

Abstract
Chronic alcohol abuse results in a variety of pathological effects including damage to the brain. The causes of alcohol-induced brain pathology are presently unclear. Several mechanisms of pathogenicity of chronic alcoholism have been proposed, including accumulation of DNA damage in the absence of repair, resulting in genomic instability and death of neurons. Genomic instability is a unified genetic mechanism leading to a variety of neurodegenerative disorders. Ethanol also likely interacts with various metabolic pathways, including one-carbon metabolism (OCM). OCM is critical for the synthesis of DNA precursors, essential for DNA repair, and as a methyl donor for various methylation events, including DNA methylation. Both DNA repair and DNA methylation are critical for maintaining genomic stability. In this review, we outline the role of DNA damage and DNA repair dysfunction in chronic alcohol-induced neurodegeneration.
Keywords: alcohol, DNA damage, neuron, cell death
Introduction
Chronic alcohol consumption is associated with an increase in the incidence of a variety of diseases, including central nervous system (CNS) degeneration.1 The brain is one of the major targets of alcohol actions. In adult human chronic alcoholics, brain damage is characterized by cerebral and cerebellar atrophy, and impaired neuronal function within the hippocampus and frontal cortex.2,3 Besides specific alcohol-related disorders, such as Wernicke–Korsakoff syndrome, hepatic encephalopathy and pellagra, heavy alcohol consumers exhibit cognitive and motor impairments, cholinergic deficits and dementia. It is estimated that 50–75% of long-term alcoholics show cognitive impairment and structural damage to the brain, making chronic alcoholism the second leading cause of dementia behind Alzheimer’s disease.4–6 These changes may be caused by loss of neurons, shrinkage of neuronal cell bodies, or reduction in the number and extent of dendrites. Careful neuropathological analyses have provided evidence for loss of neurons in certain regions of the brain of alcoholics.2,7 A direct toxic effect of ethanol on the brain has been suggested as the primary cause of alcohol-related neuronal loss.8 The effects of alcohol on the brain are well documented. However, the mechanisms are poorly understood. Several mechanisms have been proposed to explain ethanol-related brain damage. The N-methyl-D-aspartate (NMDA)-glutamate receptor could contribute to alcohol-induced brain damage. Ethanol, when administered acutely, potently inhibits NMDA receptors, while chronic exposure causes adaptive upregulation in the sensitivity of NMDA receptors and can result in an increased vulnerability for glutamate-induced cytotoxic response (excitotoxicity).9 Increased calcium influx through NMDA receptors, in turn, is tightly coupled to uptake into mitochondria and causes the production of reactive oxygen species (ROS). Alcohol metabolism is also associated with ROS production10,11 (Figure 1). Ethanol oxidation by cytochrome P450 2E1 (CYP2E1) and catalase is particularly relevant to ethanol metabolism in the brain.12 It is well known that ROS cause damage to DNA.13 Acetaldehyde, the primary metabolite of ethanol, can also damage DNA.14,15
Figure 1.
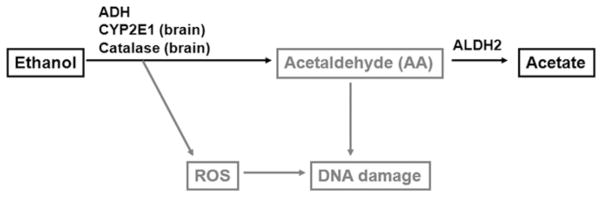
How alcohol may impact DNA. Ethanol is metabolized to acetaldehyde (AA) by alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1) and catalase. AA is metabolized to acetate by aldehyde dehydrogenase 2 (ALDH2). Catalase and CYP2E1 are particularly relevant to ethanol metabolism in brain tissue. The CYP2E1-dependent pathway is a major contributor to ethanol-generated reactive oxygen species (ROS). ROS induce DNA damage
Chronic ethanol exposure has been shown to be more harmful than acute exposure.16,17 Chronic alcoholism is mutagenic and carcinogenic in humans and is also associated with brain dysfunction.1,6,10,13,18–20 However, the mechanisms by which long-term chronic excessive alcohol consumption leads to a variety of pathologies are unclear.
The sulfur-containing amino acid homocysteine (Hcy) has been suggested to be toxic in alcoholism.6,21 Chronic alcoholic patients often have sustained hyperhomocysteinemia, a marker of one-carbon metabolism (OCM) impairment and a role for alcohol in disrupting OCM is strongly supported by the literature.10,21 OCM impairment directly impacts the brain in the conditions of ethanol exposure, as evidenced by increased Hcy concentrations in the cerebellum of rats22 and increased risk of alcohol withdrawal seizures with the increase of Hcy plasma concentrations.23 Also, plasma Hcy concentrations show the most significant correlation to hippocampal volume reduction in patients with alcoholism.3 Such effects of OCM impairment can be explained by the critical importance of OCM for the synthesis of DNA nucleotides (dNTPs), precursors for DNA biosynthesis and repair, and for methylation reactions.24,25 DNA repair and methylation of DNA play essential roles in maintaining genomic stability. Given that ethanol (or its metabolites) is genotoxic14,17 and OCM is critical for maintaining genomic stability,26 alcohol-induced OCM impairment may play a significant role in alcohol pathogenicity. In addition, Hcy itself may act as a convulsant through its agonism at NMDA receptors.27
Here, we link chronic alcohol-induced neurotoxicity to DNA damage, which suggests far-reaching implications for the understanding of the pathogenesis of alcoholism in humans.
