

Abstract
Background
Cystic fibrosis (CF) is a monogenic disease caused by CFTR gene mutations, with clinical expression similar to complex disease, influenced by genetic and environmental factors. Among the possible modifier genes, those associated to metabolic pathways of glutathione (GSH) have been considered as potential modulators of CF clinical severity. In this way it is of pivotal importance investigate gene polymorphisms at Glutamate-Cysteine Ligase, Catalytic Subunit (GCLC), Glutathione S-transferase Mu 1 (GSTM1), Glutathione S-transferase Theta 1 (GSTT1), and Glutathione S-transferase P1 (GSTP1), which have been associated to the GSH metabolic pathway and CF clinical severity.
Method
A total of 180 CF’s patients were included in this study, which investigated polymorphisms in GCLC and GST genes (GCLC -129C>T and -3506A>G; GSTM1 and GSTT1 genes deletion, and GSTP1*+313A>G) by PCR and PCR-RFLP associating to clinical variables of CF severity, including variables of sex, clinical scores [Shwachman-Kulczycki, Kanga e Bhalla (BS)], body mass index, patient age, age for diagnosis, first clinical symptoms, first colonization by Pseudomonas aeruginosa, sputum’s microorganisms, hemoglobin oxygen saturation in the blood, spirometry and comorbidities. The CFTR genotype was investigated in all patients, and the genetic interaction was performed using MDR2.0 and MDRPT0.4.7 software.
Results
The analysis of multiple genes in metabolic pathways in diseases with variable clinical expression, as CF disease, enables understanding of phenotypic diversity. Our data show evidence of interaction between the GSTM1 and GSTT1 genes deletion, and GSTP1*+313A>G polymorphism with CFTR gene mutation classes, and BS (Balance testing accuracy= 0.6824, p= 0.008), which measures the commitment of bronchopulmonary segments by tomography.
Conclusion
Polymorphisms in genes associated with metabolism of GSH act on the CF’s severity.
Background
Cystic Fibrosis (CF) is a common disease in Caucasian population, with a prevalence of 1:2,500 live births [1]. CF shows an autosomal recessive pattern of inheritance [2], which is caused by mutations in Cystic Fibrosis Transmembrane Regulator (CFTR) gene. More than 1,900 mutations in CFTR gene have been associated to CF clinical severity [3-6]. However, in the pulmonary disease, which is the main cause of CF morbidity and mortality, several studies have demonstrated that clinical variability is influenced by modifier genes and environmental factors [2,7].
Most of modifier genes related to CF have been associated with chloride transportation, infection and inflammation in the lungs [2,7-10]. In our group, as previously published, multiple genes are associated with CF clinical severity, including Transforming growth factor beta 1 (TGF-β1) [11], Glutathione S-transferase Mu 1 (GSTM1), Glutathione S-transferase Theta 1 (GSTT1) [12], Angiotensin-converting enzyme (ACE) [13] and Beta-2-Adrenergic Receptor (ADBR2) [14] genes as possibly modifier genes. In the same population the mannose-binding lectin (protein C) 2 (MBL2) and monocyte differentiation antigen CD14 (CD14) genes were not associated with the CF clinical severity [11].
Glutathione (GSH) is a tripeptide involved in the intracellular defense system, which protect the epithelium against injuries and inflammation, both common events of CF [15]. Polymorphisms in regulator genes of GSH metabolic pathway have been described in CF, including Glutathione S-transferase P1 (GSTP1), GSTT1, GSTM1 and Glutamate-Cysteine Ligase, Catalytic Subunit (GCLC) genes, which have been associated with greater CF clinical severity [7,12,16-23]. In the other hand, GCLC [129C>T and -3506A>G] polymorphisms gene has not been yet evaluated for pulmonary disease variability in CF.
GCLC gene encodes the catalytic subunit of glutamate-cysteine ligase (GCL) [24], which is the first limiting enzyme in the GSH synthesis [25]. Polymorphisms -129C>T and -3506A>G, located in GCLC gene promoter region, have been associated to reduced GSH production [15,25].
