vol/vol) FBS versus cells that were serum starved overnight. The glycolytic intermediates from 293T cells are highly responsive to serum (glucose uptake), whereas RPMI-8226 and H929 multiple myeloma cell lines show lower glycolytic response. Compared with 293T cells, phosphoenolpyruvate does not increase with FBS stimulation with little to no AKT activation. Western blots are shown for pAKT (S473) and pERK1/2 with relevant controls for serum-fed conditions. The glycolytic responses directly correlate with pAKT expression levels, but not necessarily with pERK levels in the multiple myeloma cells tested. It has been established in the literature that glycolysis is closely related to AKT activation33-35. Although RPMI-8226 and H929 cells do not perform glycolysis to a marked extent upon acute serum stimulation, the cells still produce both pyruvate and lactate to maintain their growth in culture. In addition to cancer cells, we have developed methods for profiling tumor tissue, bodily fluids and FFPE tissue samples16,23. Figure 3a shows a PCA cluster plot from nearly 150 polar metabolites extracted from CSF from ten cancer patients and ten patients without cancer. Note that we can distinguish between different patient groups using metabolic signatures according to PCA, including advanced disease, early-stage disease, and two different groups of patients without cancer from whom CSF was extracted at two different locations. Figure 3b shows hierarchical clustering via the Ward method in MetaboAnalyst from FFPE-extracted polar metabolites from normal lung and kidney tissue as well as acute myeloid leukemia from kidney and lymphangioleiomyomatosis lung disease. By using ~135 robustly detected metabolites from 1-mm2 cores extracted from fixed tissue blocks, the clustergram shows that the metabolites mostly cluster according to disease state and tissue type. The data show that stored tissue samples can be used for patient classification and potentially for interrogating metabolic pathways.
Figure 2.
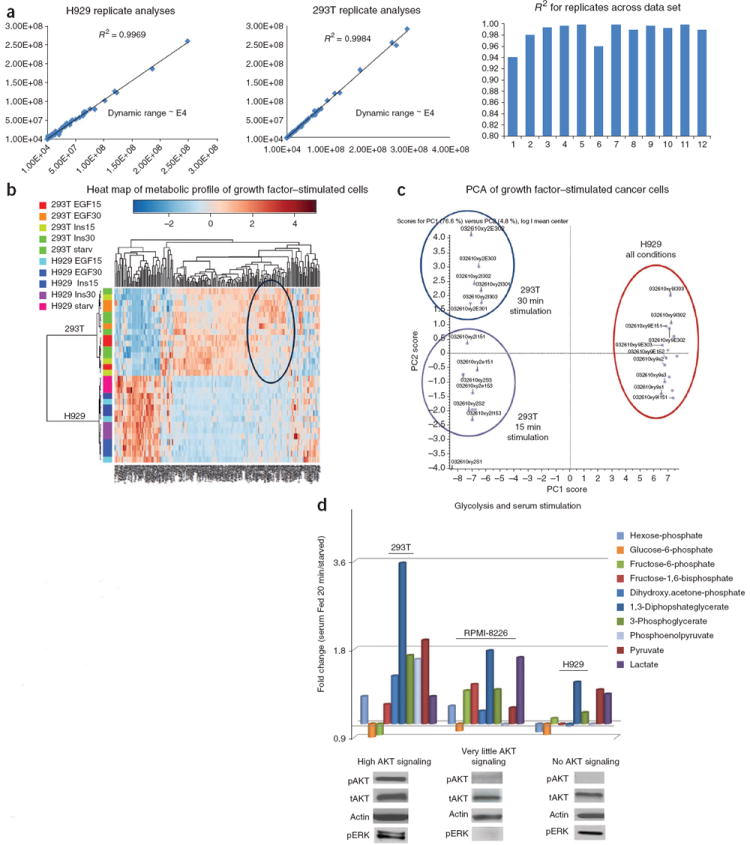
Reproducibility and heat maps from cancer cell metabolomics. (a) Reproducibility of cancer cell extracts by polar metabolomics profiling platform. Typical R2 values across biological replicates in a data set from 293T embryonic kidney and H929 multiple myeloma cells are shown. (b) Unsupervised hierarchical clustering heat map of metabolites from both H929 and 293T cancer cells stimulated with the growth factors EGF and insulin across different time points (0, 15, 30 min). (c) PCA clustering of polar metabolites from the same 293T and H929 human cancer cell extracts with EGF and insulin stimulation, showing different clustering groups according to incubation time of growth factor stimulation. (d) Fold changes of individual glycolytic intermediates upon acute serum stimulation in 293T and H929 cells. Western blots of key signaling nodes such as phosphorylated AKT (pAKT) correlate with glucose uptake in cancers.