Potentially toxic metabolites of ethanol
As shown in Figure 1, ethanol is metabolized mainly through oxidation catalyzed by alcohol dehydrogenase (ADH), cyto-chrome P450 2E1 (CYP2E1) and catalase enzymes.28–30 Due to the low capacity and the relatively high affinity (Km = 0.05–0.1 g/L) of ADH, the enzyme gets saturated after only a few drinks. Once formed, acetaldehyde is oxidized by the mitochondrial isoform of aldehyde dehydrogenase (ALDH), ALDH2, to acetate, in an irreversible reaction. ALDH2 has a very low Km value, which makes the elimination of toxic acetaldehyde highly efficient. The rates of ethanol metabolism by ADH and ALDH2 are critical in determining its toxicity because the alcohol metabolite acetaldehyde is highly toxic.29,31 ADH and ALDH2 use the co-factor nicotinamide adenine dinucleotide (NAD+), which converts to NADH and significantly changes the ratio NADH/NAD+, resulting in an altered cellular redox and overall energy metabolism states.32 ALDH2 is one of 19 members of the ALDH gene family in humans that play a crucial role in the oxidation and detoxification of numerous reactive aldehydes including 4-hydroxy-2-nonenal (4-HNE), a well-known, highly toxic by-product of lipid peroxidation, and nitroglycerin.32–34 Consistent with the critical importance of ALDH2 for detoxification of acetaldehyde, a risk of alcohol-induced toxicity in individuals with mutant ALDH2 is remarkably increased. Approximately 500 million Asians are heterozygous for the ALDH2 gene and, therefore, possess one normal and one mutant copy of the ALDH2 gene, termed ALDH2*2. The mutant copy encodes an inactive isozyme.35,36
Although ADH activity is not present in the brain, there is evidence of brain ethanol metabolism through ethanol oxidation into acetaldehyde by catalase37,38 and CYP2E139 (Figure 1). It is estimated that catalase accounts for 60–70% of ethanol-generated acetaldehyde in the brain, while CYP2E1 accounts for 10–20%.39,40 Acetaldehyde is then readily oxidized into acetate by ALDH.38 A number of studies based on the analysis of brain homogenates39,41 and at least one study based on the in vivo microdialysis42 provide evidence for the presence of acetaldehyde in the brain following ethanol intake. The levels of acetaldehyde were significantly increased under conditions of ALDH2 deficiency in ALDH2-knockout mice and were consistent with the dose of ethanol.42 ALDH2 transgenic overexpression alleviated chronic alcohol-induced cell death in the cerebral cortex of mice.43 Since ALDH2 is essential for detoxification of acetaldehyde, ALDH2 deficiency can directly contribute to excess acetaldehyde accumulation, while its overexpression can reduce acetaldehyde concentrations (and toxicity). Other indications of acetaldehyde presence in the brain include a reduction in acetaldehyde accumulation under conditions of pharmacological38 or genetic (via injection of anti-catalase shRNA construct into the CNS)44 inhibition of catalase. Thus, acetaldehyde can be produced in the brain by metabolic transformation of ethanol, and neurotoxic effects of ethanol may be associated with its toxicity.
Alcohol and DNA damage
Preservation of genetic information is of prime importance to all living systems. However, the integrity of the genome is continuously threatened by environmental agents (e.g. the ultraviolet [UV] component of sunlight, ionizing radiation and genotoxic chemicals) or intrinsic sources of DNA damage (metabolic by-products, spontaneous disintegration of DNA structure). DNA damage can lead to mutations, a primary step into cancer initiation. DNA damage may also result in cellular malfunction, senescence or death. Together, these changes may cause the progressive loss of tissue homeostasis and organismal decline. Oxidative stress, ionizing radiation, UV light and numerous chemicals induce a wide verity of DNA lesions.45 To cope with this permanent challenge, cells are equipped with an efficient genome defense mechanism responsible for detecting and removing DNA lesions, as well as eliminating the irreparably damaged cells. Alcohol metabolism generates potentially genotoxic ROS and acetaldehyde, which have been shown to induce DNA damage, including oxidative modifications, acetaldehyde-derived DNA adducts and cross-links.14,17,18,46–48 One of the most abundant ROS-induced DNA lesions is the oxidative DNA damage, 7,8-dihydro-8-oxo-2′-deoxyguanosine (oxo8dG),49 which is repaired by the base excision repair (BER) pathway. The primary acetaldehyde-derived DNA adduct is N2-ethylidene-deoxyguanosine,50 which may be converted in vivo to N2-ethyldeoxyguanosine (N2-ethyl-dG). N2-ethyl-dG has been found in the DNA of ethanol-treated mice51 and human alcoholics,52 suggesting that N2-ethyl-dG is a potential biomarker of acetaldehyde-induced DNA damage. This DNA adduct blocks translesion DNA synthesis (a DNA damage tolerance process that allows the DNA replication machinery to replicate past DNA lesions) catalyzed by a variety of DNA polymerases, and induces mutations.53,54 DNA polymerase η can bypass N2-ethyl-dG in an error-free manner.55 The DNA repair pathway for N2-ethyl-dG has not been identified. N2-ethyl-dG is not the only acetaldehyde-generated DNA damage. Acetaldehyde can also form DNA–DNA and DNA–protein cross-links.46,47 The major mechanism responsible for efficient cross-link removal is the Fanconi anemia (FA) pathway. Cross-link repair also involves the coordinated activities of other DNA repair pathways, such as nucleotide excision repair (NER) and homologous recombination (HR).56 Defects in a cluster of FA proteins leading to inactivation of associated DNA repair pathways are associated with hereditary disease FA that causes developmental defects, sterility, bone-marrow failure and a highly elevated risk of cancer.15 Langevin et al.15 recently provided strong evidence that metabolically produced acetaldehyde is indeed a DNA-damaging agent normally counteracted by the FA network. They demonstrated that simultaneous knockout of the Aldh2 gene (which encodes Aldh2, the main detoxifying enzyme of acetaldehyde) and the Fancd2 gene (a key player in the FA system) in double-mutant (Aldh2−/− Fancd2−/−) mice make these mice unusually sensitive to ethanol exposure. Although many studies of alcohol-mediated DNA damage have been conducted in the liver and other proliferating tissues in association with carcinogenesis, evidence exists that alcohol can produce DNA damage in the brain. Brains of mice exposed to ethanol exhibited FA activation, suggesting formation of acetaldehyde-induced DNA damage in the brain, although DNA damage was not determined directly.14 Importantly, acute alcohol intoxication induces reversible DNA lesions which do not exceed the capacities of the cellular repairing systems, while chronic alcohol exposure is