The Glutathione S-transferase (GST) is a family of enzymes, which associates GSH and causative compounds of oxidative stress, to a wide variety of endogenous (e.g. by-products of reactive oxygen species activity) and exogenous (e.g. polycyclic aromatic hydrocarbons) electrophilic substrates [26]. Polymorphisms in GST family genes can also be involved in CF’s severity [15,26], especially in pulmonary disease, including GSTM1, GSTT1 and GSTP1 genes [15,19,20,26-28].
In our referral center, we have observed clinical variability among CF’s patients. However, the patients present similar socioeconomic status, none severe malnutrition, mutations of classes I, II and/or III, similar support of the Parents Association (http://www.fibrocis.org.br), and receive free medication by the government. Therefore, we postulate that clinical variability could be associated with interaction of polymorphisms in modifier genes. In this context, we aim to analyze polymorphisms in GCLC and GST genes (GCLC -129C>T and -3506A>G; GSTM1 and GSTT1 genes deletion, and GSTP1*+313A>G) associating to clinical markers of CF severity.
Methods
Ascertainment of patients
It was conducted a cross-sectional study and patients were selected during the period of 2010 and 2011, in a university center for CF patients care. This study was approved by Ethics Research Board of the Faculty of Medical Sciences at University of Campinas - São Paulo – Brazil (#528/2008). The study was in accordance to the Helsinki declaration and all patients signed a consent form before beginning the study. For patients below the age of 18 the consent was granted by their parents or guardians.
Diagnosis of CF was confirmed in patients with two doses of sodium and chloride from the sweat with values greater than 60 mEq/L. In a group of patients we identified two CFTR gene mutations. No patient had received neonatal screening test performed for CF. A total of 215 patients were selected for the study. Among them, 35 patients without clinical data for statistical analysis and those who did not sign the consent form were excluded from the study, and 180 CF patients were included at the study.
Clinical variables
The following clinical variables were employed: clinical scores [Shwachman-Kulczycki (two groups: ≤ 65 and > 65), Kanga (two groups: ≤ 17 and > 17) and Bhalla (BS) (two groups: ≤ 8 and > 8)] [29]; body mass index (BMI) [for the patients older than 19 years the BMI= weight/(height)2 formula was used; for the remaining patients the WHO ANTHRO programs (children under 5 years old) and WHO ANTHRO PLUS (children 5 - under 19 years old) [30,31] were used; patient age (group: ≤ 154 and > 154 months) and age at diagnosis (according to the sodium and chloride in altered perspiration: ≤ 25 and > 25 months); first clinical symptoms [(digestive: ≤ 4 and > 4 months; pulmonary: ≤ 7 and > 7months)]; the period up to first colonization by Pseudomonas aeruginosa (≤ 31 and > 31 months); sputum’s microorganisms [P. aeruginosa mucoid and non mucoid, Achromobacter xylosoxidans, Burkolderia cepacia and Staphylococcus aureus]; hemoglobin oxygen saturation in the blood (≤ 96 and > 96); spirometry and comorbidities: nasal polyps, osteoporosis, meconium ileus, diabetes mellitus and pancreatic insufficiency (PI).
