

Abstract
The embryonic stem cell–specific cell cycle–regulating (ESCC) family of microRNAs (miRNAs) enhances reprogramming of mouse embryonic fibroblasts to induced pluripotent stem cells1. Here we show that the human ESCC miRNA orthologs hsa-miR-302b and hsa-miR-372 promote human somatic cell reprogramming. Furthermore, these miRNAs repress multiple target genes, with downregulation of individual targets only partially recapitulating the total miRNA effects. These targets regulate various cellular processes, including cell cycle, epithelial-mesenchymal transition (EMT), epigenetic regulation and vesicular transport. ESCC miRNAs have a known role in regulating the unique embryonic stem cell cycle2,3. We show that they also increase the kinetics of mesenchymal-epithelial transition during reprogramming and block TGFβ-induced EMT of human epithelial cells. These results demonstrate that the ESCC miRNAs promote dedifferentiation by acting on multiple downstream pathways. We propose that individual miRNAs generally act through numerous pathways that synergize to regulate and enforce cell fate decisions.
Dedifferentiation of a somatic cell to an induced pluripotent stem cell (iPSC) requires global epigenetic reprogramming and a shift in the expression of thousands of genes4. A number of small molecules and genes have been identified that increase the efficiency of reprogramming, but whether and how they converge into a common set of pathways is poorly understood5,6. MiRNAs function by suppressing many mRNA targets simultaneously7. A large family of miRNAs highly expressed in mouse embryonic stem cells (ESCs) targets multiple inhibitors of the CyclinE-Cdk2 pathway, thereby promoting the unique cell cycle program of these cells3. This family, termed ESCC miRNAs, greatly enhances the reprogramming efficiency of mouse embryonic fibroblasts into iPSCs, as does the closely related miR-106 family1,8. Together, these data suggest a pathway for downregulation of cell cycle inhibitors by the ESCC miRNAs during reprogramming, resulting in increased efficiency of dedifferentiation. However, as miRNAs target hundreds of mRNAs, it is likely that cell cycle regulation is only part of the mechanism.
The human orthologs of the ESCC miRNAs are also highly expressed in ESCs, are downregulated upon differentiation and regulate the cell cycle2,9–12. These miRNAs include members of the human miR-302 cluster (hsa-miR-302a–d), orthologous to the mouse miR-302s, and hsa-miR-372 and 373, orthologous to the mouse miR-291, miR-294 and miR-295 miRNAs. To determine whether these miRNAs play an analogous role in human somatic cell reprogramming, we introduced synthesized mimics of mature hsa-miR-302b and/or 372 into human foreskin (BJ) and lung fibroblasts (MRC-5) on days 3 and 10 after infection with combinations of retroviruses expressing OCT4, SOX2, KLF4, cMYC and Venus (4Y = OCT4, SOX2, KLF4, cMYC and Venus, whereas 3Y = OCT4, Sox2, KLF4 and Venus). An increase in number of colonies with human ESC-like morphology was observed in all wells transfected with mimics (Fig. 1a). Most of the colonies showed retroviral silencing (as indicated by silencing of Venus expression), a strong indicator of full reprogramming to iPSCs13.
Figure 1.
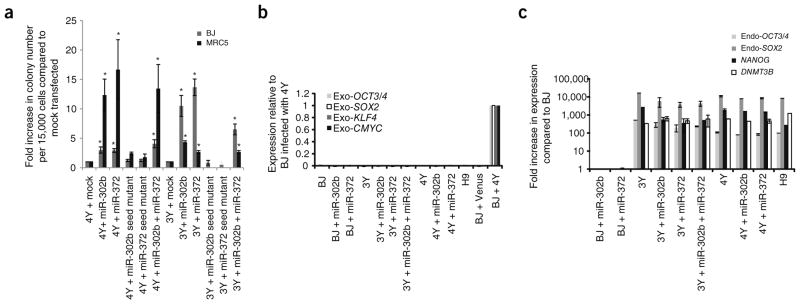
Hsa-miR-302b and/or hsa-miR-372 enhances reprogramming efficiency of human somatic cells. (a) Fold increase in number of human ESC-like colonies obtained per 15,000 cells compared to mock-transfected cells. Cells infected with 4Y ± miRNA were counted on day 21 after infection, whereas cells infected with 3Y ± miRNA were counted on day 31 after infection. *, a significant difference when compared to mock-transfected; P < 0.05; N = 6. Error bars represent mean ± s.e.m. (b) Expression of exogenous factors in iPSC lines that were picked and expanded after 3Y or 4Y infection and the indicated miRNA. Expression was determined by RT-qPCR using primers specific to only the exogenous factors. Data were normalized to BJ cells 3 d after retroviral infection with 4Y. (c) Expression of pluripotency markers in iPSC lines that were picked and expanded after 3Y or 4Y infection and the indicated miRNA. H9 hESCs shown as control. Expression was determined by RT-qPCR. Data were normalized to expression observed in BJ cells.