associated with extensive DNA damage17,18 associated with genomic instability. Genomic instability refers to a set of events capable of causing unscheduled alterations, either of a temporary or permanent nature, within the genome and encompasses diverse genetic changes. Genomic instability usually results from an aberrant cellular response to DNA damage. DNA damage response is an extremely important mechanism of preserving genomic stability which includes DNA repair machinery. Damage to the genome is not only caused by exogenous and endogenous chemical and physical agents but can also occur due to inherited or acquired defects in DNA repair,57–59 conditions where the rate of DNA damage exceeds DNA repair capacity of the cell. As a result, fundamental changes to the genetic code and gene expression may cause serious defects in cellular function and tissue physiology. This can be a consequence of dysfunctional DNA repair, epigenetic changes leading to disturbed DNA damage response and accumulation of genetic aberrations in cells. These changes can be classified into the two major groups: instability occurring at the chromosomal and at the nucleotide levels. Instability at the nucleotide level occurs due to faulty DNA repair pathways such as BER and NER. The chromosomal instability represents alterations, which result in gains or losses of whole chromosomes as well as inversions, deletions, duplications and translocations of large chromosomal segments.60 DNA repair processes play critical roles in repairing damaged DNA, and in ensuring accurate transmission of genetic material. Inherited defects of genes in these pathways lead to disorders, most of which significantly increase susceptibility to cancer and neurodegeneration.26,61 Epigenetic modifications such as methylation and histone modification have also been shown to affect genomic stability.60 Global hypomethylation and genomic instability are seen in many cancers.62 The classic model for neurodegeneration due to dysfunctional DNA repair represents the idea that DNA damage accumulates in the absence of repair, resulting in the death of neurons.5 According to this mechanism, which presently is generally accepted, endogenous DNA damage is constantly being produced in normal conditions but also repaired, resulting in a low-steady-state level of damage which is compatible with normal cellular function. However, under conditions of DNA repair deficiency, endogenous damage is not repaired and therefore accumulates over time, ultimately leading to neuronal death as a result of impaired transcription. Indeed, defects in DNA damage response/DNA repair observed in patients with a variety of hereditary DNA repair diseases are associated with neurological abnormalities.61 Therefore, alcohol-induced DNA damage, if not repaired, may play a key role in alcohol-induced neurotoxicity.
Alcohol and OCM
Hyperhomocysteinemia, which is an indication of OCM dysfunction, is often observed in patients suffering from chronic alcoholism.10,21 It has been known for many years that ethanol has an effect on folate metabolism. The etiology of folate deficiency in alcoholism can be caused by any or all of the following: poor absorption, decreased uptake and retention, and increased urinary excretion.21 Furthermore, increased Hcy in patients suffering from alcohol dependence may be due to direct interactions of ethanol or its metabolites with OCM.63 Indeed, ethanol has been shown to directly affect OCM by inhibiting a key OCM enzyme, methionine synthase (MS), by 50% as early as six days of ethanol exposure.64,65 These findings have been corroborated by other investigators using different animal models of ethanol injury.21,66,67 Ethanol treatment has been demonstrated to reduce MS activity in the brain.68 Based on in vitro experiments, it has been suggested that acetaldehyde, but not ethanol, causes inhibition of MS activity.69 In spite of supraphysiological inhibitory levels of acetaldehyde in these experiments, the actual concentrations may be much lower than the reported values because acetaldehyde is highly volatile. Thus, it may be reasonable to assume that the in vitro assay could represent an acute treatment for a short time and the in vitro data may not reflect the true situation occurring in vivo. Given that MS is critical for OCM function, its down-regulation or direct inhibition may play a significant role in alcohol-induced OCM impairment.
OCM reactions (Figure 2) can be viewed as two intertwined cycles, one producing dNTPs, the precursors for DNA biosynthesis and repair, and the other producing and utilizing S-adenosyl methionine (SAM), the universal donor in cellular methylation reactions.24,25 5,10- methylene-tetrahydrofolate (5,10-methylene-THF) is a critical OCM junction where one-carbon groups can either be used to produce dNTPs for DNA synthesis or serve for the methylation cycle via irreversible synthesis of 5-methyl-THF catalyzed by methylene THF reductase. Following SAM-dependent transmethylation and hydrolysis of the ensuing S-adenosyl homocysteine (SAH), the resulting Hcy can be re-methylated back to methionine by the folate-mediated MS reaction. OCM dysfunction causes alteration in the de novo synthesis of DNA precursors, purines and thymidylate, negatively affecting the intracellular dNTP pool and, consequently, DNA synthesis (and cell proliferation) and repair (and genomic stability).70 The dNTP supply via the salvage pathway, whose purpose is to provide the cell with a low supply of dNTPs, cannot compensate DNA precursor pool imbalance from aberrant de novo dNTP synthesis. As a result, the dNTP synthesis pathway is the predominant mechanism for supplying dNTPs in the brain.71,72 The DNA repair pathway for removal of an incorrect DNA base, such as one of the most abundant alcohol-induced DNA base lesions oxo8dG, is BER.73 Acetaldehyde-derived cross-links are repaired by the FA-linked pathway, coordinated with classical NER, applicable to non-replicating cells such as neurons.56,74 Both DNA excision repair pathways, BER and NER, involved in the repair of alcohol-generated DNA damage, operate through the removal of damaged bases and their substitution with correct ones by the pathway-specific polymerases. The availability of dNTPs from OCM is important for BER and NER because DNA polymerases involved in these pathways are not functional when nucleotides are depleted. This leads to compromised DNA repair and accumulation of DNA lesions.75 Thus, OCM dysfunction is associated with compromised DNA excision repair pathways due to a decrease of synthesis of dNTPs and ensuing dNTP pool imbalance. Particularly, chronic alcohol administration has been reported to significantly reduce BER.76 Dysfunctional DNA repair results in accumulation of DNA damage and death of neurons.5
Figure 2.