The clinical score evaluation was performed by two specialists in double-blind analyses, and in discordance case, another one was consulted. The Shwachman-Kulczycki score compares clinical manifestations among patients, detect treatment effects and aid in the determination of diagnostic criteria. To that end, the system evaluates 4 major parameters: general activity, nutrition, chest radiographic findings and physical examination. The score for each parameter ranges from 5 to 25. Lower score’s values are associated with CF severity [29]. The BS is a tomographic scoring system to assess pulmonary involvement, determine therapeutic effects and aid selection of patients for transplants, with small variation between the scores given by the various examiners and the score proved to have good reproducibility and high correlation with pulmonary function test results. A total of 9 categories, worth 3 points each, are scored, and a maximal score equals a high degree of severity. The final score must be subtracted from 25. Lower score’s values are associated with CF severity [29]. Kanga score is a system designed to assess acute exacerbations of the disease, to predict improvement or worsening of pulmonary function and to evaluate therapeutic effects with little variability between examiners and correlated significantly with pulmonary function (FEV1 and forced vital capacity) test results. The system helps identify daily clinical changes and includes 5 common symptoms (cough, fluid secretion, appetite loss, dyspnea and frailty) and 5 physical signs (temperature, weight, respiratory frequency, wheezing and respiratory sounds). Each criterion is worth 1 to 5 points. Higher score’s values are associated with CF severity [29].
Spirometry was performed in patients older than 7 years, using the CPFS/D spirometer (MedGraphics, Saint Paul, Minnesota, USA). Data was recorded by the PF BREEZE software version 3.8B for Windows 95/98/NT [32] and the following markers were included: forced vital capacity [FVC(%) - ≤ 82 and > 82], forced expiratory volume in the first second [FEV1(%) - ≤ 72 and > 72], FEV1/FVC(%) - ≤ 86 and > 86] and forced expiratory flow between 25 and 75% of the FVC [FEF25-75% - ≤ 57 and > 57].
DNA extraction and polymorphisms genotyping
DNA sample was obtained from peripheral blood using phenol-chloroform standard procedure. In order to evaluate DNA concentration, we quantified the entire sample using GE NanoVue™ Spectrophotometer (GE Healthcare Biosciences, Pittsburgh, USA).
The CFTR gene mutations were investigated by PCR technique (F508del) and the restriction fragment length polymorphism (RFLP) method (G542X, R1162X, R553X, G551D and N1303K). Some mutations in CF patients were obtained by sequencing or MLPA (Multiplex Ligation - dependent Probe Amplification) analysis: S4X, 2183A>G, 1717-G>A and I618T. For sequencing and MLPA, we used the same MegaBace1000® (GE Healthcare Biosciences, Pittsburgh, USA) [33]. The CFTR genotype separated the patients in 3 groups: (1) patients with two mutations identified classes I, II and/or III, (2) patients with one mutations identified classes I, II or III, (3) patients with no identified mutation. All mutations identified were included in the class one, two or three of the CFTR gene. Other identified mutations, class IV (P205S e R334W) were not included in the statistical analysis.
For the GSTM1 and GSTT1 genes deletion, a multiplex PCR reaction was performed and the CYP1A1 gene was included as an internal reaction control [34]. In the assay tests for the wild type allele, the samples for which no signal is obtained are homozygous for the deletion. Heterozygotes and homozygotes for the wild-type allele cannot be differentiated by the multiplex technique used. As GSTM1 and GSTT1 deletion analyze not provide complete genotypes, minor allele frequencies and Hardy-Weinberg Equilibrium cannot be evaluated for these loci in our study. By the method realized we have two groups of GSTM1 and GSTT1 gene: (i) patients homozygous to gene deletion; (2) patients with at least an allele expressed. The RFLP was performed for polymorphisms GCLC*-129C>T, GCLC*-3506A>G [10,15] and GSTP1*+313A>G [28].
Other genes were studied in the same CF reference center with same population as previously published [11,13,14]. The GSTM1 and GSTT1 genes were studied in our center considering other CF patients as recent published data showed [12]. The patient’s studied by Lima et al. (2012) [12] did not include the same CF patients, and in the previously study, only the follow clinical variables were employed: age, sex, ethnic and Shwachman-Kulczycki.
Statistical analysis
To develop statistical analyses, patients were divided into two subgroups according to clinical variables distribution. We evaluate minor allele frequency (MAF) and Hardy-Weinberg equilibrium (HWE) using OEGE software (Online Encyclopedia for Genetic Epidemiology studies, 2008). In order to evaluate genetic interaction among the polymorphisms in our sample, we used the Multifactor Dimensionality Reduction (MDR) model, which is a nonparametric and genetic model-free data mining for nonlinear interaction identification among genetic and environmental attributes [35-37]. To adjust results for multiple comparisons, we performed a MDR permutation test in our sample, totalizing 100,000 permutations.