Representative BJ iPSC colonies were expanded and molecularly characterized. Reverse transcriptase–quantitative PCR (RT-qPCR) analysis of gene expression confirmed silencing of the exogenous factors (Fig. 1b), as well as endogenous expression of pluripotency markers, such as OCT3/4, SOX2, NANOG and DNMT3B, similar to that of H9 human embryonic stem cells (hESCs) (Fig. 1c). The colonies also showed immunostaining for pluripotency markers including OCT3/4 and TRA-1-60 (Supplementary Fig. 1). Together, these characteristics are indicative of a fully reprogrammed state14. Thus, the introduction of ESCC miRNAs along with the reprogramming factors enhanced the reprogramming of the human cells into pluripotent stem cell colonies, similar to what was previously described for mouse cells1.
MiRNAs regulate their targets by incomplete complementation to nucleotides within the 3′ untranslated region (UTR) or open reading frame (ORF) of coding mRNAs15. Of particular importance is base pairing between a specific portion of the miRNA called the seed sequence (bases 2 through 8) with a complementary sequence in the transcript7. Introduction of mimics with seed sequence mutations together with either 3Y or 4Y resulted in abrogation of the miRNA-induced enhancement of colony formation (Fig. 1a), confirming the essential role of seed sequence–based targeting in the enhancement of reprogramming.
We sought to identify the genes and mechanisms by which miR-302b and miR-372 enhance human iPSC production. Previous mRNA profiling after introduction of the miRNAs into miRNA-deficient mouse ESCs, together with bioinformatic analysis for seed matches in the mRNAs, had identified a set of putative miR-294 target genes16. As miR-302b and 372 share the same seed sequence as miR-294, this compilation likely represents a set of putative targets for the entire ESCC miRNA family. From this list, we chose a subset of target genes for further testing to determine whether they were targeted during the reprogramming process (Supplementary Table 1). Transcript levels of 34 putative targets were quantified by RT-qPCR on day 7 of 4Y- or 3Y-mediated reprogramming in the presence or absence of the miR-NAs (Fig. 2a and Supplementary Fig. 2). Among these, 12 transcripts (labeled in red) showed statistically significant decreases in levels after transfection of the miRNA in cells that had been infected with either 4Y or 3Y (P < 0.05). An additional 15 transcripts were downregulated at levels that did not reach our significance cut-off but are still probably meaningful as cells undergoing reprogramming are heterogeneous and miRNA effects on mRNA stability are typically slight. Notably, after miRNA transfection into uninfected BJ fibroblasts only a few of the putative targets were downregulated, suggesting context-dependent effects. The 12 target genes that were significantly downregulated during reprogramming were further characterized. The mRNAs fell into several functional modules: (i) cell cycle regulation (CDKN1A, RBL2, CDC2L6); (ii) epigenetic regulation (MECP2, MBD2, SMARCC2); (iii) vesicular transport (RAB5C, RAB11FIP5); (iv) cell signaling (AKT1, ARHGAP26); and (v) epithelial-mesenchymal transition (RHOC, TGFBR2). Of these, CDKN1A, RBL2 and MECP2 have been previously described as targets of miR-302b/372 (refs. 2,3,17–19).
Figure 2.

Hsa-miR-302b and hsa-miR-372 regulate expression of a number of targets that influence reprogramming of human somatic cells. (a) Heat map showing average expression from three independent experiments of 34 predicted targets of hsa-miR-302b and hsa-miR-372 on day 7 in the process of reprogramming. Expression was determined by qRT-PCR and was first normalized to GAPDH followed by normalization to mock-transfected cells. Statistically significant genes (P < 0.05; ANOVA) are labeled red. (b) Fold increase in reprogramming of human somatic cells infected with 4Y upon introduction of siRNAs against specific targets. Human ESC-like colonies were counted on day 21 after infection. N = 4. Error bars represent s.d. *, significant difference when compared to mock transfected; P < 0.05; measured by Kruskal-Wallis test. i, siRNA; ROCKi, ROCK inhibitor. (c ) Fold increase in reprogramming of human somatic cells infected with 3Y upon introduction of siRNAs against specific targets. Human ESC-like colonies were counted on day 31 after infection. N = 3. Error bars represent s.d. *, significant difference when compared to mock-transfected; P < 0.05; measured by Kruskal-Wallis test. i, siRNA; ROCKi, ROCK inhibitor.