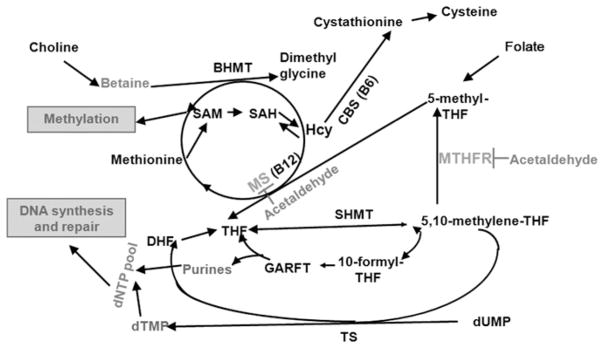
One-carbon metabolism as a target for ethanol. BHMT, betaine-homocysteine methyltransferase; CBS, cystathionine β-synthase; DHF, dihydrofolate; dNTP, deoxyribonucleotide triphosphate; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; GARFT, glycinamide ribonucleotide formyltransferase; Hcy, homocysteine; MS, methionine synthase; MTHFR, methylene THF reductase; SAH, S-adenosyl homocysteine; SHMT, serine hydroxy-methyltransferase; THF, tetrahydrofolate; TS, thymidylate synthase
OCM is essential for DNA methylation and its disturbance can result in global DNA hypomethylation.77 DNA methylation is a major epigenetic mechanism central to regulation of chromatin structure, genomic stability, gene transcription, and ultimately cell function and development. Functionally, proper DNA methylation can restrain the inappropriate expression of genes, thereby shaping cellular fate and function. In higher organisms, a methyl moiety is preferentially targeted to the DNA base cytosine in the context of a CpG dinucleotide, with the exception being X-inactivation. DNA is highly methylated in CpG-rich sequences (>50%), which has been suggested to represent a defense mechanism for silencing parasitic DNA elements such as transposons and retrotransposons.78 DNA methylation leads to a condensed structure and transcriptional repression. Aberrant methylation is associated with increased genomic instability79 and carcinogenesis,80 as well as brain disorders such as ischemia.81 At least three hereditary diseases with aberrant methylation, immunodeficiency/centromeric instability/facial anomalies (ICF), Rett and fragile X syndromes,82 are characterized by mental impairment, indicating the importance of methylation for brain function. DNA methylation depends on OCM as a source of methyl groups and dNTP methyltransferases which methylate cytosine residues at CpG sites in DNA. For this reason, hypomethylation can be caused by OCM impairment or mutations/inactivation of methyltransferases. Indeed, DNA hypomethylation has been demonstrated experimentally as a result of dietary folate deficiency,83 or mutations of enzymes, important for DNA methylation.83,84 Given that alcohol interferes with OCM,63,85,86 alcohol-induced aberrant DNA methylation87 is likely mediated by OCM impairment. Interestingly, elevated Hcy (OCM impairment) is considered an independent risk factor for numerous neurodegenerative diseases.26,78
In summary, alcohol affects OCM function. OCM is critical for synthesis of DNA precursors and methylation reactions (including DNA methylation). OCM impairment may cause DNA precursor imbalance and ensuing disturbance of DNA synthesis, which is important for cell proliferation and DNA repair. Dysfunctional DNA repair leads to genomic instability, and is a unified genetic mechanism causing a variety of neurological and neurodegenerative disorders.88–90 There has been compelling evidence accumulated that disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases and Down’s syndrome result from dysfunctional DNA repair and increased DNA damage.90 Different brain regions are affected in these neurodegenerative diseases, as well as hereditary diseases with DNA repair deficiencies: cerebellar Purkinje neurons in ataxia telangiectasia, dopaminergic neurons in the substantia nigra in Parkinson’s disease, neuronal loss in the striatum and cerebral cortex in Huntington’s disease, and loss of neurons in the cerebral cortex in Alzheimer’s disease.90 It is unclear, however, why different brain regions are affected in these disorders. Analysis of the types of neurons lost from the frontal cortex of alcoholics revealed that this population of neurons is also more vulnerable in both Alzheimer’s disease91 and normal aging92 in which DNA repair disturbance plays an important role.26 Atrophy of the cerebellum is commonly associated with alcoholism. Torvik et al.93 reported that 26.8% of alcoholics with Wernicke–Korsakoff syndrome had cerebellar atrophy with a loss of Purkinje cells that correlated with clinical ataxia/unsteadiness.94 This is especially interesting given the ataxia and death of Purkinje neurons in patients with ataxia telangiectasia, a hereditary disease with impaired DNA damage response.90 Understanding the causes and time-courses of DNA damage in the brain generally and in specific regions following chronic ethanol exposure will help to understand how DNA repair dysfunction and ensuing DNA damage may cause the damage of particular brain regions.
The phenotype of OCM impairment includes reduced tolerance to DNA-damaging agents, and genomic instability.70,75,95,96 Genomic instability results in mutagenesis and carcinogenesis.97 At the same time, it is associated with neuronal cell death and neurodegeneration.88–90 Alcohol abuse is known to increase the risk for various types of cancer,10,20,97–99 and contributes to neurodegeneration.88–90 The contribution of genomic instability to both carcinogenesis and neurodegeneration is illustrated by the fact that DNA repair defects in various human syndromes are usually characterized by both cancer predisposition and neurological abnormalities.90 Since OCM impairment is associated with DNA repair dysfunction and aberrant methylation, both linked to genomic instability, chronic alcohol-induced genomic instability and associated neuro-degeneration are likely mediated, at least in part, by alcohol’s impact on OCM.