Results and discussion
The CF is a disease with a high phenotypic variability, resulting from the interaction between genetic and environmental factors, which could affect differential expression between patients with similar CFTR gene mutations [2]. Despite CF’s severity is difficult to characterize, we describe 28 clinical variables (Table 1), consistent with the phenotype feature of the disease.
Table 1.
Clinical features of cystic fibrosis patients included in the study
| Sex (male) |
50% (80)# |
| Age |
17.72 ± 15.75 years (0.6 – 24 years)* |
| BMI - thinness and accentuated thinness |
22.22% (40)# |
| One Class I. II or III identified mutation |
28.33% (51)# |
| Two Class I. II or III identified mutation |
47.22% (85)# |
| First clinical manifestation |
2.90 ± 8.89 years (0 – 13 years)* |
| Age at diagnosis |
7.62 ± 13.63 years (0 – 14.23 years)* |
| Onset of digestive symptoms |
3.39 ± 9.11 years (0 – 12.45 years)* |
| Onset of pulmonary symptoms |
2.90 ± 9.89 years (0 – 13 years)* |
| SpO2 |
94.92 ± 4.26 (66 – 99)* |
| Bhalla score |
8.74 ± 5.724 (0 – 25)* |
| Kanga score |
18.85 ± 5.84 (10 – 40)* |
| Shwachman-Kulczycki score |
65.85 ± 16.77 (20 – 95)* |
| FVC (%) |
79.29 ± 23.55 (19 – 135)* |
| FEV1 (%) |
71.29 ± 27.47 (17 – 132)* |
| FEV1/FVC (%) |
83.46 ± 15.95 (37 – 137)* |
| FEF25-75% |
59.05 ± 35.55 (7 – 150)* |
| Nasal Polyps |
18.33% (33)# |
| Diabetes mellitus |
18.33% (33)# |
| Osteoporosis |
16.11% (29)# |
| Pancreatic insufficiency |
80.0% (144)# |
| Meconium ileus |
15.00% (27)# |
| First isolated P. aeruginosa |
8.55 ± 14.45 years (2 – 15 years) |
|
P. aeruginosa status |
56.67% (102)# |
|
P. aeruginosa mucoid status |
42.22% (76)# |
|
B. cepacia status |
13.88% (25)# |
|
A. xylosoxidans status |
10.00% (18)# |
| S. aureus status | 78.88% (142)# |
BMI body mass index, SpO2 Hemoglobin oxygen saturation in the blood, % percentage, FVC forced vital capacity, FEV1 forced expiratory volume in the first second, FEF25-75 forced expiratory flow between 25 and 75% of FVC. 2. Based on 3 Consecutive positive respiratory cultures.
# Percentage (Number of patients).
* Continuous variables expressed as mean ± SD (range).
Analyses of GCLC, GSTM1, GSTT1, and GSTP1 genes polymorphism is shown in Table 2. The CFTR mutation showed two different mutations in 85 (47.22%) CF patients; only one mutation in 51 (28.33%) CF patients and none mutation in 44 (24.44%) CF patients (Table 2).
Table 2.