Next, we evaluated the potential contribution to reprogramming of the individual target genes that are significantly downregulated by the miRNAs. Inhibitors to the type I TGF-β receptor (TβRI) kinase have been shown to enhance reprogramming to mouse and human iPSCs20–22. Consistent with these reports, we observed an increase in reprogramming efficiency with both 4Y and 3Y upon addition of RepSox, a TβRI inhibitor (Fig. 2b,c). Furthermore, expression of the type II TGF-β receptor cDNA (TβRII) without its 3′ UTR or addition of TGF-β ligand abolished iPSC generation (Supplementary Fig. 3), overriding the positive effects of miR-302b and miR-372. For the remaining 11 significantly downregulated target genes, siRNA pools against each target were transfected together with transduction of 4Y or 3Y. All siRNA pools robustly suppressed their target gene (Supplementary Fig. 4). Five of the 11 knockdowns significantly increased the number of iPSC colony numbers in combination with 4Y, and four did so with 3Y, totaling six independent gene knockdowns enhancing reprogramming (Fig. 2b,c). However, the increases were modest, with few reaching levels seen with miR-372. As downregulation of RHOC expression significantly enhanced the reprogramming efficiencies of both 4Y and 3Y, we tested an alternative means of repressing RHO functions. Y-27632 (ROCKi) is a small-molecule inhibitor of the RHO-dependent protein kinase ROCK, which acts immediately downstream of RHO23. ROCKi enhances survival of dissociated hESCs24–26 but has not been previously shown to enhance reprogramming. We found that the combination of ROCKi together with 4Y or 3Y indeed increased colony number (Fig. 2b,c).
As inhibition of RHOC and TβRI signaling enhanced reprogramming, we explored whether these two pathways synergized to regulate this process by conducting 3Y and 4Y reprogramming in the presence of ROCKi, RepSox or both molecules. Indeed, various combinations of ROCKi, RepSox and miR-372 also resulted in a significant increase in colony number compared to mock-transfected cells (Fig. 2b,c). The increase in efficiency observed upon using combinations of inhibitors in 3Y-infected cells was significantly higher than when a single inhibitor was used and reached levels seen with miR-372 (Fig. 2c). Together, these results are consistent with the ESCC miRNAs acting through multiple targets and/or pathways to enhance reprogramming.
To verify that the ESCC miRNAs enhance reprogramming through multiple transcripts, we evaluated the two novel targets, RHOC and TGFBR2. Consistent with the RT-qPCR data, western blots showed that the level of TβRII protein was suppressed by miR-302b and miR-372 during reprogramming (Fig. 3a). TGFBR2 mRNA has three 7-mer seed matches and three 6-mer seed matches in its 3′UTR (Fig. 3b, schematic). The 3′UTR was fused to a luciferase reporter and introduced into miRNA-deficient mouse ESCs in the presence and absence of the miRNA mimics. Luciferase activity was significantly reduced in the presence of the ESCC miRNAs (Fig. 3b, top panel) but not when mutations were introduced into the seed sequence of the mimics (Fig. 3b, middle panel). Furthermore, mutation of the 7-mer seed matches in the TGFBR2 mRNA 3′UTR resulted in a significant recovery of luciferase activity (Fig. 3b, top panel, and Supplementary Fig. 5). Incomplete additional target sites in the TGFBR2 3′UTR (Fig. 3b). In contrast to the TGFBR2 mRNA, RHOC mRNA has only one 7-mer seed match in its 3′UTR (Fig. 3b, schematic). The 3′UTR still suppressed luciferase activity, albeit to a lesser degree, and was fully reversed by inserting mutations into the predicted site (Fig. 3b, bottom panel). Together, these data illustrate that the genes encoding RHOC and TGFBR2 are direct targets of the ESCC miRNAs. miR-302 was previously shown to enhance Nodal signaling, another member of the TGF-β family, by targeting Lefty1 and Lefty2 transcripts during early hESC differentiation, promoting mesoendodermal cell fates27. However, the genes encoding Lefty1 and Lefty2 were expressed at low levels during reprogramming to iPSCs and were not downregulated by the ESCC miRNAs (data not shown), suggesting cell context–specific effects.
Figure 3.