Conclusion
Throughout this paper, we have emphasized the role of genomic instability in the adverse effects of chronic alcohol abuse on the brain. Ethanol metabolism, including those in brain, elicits the formation of genotoxic ROS and acetaldehyde.5,14,42 If not efficiently detoxified, these metabolites induce DNA damage, which is reversible in conditions of an acute alcohol exposure.14,17,18,48 However, chronic alcohol abuse is characterized by extensive DNA damage and genomic instability,14,17,18 which is a risk factor for different types of cancer10,20,98,99 and neurodegeneration.88–90 Alcoholism is known to increase the risk for cognitive impairment and dementia and is characterized by structural damage to the brain.4–6 The present review describes the current knowledge concerning alcohol-induced genomic instability with alcohol-induced OCM impairment. Increased Hcy concentrations, a marker of OCM disturbance is common in alcoholics,21,100 and at the same time is a risk factor for various neurodegenerative diseases.101,102 OCM is vital for cellular homeostasis, providing cells with DNA precursors, essential for cell proliferation and DNA repair, as well as methyl groups for DNA methylation. Thus, OCM dysfunction can lead to a shortage of DNA precursors, resulting in impaired DNA repair, as well as aberrant DNA methylation. Both DNA repair dysfunction and aberrant DNA methylation cause genomic instabilty.79,97 OCM impairment is among the factors that promote genomic instability.67,95 Different factors can affect the sensitivity of brain to alcohol in this context. Two examples are age and gender. Accumulated DNA damage is thought to be an important factor underlying aging.103 Defective DNA repair causes an accelerated aging-like phenotype of the brain.104 Hcy concentrations (and OCM impairment) increase with age,101 and age-related brain atrophy in healthy elderly people correlates with plasma Hcy concentrations.105,106 Among the factors that influence vulnerability of the brain to alcohol is gender. Women show a greater susceptibility to alcohol-related diseases, including an increased risk for brain damage with significantly lower alcohol exposure compared with men.107 The mechanism involving increased DNA damage and impaired DNA repair may contribute to the gender-related differences in vulnerability to alcohol. It is known, for example, that poly (ADP-ribose) polymerase-1 (PARP-1) deletion reduces ischemic brain injury in men but exacerbates injury in women.108 Since PARP-1 is a factor that plays an important role in the DNA damage response and DNA damage-induced cell death signaling,109 this difference suggests that the DNA damage response is different in men and women, and that the gender-related differences in vulnerability to alcohol may be caused by different responses to DNA damage. Further investigation of the cellular and molecular mechanisms involved in the genotoxicity of chronic alcohol, its interference with OCM, and the impact on DNA repair and DNA methylation will help to understand the mechanisms of ethanol-induced brain damage and will likely contribute to the development of treatments and/or therapeutic agents that could reduce or eliminate the deleterious effects of alcohol on the brain.
Acknowledgments
This work was supported by the Foundation for Alcohol Research, the South Plains Alcohol and Addiction Research Center (SPAARC) and NIH R01 AA010114.
Footnotes
Author contributions: All authors participated in the design, interpretation and writing of the review.
References
- 1.Brooks PJ. DNA damage, DNA repair, and alcohol toxicity. Alcohol Clin Exp Res. 1997;21:1073–82. [PubMed] [Google Scholar]
- 2.Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–10. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Bleich S, Wilhelm J, Graesel E, Degner D, Sperling W, Rössner V, Javaheripour K, Kornhuber J. Apolipoprotein E epsilon 4 is associated with hippocampal volume reduction in females with alcoholism. J Neural Transm. 2003;110:401–11. doi: 10.1007/s00702-002-0789-1. [DOI] [PubMed] [Google Scholar]
- 4.Arendt T. Impairment in memory function and neurodegenerative changes in the cholinergic basal forebrain system induced by chronic intake of ethanol. J Neural Transm Suppl. 1994;44:173–87. doi: 10.1007/978-3-7091-9350-1_13. [DOI] [PubMed] [Google Scholar]
- 5.Brooks PJ. Brain atrophy and neuronal loss in alcoholism: a role for DNA damage? Neurochem Int. 2000;37:403–12. doi: 10.1016/s0197-0186(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 6.Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5:73–8. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Harper C, Kril J. If you drink your brain will shrink. Neuropathological considerations. Alcohol Alcohol Suppl. 1991;1:375–80. [PubMed] [Google Scholar]
- 8.Sun AY, Sun GY. Ethanol and oxidative mechanisms in the brain. J Biomed Sci. 2001;8:37–43. doi: 10.1007/BF02255969. [DOI] [PubMed] [Google Scholar]
- 9.Krystal JH, Petrakis IL, Mason G, Trevisan L, D’Souza DC. N-methyl-D-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99:79–4. doi: 10.1016/s0163-7258(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 10.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 11.Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet. 2011;26:30–46. doi: 10.2133/dmpk.dmpk-10-rv-087. [DOI] [PubMed] [Google Scholar]
- 12.Deng XS, Deitrich RA. Putative role of brain acetaldehyde in ethanol addiction. Curr Drug Abuse Rev. 2008;1:3–8. doi: 10.2174/1874473710801010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasai H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res. 1997;387:147–63. doi: 10.1016/s1383-5742(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 14.Rulten SL, Hodder E, Ripley TL, Stephens DN, Mayne LV. Alcohol induces DNA damage and the Fanconi anemia D2 protein implicating FANCD2 in the DNA damage response pathways in brain. Alcohol Clin Exp Res. 2008;32:1186–96. doi: 10.1111/j.1530-0277.2008.00673.x. [DOI] [PubMed] [Google Scholar]