Genotypic characteristic of gene polymorphisms at GCLC, GSTM1, GSTT1, and GSTP1 genes and CFTR gene mutation among cystic fibrosis patients
| Gene | Chromosome position | Location | Variation | Genotype | MAF | p* | ||
|---|---|---|---|---|---|---|---|---|
| |
|
|
|
C/C |
C/T |
T/T |
|
|
|
GCLC, rs17883901 |
6p12 |
Promoter region |
C/T |
144 (80%) |
29 (16.11%) |
7 (3.89%) |
0.12 |
<0.005 1 |
| |
|
|
|
A/A |
A/G |
G/G |
|
|
|
GCLC, rs137852340 |
6p12 |
Promoter region |
A/G |
118 (65.56%) |
56 (31.11%) |
6 (3.33%) |
0.19 |
>0.05 |
|
GSTP1, rs1695 |
11q13 |
Exon 5 |
A/G |
97 (53.89%) |
74 (41.11%) |
9 (5%) |
0.26 |
>0.05 |
| |
|
|
|
Wt/Wt + Wt/del |
del/del |
|
|
|
|
GSTM1 |
1p13.3 |
|
Deletion |
108 (60%) |
72 (40%) |
|
|
|
|
GSTT1 |
22q11.23 |
|
Deletion |
117 (65%) |
63(35%) |
|
|
|
|
CFTR mutation genoytpe |
N |
Frequency |
||||||
| F508del/F508del |
57 |
31.67% |
||||||
| F508del/G542X |
12 |
6.67% |
||||||
| F508del/R1162X |
5 |
2.78% |
||||||
| F508del/N1303K |
4 |
2.22% |
||||||
| F508del/R553X |
1 |
0.56% |
||||||
| F508del/S4X |
1 |
0.56% |
||||||
| F508del/1717-1G>A |
1 |
0.56% |
||||||
| G542X/R1162X |
1 |
0.56% |
||||||
| G542X/I618T |
1 |
0.56% |
||||||
| G542X/2183A>G |
1 |
0.56% |
||||||
| R1162X/R1162X |
1 |
0.56% |
||||||
| F508del/- |
45 |
25.00% |
||||||
| G542X/- |
5 |
2.78% |
||||||
| R1162X/- |
1 |
0.56% |
||||||
| −/− | 44 | 24.45% | ||||||
GCLC glutamate-cysteine ligase catalytic subunit, GSTM1 Glutathione S-transferase Mu 1, GSTT1 Glutathione S-transferase theta 1, GSTP1 Glutathione S-transferase P1, CFTR Cystic fibrosis transmembrane conductance regulator, C Cytosine, T Thymine, A Adenine, G Guanine, < minor than, > bigger than, MAF minor allele frequency, % percentage, *p value for Hardy-Weinberg Equilibrium, N number of patients, Wt Wild allele, del deleted allele, (−) CFTR mutation no identified. 1- GCLC, rs17883901 is not in Hardy-Weinberg Equilibrium in our sample.
The polymorphism GCLC-129C>T is not in Hardy-Weinberg equilibrium. The MAF observed in present data is 0.012, and the data achieved at National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), by HapMap project, shows a MAF of 0.076. The observed difference can be a strong factor to consider the polymorphism as an important modifier of CF severity, but we did not find in our data genotype-phenotype-association. Maybe, with a bigger population and other clinical variables can be possible to determine the polymorphism influence. Another factor should be considered that is the Brazilian population is an admixture population, what can provide changes in the clinical associations.
The MDR analysis showed evidence of interaction of GSTM1 and GSTT1 genes deletion, GSTP1*+313A>G, and CFTR mutations (p= 0.008) and BS clinical score (Table 3). All data was previously associated with a point-point analysis considering which gene polymorphism in association with one clinical manifestation at a time by CFTR groups, and after Bonferroni correction, we did not observed positives p-value (Additional file 1: Table S4, Additional file 2: Table S5, Additional file 3: Table S6, Additional file 4: Table S7, Additional file 5: Table S8). In this context, the data by gene-gene interaction provides a better tool to contribute in the clinical manifestation association. The association observed cannot identify if those patients who carry risk alleles at all four loci will have the most severe clinical phenotype reflected by the BS.
Table 3.