Hsa-miR-302b and hsa-miR-372 enhance reprogramming by regulating mesenchymal-epithelial transition. (a ) Western blot showing levels of TβRII from lysates prepared from BJ cells infected with either 4Y or 3Y, or uninfected cells plus the indicated miRNAs. N= 3. (b) luciferase analysis of TGFBR2 and RHOC 3′UTRs. Seed matches for ESCC miRNAs in the 3′UTRs along with different mutant constructs are shown in the top panel. luciferase results after co-transfection with ESCC miRNAs relative to mock transfection are shown in the lower panel after normalization to firefly luciferase values. All data are represented as mean ± s.d. *P < 0.05 by t-test. (c) RT-qPCR showing relative expression levels of mesenchymal (ZEB1 and SLUG) and epithelial (E-cadherin, CDH1, and occludin, OCLN) markers at day 7 after infection in the process of reprogramming normalized to GAPDH. N = 3. Error bars represent s.e.m. *, significant difference when compared to mock-transfected cells within each group (P < 0.05) by t-test. (d) Immunocytochemistry performed at different days during the course of reprogramming with 4Y or 3Y ± hsa-miR-372. Representative portions of the well are shown in each image. N = 2. Scale bars, 25 μm.
The finding that miR-302b and miR-372 inhibited TGFBR2 and RHOC expression, and to a lesser extent, the expression of SMAD2, INHBB, GAPs and GEFs (Fig. 2a) is intriguing as these signaling mediators regulate the choice between epithelial and mesenchymal cell fates28. Recent work has shown that mouse fibroblasts undergo a mesenchymal-epithelial transition (MET) early in the process of reprogramming29,30. Therefore, we hypothesized that miR-302b and miR-372 may increase reprogramming efficiency by accelerating MET. To test this hypothesis, we followed marker expression during reprogramming with or without the addition of miR-302b and miR-372. RT-qPCR was performed on RNA isolated 7 d after transduction (4 d after transfection of miRNAs) of human fibroblasts. The addition of miR-302b and/ or miR-372 decreased the levels of the mesenchymal markers ZEB1 and FN1, independent of the introduction of reprogramming factors, and consistent with the presence of seed matches in their ORF or 3′UTR (Fig. 3c, Supplementary Fig. 6 and Supplementary Table 1). ZEB2 expression was suppressed by the miRNAs only in the context of reprogramming and SLUG showed a decrease with miR-372 during reprogramming. The transcription factors ZEB1, ZEB2 and SLUG are not only markers of the mesenchymal fate but also directly inhibit the epithelial fate in part by inhibiting the expression of E-cadherin31. Indeed, E-cadherin (CDH1) expression and, to a lesser extent, the expression of the epithelial markers, occludin (OCLN) and EP-CAM, were upregulated at day 7 of reprogramming with the addition of miRNAs (Fig. 3c and Supplementary Fig. 6). In contrast, the introduction of miRNAs alone into uninfected fibroblasts was unable to induce expression of the epithelial markers (Fig. 3c and Supplementary Fig. 6).
To more closely follow the kinetics of MET with and without the introduction of the miRNAs, we examined the appearance of epithelial markers by immunoblotting and immunocytochemistry at different time points during reprogramming. Immunoblotting clearly showed earlier and stronger expression of E-cadherin in the presence of miR-372 (Supplementary Fig. 7a). At day 10, E-cadherin was robustly expressed in 4Y-infected cells plus miR-372 but barely visible in 4Y alone or 3Y plus miR-372. At day 18, E-cadherin expression was higher in 4Y cells plus miR-372, relative to 4Y alone, and could be seen in 3Y cells only in the presence of miR-372. Similar enhancement in E-cadherin levels was also observed upon treatment of cells with either ROCKi (Supplementary Fig. 7b) or RepSox (Supplementary Fig. 7c). Immunocytochemistry was performed to detect the expression of the epithelial markers JAM-1 and E-cadherin (Fig. 3d), and the organization of F-actin, whose distribution changes dramatically during MET (Supplementary Fig. 8). Cells transduced with 4Y alone began expressing JAM-1 at day 8 and E-cadherin at day 15 after infection. When supplemented with miR-372, the expression of JAM-1 and E-cadherin was accelerated to days 5 and 8, respectively. Similar to 4Y infection of cells, miR-372 accelerated the initiation of JAM-1 expression from day 15 to day 10 in 3Y-infected cells. E-cadherin expression was not detected in this time course with 3Y-infected cells but was robust at day 18 with the addition of miR-372 (Fig. 3d). These changes coincided with the relocalization of F-actin in the cells. That is, the actin filaments acquired a cortical organization during dedifferentiation, which occurred earlier in 3Y with addition of the miRNAs (Supplementary Fig. 8). Importantly, the appearance of E-cadherin expression coincided with the emergence of completely reprogrammed colonies in the presence of the miRNAs (Supplementary Fig. 9). Together, these results support a role for the occurrence of MET during the course of reprogramming of human cells, similar to what was described for mouse cells29,30, and illustrate that the ESCC miRNAs increase the efficiency of reprogramming in part by inducing more efficient MET.