- 15.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–8. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 16.Eysseric H, Gonthier B, Soubeyran A, Bessard G, Saxod R, Barret L. There is not simple method to maintain a constant ethanol concentration in long-term cell culture: keys to a solution applied to the survey of astrocytic ethanol absorption. Alcohol. 1997;14:111–5. doi: 10.1016/s0741-8329(96)00112-7. [DOI] [PubMed] [Google Scholar]
- 17.Lamarche F, Gonthier B, Signorini N, Eysseric H, Barret L. Acute exposure of cultured neurones to ethanol results in reversible DNA single-strand breaks; whereas chronic exposure causes loss of cell viability. Alcohol Alcohol. 2003;38:550–8. doi: 10.1093/alcalc/agg118. [DOI] [PubMed] [Google Scholar]
- 18.Singh NP, Lai H, Khan A. Ethanol-induced single-strand DNA breaks in rat brain cells. Mutat Res. 1995;345:191–6. doi: 10.1016/0165-1218(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 19.Tavares DC, Cecchi AO, Jordão AA, Jr, Vannucchi H, Takahashi CS. Cytogenetic study of chronic ethanol consumption in rats. Teratog Carcinog Mutagen. 2001;21:361–8. doi: 10.1002/tcm.1024. [DOI] [PubMed] [Google Scholar]
- 20.Maehara Y, Egashira A, Oki E, Kakeji Y, Tsuzuki T. DNA repair dysfunction in gastrointestinal tract cancers. Cancer Sci. 2008;99:451–8. doi: 10.1111/j.1349-7006.2007.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132:2367S–72S. doi: 10.1093/jn/132.8.2367S. [DOI] [PubMed] [Google Scholar]
- 22.Alirezaei M, Jelodar G, Niknam P, Ghayemi Z, Nazifi S. Betaine prevents ethanol induced oxidative stress and reduces total homocysteine in the rat cerebellum. J Physiol Biochem. 2011;67:605–12. doi: 10.1007/s13105-011-0107-1. [DOI] [PubMed] [Google Scholar]
- 23.Bleich S, Hillemacher T. Homocysteine, alcoholism and its molecular networks. Pharmacopsychiatry. 2009;42(Suppl 1):S102–9. doi: 10.1055/s-0029-1214396. [DOI] [PubMed] [Google Scholar]
- 24.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 25.Stover PJ. One-carbon metabolism-genome interactions in folate-associated pathologies. J Nutr. 2009;139:2402–5. doi: 10.3945/jn.109.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fenech M. Folate, DNA damage and the aging brain. Mech Ageing Dev. 2010;131:236–41. doi: 10.1016/j.mad.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D aspartate receptor. Proc Natl Acad Sci USA. 1997;94:5923–8. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141–54. doi: 10.1055/s-0029-1214370. [DOI] [PubMed] [Google Scholar]
- 29.Manzo-Avalos S, Saavedra-Molina A. Cellular and mitochondrial effects of alcohol consumption. Int J Environ Res Public Health. 2010;7:4281–304. doi: 10.3390/ijerph7124281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Wang X, Zhou R, Cederbaum A. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem Biophys Res Commun. 2010;402:116–22. doi: 10.1016/j.bbrc.2010.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George A, Figueredo VM. Alcohol and arrhythmias: a comprehensive review. J Cardiovasc Med (Hagerstown) 2010;11:221–8. doi: 10.2459/JCM.0b013e328334b42d. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: molecular mechanism and clinical implications. Pharmacol Ther. 2011;132:86–95. doi: 10.1016/j.pharmthera.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J. Interaction between high-fat diet and alcohol dehydrogenase on ethanol-elicited cardiac depression in murine myocytes. Obesity (Silver Spring) 2007;15:2932–41. doi: 10.1038/oby.2007.350. [DOI] [PubMed] [Google Scholar]
- 34.Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med. 2009;19:158–64. doi: 10.1016/j.tcm.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishida N, Tanaka M, Hayashi N, Nagata H, Takeshita T, Nakayama K, Morimoto K, Shizukuishi S. Association of ALDH(2) genotypes and alcohol consumption with periodontitis. J Dent Res. 2004;83:161–5. doi: 10.1177/154405910408300215. [DOI] [PubMed] [Google Scholar]
- 36.Peng GS, Yin SJ. Effect of the allelic variants of aldehyde dehydrogenase ALDH2*2 and alcohol dehydrogenase ADH1B*2 on blood acetaldehyde concentrations. Hum Genomics. 2009;3:121–7. doi: 10.1186/1479-7364-3-2-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aragon CM, Rogan F, Amit Z. Ethanol metabolism in rat brain homogenates by a catalase H2O2 system. Biochem Pharmacol. 1992;44:93–8. doi: 10.1016/0006-2952(92)90042-h. [DOI] [PubMed] [Google Scholar]
- 38.Jamal M, Ameno K, Uekita I, Kumihashi M, Wang W, Ijiri I. Catalase mediates acetaldehyde formation in the striatum of free-moving rats. Neurotoxicology. 2007;28:1245–8. doi: 10.1016/j.neuro.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA. Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res. 2006;30:1500–5. doi: 10.1111/j.1530-0277.2006.00181.x. [DOI] [PubMed] [Google Scholar]
- 40.Zimatkin SM, Liopo AV, Deitrich RA. Distribution and kinetics of ethanol metabolism in rat brain. Alcohol Clin Exp Res. 1998;22:1623–7. [PubMed] [Google Scholar]
- 41.Isse T, Matsuno K, Oyama T, Kitagawa K, Kawamoto T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol Clin Exp Res. 2005;29:1959–64. doi: 10.1097/01.alc.0000187161.07820.21. [DOI] [PubMed] [Google Scholar]
- 42.Jamal M, Ameno K, Miki T, Wang W, Kumihashi M, Isse T, Kawamoto T, Kitagawa K, Nakayama K, Ijiri I, Kinoshita H. Cholinergic alterations following alcohol exposure in the frontal cortex of Aldh2-deficient mice models. Brain Res. 2009;1295:37–43. doi: 10.1016/j.brainres.2009.07.099. [DOI] [PubMed] [Google Scholar]
- 43.Ren J, Babcock SA, Li Q, Huff AF, Li SY, Doser TA. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol Lett. 2009;187:149–56. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karahanian E, Quintanilla ME, Tampier L, Rivera-Meza M, Bustamante D, Gonzalez-Lira V, Morales P, Herrera-Marschitz M, Israel Y. Ethanol as a prodrug: brain metabolism of ethanol mediates its reinforcing effects. Alcohol Clin Exp Res. 2011;35:606–12. doi: 10.1111/j.1530-0277.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schumacher B, Hoeijmakers JH, Garinis GA. Sealing the gap between nuclear DNA damage and longevity. Mol Cell Endocrinol. 2009;299:112–7. doi: 10.1016/j.mce.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 46.Brooks PJ, Theruvathu JA. DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol. 2005;535:187–93. doi: 10.1016/j.alcohol.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Mechilli M, Schinoppi A, Kobos K, Natarajan AT, Palitti F. DNA repair deficiency and acetaldehyde-induced chromosomal alterations in CHO cells. Mutagenesis. 2008;23:51–6. doi: 10.1093/mutage/gem042. [DOI] [PubMed] [Google Scholar]