Genetic interaction of polymorphisms and CFTR mutations in association with cystic fibrosis clinical variables
| Clinical variables | Gene | Ratio | Testing ball. acc. | p-value | Cross validation consistency | |
|---|---|---|---|---|---|---|
| Male/female |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
1 |
0.5056 |
0.8506 |
10/10 |
|
| Race (caucasian/ no caucasian) |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.0909 |
0.3909 |
0.9915 |
10/10 |
|
| Age |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9889 |
0.5312 |
0.6870 |
10/10 |
|
| Age for diagnosis |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.8804 |
0.4100 |
0.9989 |
10/10 |
|
| Onset of symptoms |
geral |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.7526 |
0.4286 |
0.9945 |
10/10 |
| digestive |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9737 |
0.4614 |
0.9698 |
10/10 |
|
| pulmonary |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.7935 |
0.5022 |
0.8566 |
10/10 |
|
| Clinical scores |
Bhalla |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9552 |
0.6824 |
0.0080# |
10/10 |
| Kanga |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9571 |
0.5131 |
0.8165 |
10/10 |
|
| Shwachman-Kulczycki |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9 |
0.5562 |
0.5474 |
10/10 |
|
| Body mass index |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
3.45 |
0.5484 |
0.6260 |
10/10 |
|
| Sputum’s microbiology |
A. xylosoxidans |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.1118 |
0.4617 |
0.9418 |
10/10 |
|
P. aeruginosa mucoid |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.7379 |
0.5505 |
0.5507 |
10/10 |
|
|
P. aeruginosa no mucoid |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
1.2949 |
0.4151 |
0.9951 |
10/10 |
|
|
S. aureus |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
3.7105 |
0.5094 |
0.8317 |
6/10 |
|
|
B. cepacia |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.1548 |
0.5277 |
0.7610 |
10/10 |
|
| First isolated P. aeruginosa |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9552 |
0.4913 |
0.8903 |
7/10 |
|
| Hemoglobin oxygen saturation in the blood |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9602 |
0.5312 |
0.6904 |
10/10 |
|
| Spirometry |
FEV1 |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9848 |
0.4746 |
0.9448 |
10/10 |
| FVC(%) |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.900 |
0.4437 |
0.9833 |
10/10 |
|
| FEV1/FVC(%) |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9403 |
0.6426 |
0.0586 |
10/10 |
|
| FEF25-75% |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.9848 |
0.5054 |
0.8447 |
10/10 |
|
| Comorbidities |
Pancreatic insufficiency |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
3.977 |
0.5266 |
0.7486 |
10/10 |
| Meconium ileus |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.1776 |
0.5401 |
0.6950 |
10/10 |
|
| Nasal polyps |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.2292 |
0.5713 |
0.5139 |
10/10 |
|
| Diabetes mellitus |
CFTR*GCLC*GSTP1*GSTT1*GSTM1 |
0.2292 |
0.4527 |
0.9728 |
10/10 |
|
| Osteoporosis | CFTR*GCLC*GSTP1*GSTT1*GSTM1 | 0.1959 | 0.3815 | 0.9988 | 10/10 | |
GCLC glutamate-cysteine ligase catalytic subunit, GST glutathione S-transferase, Testing Ball. Acc. Testing Balance Accuracy, CFTRCystic Fibrosis Transmembrane Regulator, A. xylosoxidansAchromobacter xylosoxidans, B. cepaciaBurkolderia cepacia, P. aeruginosaPseudomonas aeruginosa, S. aureusStaplylococcus aures, FEV1 forced expiratory volume in one second, FVC - forced vital capacity, FEF25-75% forced expiratory flow between 25 and 75% of CVF. 1. Severe variants to polymorphism: allele T, GCLC 129C>T polymorphism, allele G - GCLC-350A>G and GSTP1+313A>G polymorphisms, deletion allele GSTM1 and GSTT1 genes. # Association of GCLC-129C>T, GCLC-350A>G, GSTP1+313A>G, GSTM1 and GSTT1 genes deletion and mutations in the CFTR gene with the Bhalla Score, p: 0.0080. Statistical analysis performed by the MDR2.0 and MDRPT0.4.7 software.