We next investigated whether these miRNAs are general stabilizers of the epithelial cell fate. TGF-β can induce an EMT in epithelial cells28. We therefore asked whether miR-302b and miR-372 could inhibit TGF-β–induced EMT in human keratinocyte HaCaT cells. HaCaT cells were treated with TGF-β 2 d after transfection with miRNA mimics. miR-302b/372 decreased TβRII levels without affecting TβRI levels in HaCaT cells (Fig. 4a). Moreover, the ESCC miRNAs inhibited TGF-β–induced SMAD2 and SMAD3 phosphorylation in response to TGF-β, compared to control untreated cells (Fig. 4a). Without TGF-β treatment, the expression of miR-302b/372 did not affect the epithelial morphology of HaCaT cells, as observed by phase contrast microscopy (Fig. 4b), cortical actin organization and the presence of E-cadherin and ZO-1 at cell-cell junctions (Fig. 4c). After 72 h of TGF-β treatment, HaCaT cells underwent EMT with dramatic morphological changes accompanied by cell individualization (Fig. 4b), actin reorganization into stress fibers, and loss of E-cadherin and ZO-1 expression (Fig. 4c). In contrast, expression of miR-302b and miR-372 substantially inhibited TGF-β–induced EMT in HaCaT cells. The cells failed to spread and maintained their cell-cell contacts resembling an epithelial phenotype (Fig. 4b). In addition, E-cadherin and ZO-1 remained localized at the cell-cell junctions, as revealed by immunofluorescence (Fig. 4c). Confirming these observations, E-cadherin expression was higher in miR-302b/372-transfected cells compared to control cells after 48 h of TGF-β treatment (Fig. 4d). The expression of the transcriptional repressor SLUG is normally upregulated in TGF-β–induced EMT, repressing CDH1 transcription28. Its expression was reduced in HaCaT cells transfected with miR-302b/372, compared to control cells after 24 h of TGF-β treatment (Fig. 4e). Notably, each of these effects on EMT was completely abrogated when the seed sequence was mutated in the mimics (Supplementary Fig. 10). These results demonstrate that miR-302b and miR-372 are potent inhibitors of TGF-β–induced EMT as well as promoters of MET during reprogramming.
Figure 4.

Hsa-miR-302b, hsa-miR-372 and mmu-miR-294 inhibit TGF-β–induced epithelial-mesenchymal transition in human cells. (a) Western blot showing levels of TGF-β receptors, phospho-SMAD2 and phospho-SMAD3 in HaCaT cells 0–60 min after TGF-β exposure in the presence of miRNA mimics. Cells were transfected with the indicated miRNAs 48 h before TGF-β treatment. miR-294m, mmu-miR-294 seed mutant mimic. Representative blot of N= 2. (b,c) HaCaT cells were transfected with the indicated miRNAs, then treated or not with TGF-β for 72 h and observed by phase contrast microscopy (b), or fixed and subjected to immunostaining for F-actin, E-cadherin and ZO-1 (c). N = 2. Scale bars, 100 μm (b), 20 μm (c). (d) HaCaT cells were transfected with the indicated miRNAs, then treated with TGF-β for 48 h before lysis and immunoblotting with the indicated antibodies. N= 2. ( e) HaCaT cells were transfected with the indicated miRNAs treated or not with TGF-β for 24 h, before RNA was extracted and analyzed by RT-qPCR. Expression was normalized to RPl19. Representative graph of two independent experiments is shown. Error bars represent mean ± s.e.m.
To determine whether these effects were conserved among the ESCC miRNAs, we included the mimic for the mouse ESCC homolog mmu-miR-294, which contains the same seed sequence as hsa-miR-302b/372 (Fig. 4a). The mmu-miR-294 mimic yielded similar results to those of the human ESCC miRNAs, and these effects were abrogated by disrupting the seed sequence, indicating a highly conserved and seed-dependent role of ESCC miRNAs in regulating EMT.