- 48.Blasiak J, Trzeciak A, Malecka-Panas E, Drzewoski J, Wojewódzka M. In vitro genotoxicity of ethanol and acetaldehyde in human lymphocytes and the gastrointestinal tract mucosa cell. Toxicol In Vitro. 2000;14:287–95. doi: 10.1016/s0887-2333(00)00022-9. [DOI] [PubMed] [Google Scholar]
- 49.Wells PG, McCallum GP, Chen CS, Henderson JT, Lee CJ, Perstin J, Preston TJ, Wiley MJ, Wong AW. Oxidative stress in developmental origins of disease: teratogenesis, neurodevelopmental deficits and cancer. Toxicol Sci. 2009;108:4–18. doi: 10.1093/toxsci/kfn263. [DOI] [PubMed] [Google Scholar]
- 50.Wang M, Yu N, Chen L, Villalta PW, Hochalter JB, Hecht SS. Identification of an acetaldehyde adduct in human liver DNA and quantitation as N2-ethyldeoxyguanosine. Chem Res Toxicol. 2006;19:319–24. doi: 10.1021/tx0502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang JL, Vaca CE. Development of a 32P-postlabelling method for the analysis of adducts arising through the reaction of acetaldehyde with 2′-deoxyguanosine-3′-monophosphate and DNA. Carcinogenesis. 1995;16:2177–85. doi: 10.1093/carcin/16.9.2177. [DOI] [PubMed] [Google Scholar]
- 52.Fang JL, Vaca CE. Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis. 1997;18:627–32. doi: 10.1093/carcin/18.4.627. [DOI] [PubMed] [Google Scholar]
- 53.Upton DC, Wang X, Blans P, Perrino FW, Fishbein JC, Akman SA. Replication of N2-ethyldeoxyguanosine DNA adducts in the human embryonic kidney cell line 293. Chem Res Toxicol. 2006;19:960–7. doi: 10.1021/tx060084a. [DOI] [PubMed] [Google Scholar]
- 54.Singh R, Sandhu J, Kaur B, Juren T, Steward WP, Segerback D, Farmer PB. Evaluation of the DNA damaging potential of cannabis cigarette smoke by the determination of acetaldehyde derived N2-ethyl-2′-deoxyguanosine adducts. Chem Res Toxicol. 2009;22:1181–8. doi: 10.1021/tx900106y. [DOI] [PubMed] [Google Scholar]
- 55.Perrino FW, Blans P, Harvey S, Gelhaus SL, McGrath C, Akman SA, Jenkins GS, LaCourse WR, Fishbein JC. The N2-ethylguanine and the O6-ethyl- and O6 methylguanine lesions in DNA: contrasting responses from the ‘bypass’ DNA polymerase η and the replicative DNA polymerase R. Chem Res Toxicol. 2003;16:1616–23. doi: 10.1021/tx034164f. [DOI] [PubMed] [Google Scholar]
- 56.Moldovan GL, D’Andrea AD. FANCD2 hurdles the DNA interstrand crosslink. Cell. 2009;139:1222–4. doi: 10.1016/j.cell.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol. 2009;41:87–95. doi: 10.1016/j.biocel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Maslov AY, Vijg J. Genome instability, cancer and aging. Biochim Biophys Acta. 2009;1790:963–9. doi: 10.1016/j.bbagen.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 61.Brooks PJ, Cheng TF, Cooper L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair (Amst) 2008;7:834–48. doi: 10.1016/j.dnarep.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capellà G, Ribas M, Peinado MA. Chromosomal instability correlates with genome wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006;66:8462–8. doi: 10.1158/0008-5472.CAN-06-0293. [DOI] [PubMed] [Google Scholar]
- 63.Giovannucci E. Alcohol, one-carbon metabolism, and colorectal cancer: recent insights from molecular studies. J Nutr. 2004;134:2475S–81S. doi: 10.1093/jn/134.9.2475S. [DOI] [PubMed] [Google Scholar]
- 64.Barak AJ, Beckenhauer HC, Junnila M, Tuma DJ. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol Clin Exp Res. 1993;17:552–5. doi: 10.1111/j.1530-0277.1993.tb00798.x. [DOI] [PubMed] [Google Scholar]
- 65.Barak AJ, Beckenhauer HC, Tuma DJ. Ethanol feeding inhibits the activity of hepatic N5-methyltetrahydrofolate: homocysteine methyltransferase in the rat. IRCS Med Sci. 1985;13:760–1. [Google Scholar]
- 66.Trimble KC, Molloy AM, Scott JM, Weir DG. The effect of ethanol on one-carbon metabolism: increased methionine catabolism and lipotrope methyl-group wastage. Hepatology. 1993;18:984–9. doi: 10.1002/hep.1840180433. [DOI] [PubMed] [Google Scholar]
- 67.Villanueva JA, Halsted CH. Hepatic transmethylation reactions in micropigs with alcoholic liver disease. Hepatology. 2004;39:1303–10. doi: 10.1002/hep.20168. [DOI] [PubMed] [Google Scholar]
- 68.Waly MI, Kharbanda KK, Deth RC. Ethanol lowers glutathione in rat liver and brain and inhibits methionine synthase in a cobalamin-dependent manner. Alcohol Clin Exp Res. 2011;35:277–83. doi: 10.1111/j.1530-0277.2010.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kenyon SH, Nicolaou A, Gibbons WA. The effect of ethanol and its metabolites upon methionine synthase activity in vitro. Alcohol. 1998;15:305–9. doi: 10.1016/s0741-8329(97)00134-1. [DOI] [PubMed] [Google Scholar]
- 70.James SJ, Miller BJ, Basnakian AG, Pogribny IP, Pogribna M, Muskhelishvili L. Apoptosis and proliferation under conditions of deoxynucleotide pool imbalance in liver of folate/methyl deficient rats. Carcinogenesis. 1997;18:287–93. doi: 10.1093/carcin/18.2.287. [DOI] [PubMed] [Google Scholar]
- 71.Sato K, Kanno J, Tominaga T, Matsubara Y, Kure S. De novo and salvage pathways of DNA synthesis in primary cultured neurall stem cells. Brain Res. 2006;1071:24–33. doi: 10.1016/j.brainres.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 72.Coucke PA, Cottin E, Decosterd LA. Simultaneous alteration of de novo and salvage pathway to the deoxynucleoside triphosphate pool by (E)-2′-deoxy-(fluoromethylene)cytidine (FMdC) and zidovudine (AZT) results in increased radiosensitivity in vitro. Acta Oncol. 2007;46:612. doi: 10.1080/02841860601137389. [DOI] [PubMed] [Google Scholar]
- 73.Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–93. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand crosslink repair. Mol Cell Biol. 2001;21:713–20. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cabelof DC, Guo Z, Raffoul JJ, Sobol RW, Wilson SH, Richardson A, Heydari AR. Base excision repair deficiency caused by polymerase beta haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003;63:5799–807. [PubMed] [Google Scholar]