Figure 3.
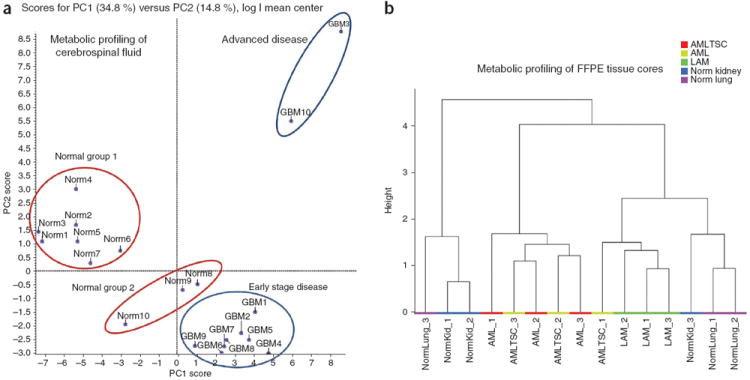
In vivo metabolomics profiling. (a) The PCA clustering from extracted polar metabolites from cerebrospinal fluid (CSF) from 20 patients, 10 of which present with gliomas. Different disease stages are highlighted. (b) Hierarchical clustergram from polar metabolites that were extracted from formalin-fixed paraffin-embedded (FFPE) tissue cores from normal (Norm) lung and kidney tissue and from acute myeloid leukemia (AML) from kidney and lymphangioleiomyomatosis (LAM) lung disease. The data show that distinct clusters can be obtained from fixed tissue samples.
Supplementary Material
Acknowledgments
We thank M. Vander Heiden, L. Cantley, J. Rabinowitz, J. Locasale, C. Lyssiotis, E. Driggers, L. Peshkin, R. Rahal and A. Sasaki for helpful and informative philosophical and technical discussions. We also thank C.-L. Wu and E. Wong for providing FFPE and CSF samples, respectively. This research was supported by grants from the US National Institutes of Health 5P01CA120964 and 5P30CA006516 (J.M.A.) and the Beth Israel Deaconess Medical Center Research Capital Fund for funding the mass spectrometry instrumentation (J.M.A.).
Footnotes
AUTHOR CONTRIBUTIONS
J.M.A. developed the platform and wrote the protocol. M.Y. and S.B.B. optimized and edited the protocol. X.Y., S.B.B. and M.Y. prepared biological samples for testing the protocol. J.M.A., M.Y. and S.B.B. performed data analysis.
Note: Supplementary information is available via the HTML version of this article.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Bayley JP, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev. 20:324–329. doi: 10.1016/j.gde.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romero-Garcia S, Lopez-Gonzalez JS, Baez-Viveros JL, Aguilar-Cazares D, Prado-Garcia H. Tumor cell metabolism: an integral view. Cancer Biol Ther. 2011;12:939–948. doi: 10.4161/cbt.12.11.18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wishart DS. Advances in metabolite identification. Bioanalysis. 2011;3:1769–1782. doi: 10.4155/bio.11.155. [DOI] [PubMed] [Google Scholar]
- 5.Mamas M, Dunn WB, Neyses L, Goodacre R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch Toxicol. 2011;85:5–17. doi: 10.1007/s00204-010-0609-6. [DOI] [PubMed] [Google Scholar]
- 6.Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev. 2011;40:387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- 7.Kitteringham NR, Jenkins RE, Lane CS, Elliott VL, Park BK. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1229–1239. doi: 10.1016/j.jchromb.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Meng Z, Veenstra TD. Targeted mass spectrometry approaches for protein biomarker verification. J Proteomics. 2011;74:2650–2659. doi: 10.1016/j.jprot.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Wei R. Metabolomics and its practical value in pharmaceutical industry. Curr Drug Metab. 2011;12:345–358. doi: 10.2174/138920011795202947. [DOI] [PubMed] [Google Scholar]
- 10.Lu W, et al. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem. 2011;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenling T, et al. The impact of delayed storage on the measured proteome and metabolome of human cerebrospinal fluid. Clin Chem. 2011;57:1703–1711. doi: 10.1373/clinchem.2011.167601. [DOI] [PubMed] [Google Scholar]
- 12.De Vos RC, et al. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat Protoc. 2007;2:778–791. doi: 10.1038/nprot.2007.95. [DOI] [PubMed] [Google Scholar]
- 13.Xia J, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc. 2011;6:743–760. doi: 10.1038/nprot.2011.319. [DOI] [PubMed] [Google Scholar]