Taken together, our findings show that the ESCC miRNAs enhance the efficiency of reprogramming to induced pluripotency by targeting multiple mRNAs enriched in various important cellular processes. Others have suggested that miR-302a-d alone can promote dedifferentiation of cancer cells and human hair follicle cells to an iPSC-like state18,32. The ESCC miRNAs regulate the unique cell cycle program of mouse ESCs by targeting inhibitors of the G1/S transition, including p21 (CdkN1A), Lats2 and Rbl2 (ref. 3), and have analogous effects on the human ESC cell cycle2. Accordingly, these miRNAs are expected to enhance reprogramming through their effects on the cell cycle. Indeed, knockdown of p21 during reprogramming has been shown to enhance induced pluripotency33–36. Here we show that RBL2 knockdown also enhances reprogramming. However, because the ESCC miRNAs have hundreds of target genes16, we tested a larger subset of these targets in reprogramming. We show that, in addition to their effects on cell cycle regulators, these miRNAs also target genes involved in epigenetic regulation, vesicular transport and MET during reprogramming, and that downregulation of many of these target genes increases the number of iPSC colonies produced. Follow-up experiments confirmed that the ESCC miRNAs function in part through MET by targeting at least TGFBR2 and RHOC, but also likely SMAD2, ZEB1 and FN1 to enhance reprogramming. Future experiments are likely to confirm the role of additional pathways downstream of these miRNAs. Indeed, our results would argue that miRNAs could be used as a powerful means to uncover many if not all the pathways important for the induction of pluripotency or any cell fate transition of interest.
ONLINE METHODS
Reprogramming
Human foreskin fibroblasts (BJ) and fetal lung fibroblasts (MRC-5) were cultured in DMEM and 10% FBS supplemented with nonessential amino acids, L-glutamine, penicillin/streptomycin and beta-mercaptoethanol (BJ medium). We plated 15,000 cells on 0.2% gelatin in 12-well plates. The following day, cells were infected with amphotropic retroviruses encoding OCT3/4, SOX2, KLF4, cMYC and Venus (gift from K. Eggan and packaged by Harvard Gene Therapy Initiative). cMYC retrovirus was added at one-tenth the concentration of the other viruses. Infections were done in 0.5 ml human ESC medium (knockout DMEM containing 15% knockout serum replacement; 10 ng/ml bFGF and supplemented with nonessential amino acids, L-glutamine and beta-mercaptoethanol). The following day, 0.5 ml BJ medium was added to the wells and the medium was changed completely to human ES medium 24 h later. Cells were transfected with miRNA mimics at a final concentration of 50 nM using Dharmafect1 (ThermoFisher) following manufacturer’s protocol on days 3 and 10. All mimics were purchased from ThermoFisher. Medium was changed every 24 h and colonies with human ESC-like morphology were counted at 21 d (in the case of reprogramming using 4Y) or at 31 d (for 3Y reprogramming). Colonies were also examined for prevalence of Venus fluorescence, which was used as a marker for silencing of exogenous factors. For picking and expansion, colonies were picked onto irradiated mouse embryonic fibroblasts and were passaged manually, until stable lines were obtained. ROCK inhibitor (10 μM final concentration, Y-27632 from EMD Biosciences) or TGF-β1 (2 ng/ml) was added to cells starting on day 3 after infection; the TβRI inhibitor (25 μM final concentration, RepSox from Sigma) was added to cells from days 6–10 after infection. For overexpression of TβRII, the ORF was cloned downstream of the EF1α promoter in pSin-IRES-Pac (puromycin resistance)3, packaged into lentiviruses followed by infection of fibroblasts on day 3.
HaCaT cell culture and EMT
HaCaT cells were cultured in DMEM with glucose (4.5 g/l) and 10% FBS. Cells were plated at 100,000 cells per well of a 6-well plate and transfected the next day with miRNA mimics (ThermoFisher) using Dharmafect 1 (ThermoFisher) according to manufacturer’s protocol. Mimics were transfected at a final concentration of 40 nM. On day 2 after transfection, cells were analyzed as follows. For signaling pathway protein quantification, cells were starved of serum with 0.5% FBS overnight, treated with 2 ng/ml of TGF-β1 (HumanZyme) for indicated times and lysed. For RNA quantification, cells were split, then treated with TGF-β1 for 24 h and lysed. For morphological and immunocytochemical analysis of EMT, cells were split into chamber slides, and treated with TGF-β1 for 72 h before fixing and imaging. To view the cell morphology or to monitor TGF-β–induced EMT, cells were observed using a Leica DMI 4000B microscope, and brightfield pictures were taken using a Leica DFC 350FX camera. Images were analyzed using the Leica Application Suite and Photoshop (Adobe) software. The TβRI kinase inhibitor SB431542 (Sigma-Aldrich) was used at 5 μM in conditions without TGF-β treatment to inhibit secreted autocrine TGF-β.