- 76.Bhupanapadu Sunkesula SR, Swain U, Babu PP. Cell death is associated with reduced base excision repair during chronic alcohol administration in adult rat brain. Neurochem Res. 2008;33:1117–28. doi: 10.1007/s11064-007-9560-1. [DOI] [PubMed] [Google Scholar]
- 77.Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med. 2004;229:988–95. doi: 10.1177/153537020422901002. [DOI] [PubMed] [Google Scholar]
- 78.Kronenberg G, Endres M. Neuronal injury: folate to the rescue? J Clin Invest. 2010;120:1383–6. doi: 10.1172/JCI40764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR, Summers RG, Chun J, Lee KF, Gage FH. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci USA. 2003;100:6777–82. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hatziapostolou M, Iliopoulos D. Epigenetic aberrations during oncogenesis. Cell Mol Life Sci. 2011;68:1681–702. doi: 10.1007/s00018-010-0624-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–81. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–9. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 83.Balaghi M, Wagner C. DNA methylation in folate deficiency: use of CpG methylase. Biochem Biophys Res Commun. 1993;193:1184–90. doi: 10.1006/bbrc.1993.1750. [DOI] [PubMed] [Google Scholar]
- 84.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 85.Min H, Im ES, Seo JS, Mun JA, Burri BJ. Effects of chronic ethanol ingestion and folate deficiency on the activity of 10-formyltetrahydrofolate dehydrogenase in rat liver. Alcohol Clin Exp Res. 2005;29:2188–93. doi: 10.1097/01.alc.0000191756.02856.a8. [DOI] [PubMed] [Google Scholar]
- 86.Kharbanda KK. Alcoholic liver disease and methionine metabolism. Semin Liver Dis. 2009;29:155–65. doi: 10.1055/s-0029-1214371. [DOI] [PubMed] [Google Scholar]
- 87.Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol Clin Exp Res. 1991;15:395–8. doi: 10.1111/j.1530-0277.1991.tb00536.x. [DOI] [PubMed] [Google Scholar]
- 88.Kulkarni A, Wilson DM., 3rd The involvement of DNA-damage and -repair defects in neurological dysfunction. Am J Hum Genet. 2008;82:539–66. doi: 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 90.Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. 2007;3:162–72. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- 91.Terry RD, Hansen LA. Neocortical cell counts in normal human adult aging. Ann Neurol. 1987;21:530–9. doi: 10.1002/ana.410210603. [DOI] [PubMed] [Google Scholar]
- 92.Terry RD, Peck A, DeTeresa R, Schechter R, Horoupian DS. Some morphological aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol. 1981;10:184–92. doi: 10.1002/ana.410100209. [DOI] [PubMed] [Google Scholar]
- 93.Torvik A, Torp S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of autopsy material. J Neurol Sci. 1986;75:43–51. doi: 10.1016/0022-510x(86)90049-3. [DOI] [PubMed] [Google Scholar]
- 94.Baker K, Harding A, Halliday G, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91:429–38. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- 95.Duthie SJ, Narayanan S, Blum S, Pirie L, Brand GM. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr Cancer. 2000;37:245–51. doi: 10.1207/S15327914NC372_18. [DOI] [PubMed] [Google Scholar]
- 96.Branda RF, Hacker M, Lafayette A, Nigels E, Sullivan L, Nicklas JA, O’Neill JP. Nutritional folate deficiency augments the in vivo mutagenic and lymphocytotoxic activities of alkylating agents. Environ Mol Mutagen. 1998;32:33–8. doi: 10.1002/(sici)1098-2280(1998)32:1<33::aid-em4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 97.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011;711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 98.Asami S, Hirano T, Yamaguchi R, Tsurudome Y, Itoh H, Kasai H. Increase in 8-hydroxyguanine and its repair activity in the esophagi of rats given long-term ethanol and nutrition-deficient diet. Jpn J Cancer Res. 2000;91:973–8. doi: 10.1111/j.1349-7006.2000.tb00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Poschl G, Seitz HK. Alcohol and cancer. Alcohol Alcohol. 2004;39:155–65. doi: 10.1093/alcalc/agh057. [DOI] [PubMed] [Google Scholar]
- 100.Cravo ML, Gloria LM, Selhub J. Hyperhomocysteinemia in chronic alcoholism: correlation with folate, vitamin B-12, and vitamin B-6 status. Am J Clin Nutr. 1996;63:220–4. doi: 10.1093/ajcn/63.2.220. [DOI] [PubMed] [Google Scholar]
- 101.Seshadri S, Beiser A, Selhub J, Jacques PP, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–83. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 102.D’Anci KE, Rosenberg IH. Folate and brain function in elderly. Curr Opin Clin Nutr Metab Care. 2004;7:659–64. doi: 10.1097/00075197-200411000-00011. [DOI] [PubMed] [Google Scholar]
- 103.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 104.Borgesius NZ, de Waard MC, van der Pluijm I, Omrani A, Zondag GC, van der Horst GT, Melton DW, Hoeijmakers JH, Jaarsma D, Elgersma Y. Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J Neurosci. 2011;31:12543–53. doi: 10.1523/JNEUROSCI.1589-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bleich S, Kornhuber J. Relationship between plasma homocysteine levels and brain atrophy in healthy elderly individuals. Neurology. 2003;60:1220. doi: 10.1212/wnl.60.7.1220. [DOI] [PubMed] [Google Scholar]
- 106.Sachdev PS, Valenzuela M, Wang XL, Looi JC, Brodaty H. Relationship between plasma Hcy levels and brain atrophy in healthy elderly individuals. Neurology. 2002;58:1539–41. doi: 10.1212/wnl.58.10.1539. [DOI] [PubMed] [Google Scholar]
- 107.Hommer D, Momenan R, Kaiser E, Rawlings R. Evidence for a gender-related effect of al coholism on brain volumes. Am J Psychiatry. 2001;158:198–204. doi: 10.1176/appi.ajp.158.2.198. [DOI] [PubMed] [Google Scholar]
- 108.Liu F, Lang J, Li J, Benashski SE, Siegel M, Xu Y, McCullough LD. Sex differences in the response to poly(ADP-ribose) polymerase-1 deletion and caspase inhibition after stroke. Stroke. 2011;42:1090–6. doi: 10.1161/STROKEAHA.110.594861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]