- 14.Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37(Web Server issue):W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodamer M, Elsinghorst PW, Kinzig M, Gutschow M, Sorgel F. Development and validation of a liquid chromatography/tandem mass spectrometry procedure for the quantification of sunitinib (SU11248) and its active metabolite, N-desethyl sunitinib (SU12662), in human plasma: application to an explorative study. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:695–706. doi: 10.1016/j.jchromb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Kelly AD, et al. Metabolomic profiling from formalin-fixed, paraffin-embedded tumor tissue using targeted LC/MS/MS: application in sarcoma. PLoS ONE. 2011;6:e25357. doi: 10.1371/journal.pone.0025357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi CH, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–620. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locasale JW, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2011;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, et al. A mass spectrometry platform to quantitatively profile cancer cell metabolism from cells, tumors, and fixed tissue. Am Assoc Cancer Res; 102nd Annu Meeting Orlando; Florida, USA. 2011. [Google Scholar]
- 21.Anastasiou D, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao D, et al. mTOR drives its own activation via SCFβrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Locasale JW, et al. Metabolomics of human cerebrospinal fluid identifies signatures of malignant glioma. Mol Cell. 2012 doi: 10.1074/mcpM111.014688. Proteomics published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koyama J, et al. Simultaneous determination of histamine and prostaglandin D2 using an LC-ESI-MS/MS method with positive/negative ion-switching ionization modes: application to the study of anti-allergic flavonoids on the degranulation of KU812 cells. Anal Bioanal Chem. 2011;401:1385–1392. doi: 10.1007/s00216-011-5200-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Ren Y, Jiao J, Li D. Ultra high-performance liquid chromatography-tandem mass spectrometry for the simultaneous analysis of asparagine, sugars, and acrylamide in Maillard reactions. Anal Chem. 2011;83:3297–3304. doi: 10.1021/ac1029538. [DOI] [PubMed] [Google Scholar]
- 26.Hara H, et al. Simultaneous analytical method for the determination of TCH346 and its four metabolites in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:377–384. doi: 10.1002/rcm.1349. [DOI] [PubMed] [Google Scholar]
- 27.Bajad SU, et al. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 28.Lu W, Bennett BD, Rabinowitz JD. Analytical strategies for LC-MS-based targeted metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:236–242. doi: 10.1016/j.jchromb.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martineau E, Tea I, Loaec G, Giraudeau P, Akoka S. Strategy for choosing extraction procedures for NMR-based metabolomic analysis of mammalian cells. Anal Bioanal Chem. 2011;401:2133–2142. doi: 10.1007/s00216-011-5310-y. [DOI] [PubMed] [Google Scholar]
- 30.Wikoff WR, Pendyala G, Siuzdak G, Fox HS. Metabolomic analysis of the cerebrospinal fluid reveals changes in phospholipase expression in the CNS of SIV-infected macaques. J Clin Invest. 2008;118:2661–2669. doi: 10.1172/JCI34138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerszten RE, Carr SA, Sabatine M. Integration of proteomic-based tools for improved biomarkers of myocardial injury. Clin Chem. 2011;56:194–201. doi: 10.1373/clinchem.2009.127878. [DOI] [PubMed] [Google Scholar]
- 32.Guo T, Gu J, Soldin OP, Singh RJ, Soldin SJ. Rapid measurement of estrogens and their metabolites in human serum by liquid chromatography-tandem mass spectrometry without derivatization. Clin Biochem. 2008;41:736–741. doi: 10.1016/j.clinbiochem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elstrom RL, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 34.Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- 35.Coloff JL, Rathmell JC. Metabolic regulation of Akt: roles reversed. J Cell Biol. 2006;175:845–847. doi: 10.1083/jcb.200610119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.