Immunocytochemistry
For hiPSC and hESC, cells were fixed with 4% paraformaldehyde for 10 min at room temperature, followed by blocking and permeabilization using 5% BSA in PBS containing 0.2% Tween 20 for 1–2 h. Cells were incubated overnight at 4 °C with Rhodamine-Phalloidin (Invitrogen); E-Cadherin (BD-Transduction Laboratories) and JAM-1 antibodies (Santa Cruz Biotechnology) and were stained with rhodamine-conjugated secondary antibodies. Cells were imaged using an inverted Leica fluorescent microscope. HaCaT cells were fixed with 4% PFA for 30 min, permeabilized in 2% PFA and 0.2% Triton X-100 for 10 min, and incubated in PBS and 3% BSA blocking solution for 1 h. The slides were incubated for 2 h with E-cadherin (BD Biosciences) and ZO-1 (Invitrogen) antibodies in PBS and 3% BSA, and were stained for 1 h with AlexaFluor-conjugated secondary antibodies and rhodamine-conjugated phalloidin (Invitrogen) to visualize actin filaments. The slides were then mounted using ProLong Gold antifade reagent with DAPI (Invitrogen). Images were viewed by epifluorescence imaging and acquired using a Nikon Ti inverted microscope with 60×/1.49 Apo TIRF objectives and Coolsnap HQ2 camera (Photometrics) controlled by NIS Elements software (Nikon).
RNA extraction and qPCR
RNA for most qRT-PCR analysis was prepared using Trizol (Invitrogen), following the manufacturer’s protocol and quantified on a Nanodrop Spectrophotometer (ThermoFisher). We treated 500 ng of RNA with DNase using DNaseI amplification grade (Invitrogen). For qRT-PCR of mRNAs, DNase-treated samples were reverse transcribed using the Superscript III first-strand synthesis system for RT-PCR (Invitrogen). qPCR reactions on resulting cDNAs were performed using Power SYBR Green Master Mix (Applied Biosystems) on either an ABI Prism 7100 or ABI 7900HT (Applied Biosystems) and data was normalized to GAPDH expression. For HaCaT cells, RNA was extracted using the RNeasy mini kit (Qiagen), following the manufacturer’s instructions. The iScript cDNA synthesis kit (Bio-Rad Laboratories) was used to generate cDNA for real-time PCR of Slug and RPL19 for normalization using iQ SYBR Green supermix (Bio-Rad Laboratories), following the manufacturer’s instructions on the CFX96 Real-Time PCR detection system (Bio-Rad Laboratories). CFX Manager software (Bio-Rad Laboratories) was used for the quantification. PCR primers are listed in Supplementary Table 2.
Western blotting
For protein quantification during reprogramming, cells were lysed on days 10 and 18 after infection in lysis buffer (50 mM Tris pH 8.0, 120 mM NaCl, 0.5% NP-40, supplemented with a commercially available mix of protease and phosphatase inhibitors). Protein lysates were separated on a 10% SDS-PAGE and were blotted onto polyvinylidene fluoride (PVDF) membrane using the Bio-Rad electrophoresis unit. Blots were blocked using LI-COR blocking buffer for 1–2 h followed by incubation with the primary and secondary antibodies diluted at the appropriate concentrations in the blocking buffer. Blots were imaged using the LI-COR Odyssey scanner. Quantification of blots was performed using the LI-COR imaging software. Anti-E-cadherin (BD-Transduction Laboratories) was used at a concentration of 1:1,000, and anti-beta-actin (Sigma, A4700) was used at 1:10,000.
For protein quantification of TGF-β signaling proteins in HaCaTs, cells were lysed in radioimmunoprecipitation (RIPA) buffer with protease and phosphatase inhibitors at noted time points after exposure to ligand. Protein concentration was determined using protein assay (Bio-Rad Laboratories), and 10 to 20 μg of proteins was separated by SDS-PAGE and transferred to 0.2 mm nitrocellulose membrane. Membranes were blocked in TBS, 0.1% Tween 20 and 5% dry milk for 1 h before overnight incubation with primary antibody diluted in TBS, 0.1% Tween 20 and 3% BSA. Antibodies used were specific for TβRI, TβRII, GAPDH (Santa Cruz Biotechnology), phospho-Smad2, phospho-Smad3, Smad2, Smad3 or E-cadherin (Cell Signaling Technology). The membranes were incubated with HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) diluted in TBS, 0.1% Tween 20. Immunoreactive proteins were detected using ECL (GE Healthcare) and BioMax film (Kodak).
Luciferase reporter assays
The 3′UTRs of TGFBR2 and RhoC were amplified from the genomic DNA of human foreskin (BJ) fibroblast cells using the TOPO (Invitrogen) vector and subcloned into the NotI and XhoI restriction sites of the psiCheck-2 vector (Promega). Mutant variants of the 3′UTR seed sequences were produced using the Quickchange XL kit (Agilent). For transfection, 8,000 miRNA-deficient Dgcr8−/− mouse ESCs were plated in ESC media onto a 96-well plate pretreated with 0.2% gelatin. The subsequent day, the cells were transfected with miRIDIAN miRNA mimics (Dharmacon, ThermoFisher) with Dharmafect1 (Dharmacon, ThermoFisher) at the manufacturer’s recommended concentration of 100 nM. Simultaneously, 200 ng of the psiCheck-2 constructs were transfected into the ESCs using Fugene6 (Roche) transfection reagent according to the manufacturer’s protocol. Transfection of each construct was performed in duplicate in each assay. The cells were lysed 18–20 h after transfection, and the luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega) on a dual-injecting SpectraMax L (Molecular Devices) luminometer according to the manufacturer’s protocol. Ratios of Renilla luciferase readings to firefly luciferase readings were averaged for each experiment. Replicates performed on separate days were median centered with the readings from the individual days.
Supplementary Material
Acknowledgments
We would like to thank A. Amiet at ThermoFisher for advising and kindly sharing miRNA and siRNA reagents, R. Shaw for access to fluorescence microscopy, and M. Yumoto for primers. This work was supported by funds to R.B. from Leona M. and Harry B. Helmsley Charitable Trust, US National Institutes of Health (K08 NS48118 and R01 NS057221), California Institute of Regenerative Medicine (CIRM) (Seed Grant RS1-00161, New Faculty Award RN2-00906), grant RO1-CA136690 to R.D., an American Heart Association scientist development award (no. 09SDG2280008) to S.L. and SFSU CIRM Bridges TB1-01194 to J.Y.L. Venus was a gift from K. Eggan, packaged by Harvard Gene Therapy Initiative.
Footnotes
Note: Supplementary information is available on the Nature Biotechnology website.
AUTHOR CONTRIBUTIONS
D.S. did the experiments described in Figures 1, 2 and 3. S.L. and R.L.J. did experiments described in Figure 4. J.Y.L. helped with experiments described in Figure 2a and performed experiments described in 3b. N.B. helped with experiments in Figure 1b,c. D.S. and R.B. wrote the manuscript with help from S.L., R.L.J. and R.D.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Judson RL, Babiarz JE, Venere M, Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat Biotechnol. 2009;27:459–461. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qi J, et al. microRNAs regulate human embryonic stem cell division. Cell Cycle. 2009;8:3729–3741. doi: 10.4161/cc.8.22.10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, et al. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–523. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez F, Boue S, Belmonte JC. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011;12:231–242. doi: 10.1038/nrg2937. [DOI] [PubMed] [Google Scholar]
- 6.Li W, Ding S. Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol Sci. 2010;31:36–45. doi: 10.1016/j.tips.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Yang CS, Nakashima K, Rana TM. Small RNA-mediated regulation of iPS cell generation. EMBO J. 2011;30:823–834. doi: 10.1038/emboj.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bar M, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Card DA, et al. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin RD, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suh MR, et al. Human embryonic stem cells express a unique set of microRNAs. Dev Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Brambrink T, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan EM, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- 15.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol. 2010;17:1169–1174. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 16.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benetti R, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:998. doi: 10.1038/nsmb0908-998b. [DOI] [PubMed] [Google Scholar]
- 18.Lin SL, et al. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res. 2011;39:1054–1065. doi: 10.1093/nar/gkq850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinkkonen L, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–267. doi: 10.1038/nsmb.1391. [DOI] [PubMed] [Google Scholar]
- 20.Ichida JK, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin T, et al. A chemical platform for improved induction of human iPSCs. Nat Methods. 2009;6:805–808. doi: 10.1038/nmeth.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maherali N, Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 25.Ohgushi M, et al. Molecular pathway and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell. 2010;7:225–239. doi: 10.1016/j.stem.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Chen G, Hou Z, Gulbranson DR, Thomson JA. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7:240–248. doi: 10.1016/j.stem.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosa A, Spagnoli FM, Brivanlou AH. The miR-430/427/302 family controls mesendodermal fate specification via species-specific target selection. Dev Cell. 2009;16:517–527. doi: 10.1016/j.devcel.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li R, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Samavarchi-Tehrani P, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Lin SL, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA. 2008;14:2115–2124. doi: 10.1261/rna.1162708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banito A, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–2139. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong H, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawamura T, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.