Genotype combinations of the genes that show association with BS clinical score are demonstrated in Figure 1. The largest association regarding to BS clinical response was given by the interaction of GSTM1 and GSTT1 genes deletion, and CFTR mutations. The polymorphism GSTP1*+313A>G presented entropy with BS clinical score, but with minor intensity (Figure 2).
Figure 1.
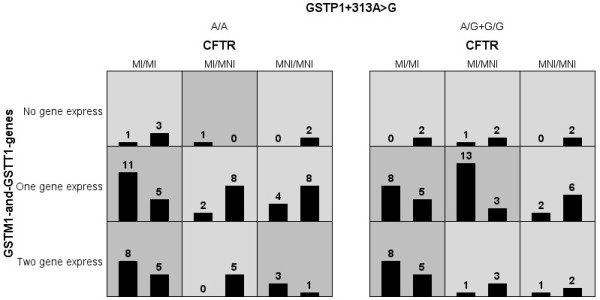
Distribution of patients according to different genotype combinations for the clustering of mutations in the CFTR gene and GCLC*-129C>T, GCLC*-350A>G, GSTP1*+313A>G, GSTM1 and GSTT1 genes deletion polymorphisms with the Bhalla score (BS). For the CFTR gene: (MI) Mutation-Identified and (MNI) Mutation-No-Identified to class I, II and/or III. Combinations of high risk are in gray and white to low-risk. In the figure only the polymorphisms with positive interaction with the BS are shown. The stronger association with the BS is observed by the genes with bigger interaction (GSTM1, GSTT1 and CFTR), and with the GSTP1*+313A>G polymorphism in a weak association. To data showed, the GCLC*-129C>T, GCLC*-350A>G polymorphisms not show association, and are not included in the figure. GST - glutathione S-transferase; CFTR - Cystic Fibrosis Transmembrane Regulator. The number in the figure represents the patients with genotype combination, for example, in the first square one patient (left column – severe BS) has the follow genotype: (i) two identified mutation in the CFTR gene, (ii) deletion of GSTM1 and GSTT1 in homozygous, and (iii) A/A genotype to GSTP1+313A>G polymorphism and three (right column- mild BS) patients without the combined genotype presence.
Figure 2.

Dendrogram and graph of entropy with the genes interaction and their polymorphisms in response to Bhalla score. A. Dendrogram with the genes interaction and their polymorphisms in response to Bhalla score (BS). The stronger interaction of response to Bhalla clinical score is given by CFTR mutations and GSTM1 and GSTT1 genes deletion. Secondly, the polymorphism GSTP1*+313A> G was associated with the clinical variable. Polymorphisms GCLC*-129C>T and GCLC*-350A>G were not associated with the BS. B. Graph of entropy measuring the power of different genotypes and the interaction between them, for the genes analyzed in the gene-gene interaction with BS. The CFTR mutation gene, GSTP1*+313A>G and GSTM1 and GSTT1 deletion polymorphisms showed significant association with BS. The CFTR mutation showed influence of 3.38% for BS, the GSTM1 and GSTT1 deletion together, showed 2.82% of influence, while the GSTP1*+313A>G showed influence of 0.5%. The CFTR mutation showed a value of 4.15% for the interaction between the deletion polymorphism with GSTM1 and GSTT1 genes and 3.57% for the association with GSTP1*+313A>G. The dendrogram construction was performed by the software MDR2.0. The analysis included 180 Cystic Fibrosis patients. # Association of GCLC-129C>T, GCLC-350A>G, GSTP1+313A>G, GSTM1 and GSTT1 genes deletion and CFTR mutations with the BS show a p-value of 0.0080. CFTR - Cystic Fibrosis Transmembrane Regulator; GCLC - glutamate-cysteine ligase, catalytic subunit; GSTP1 - glutathione S-transferase P1; GSTM1 - glutathione S-transferase M1; GSTT1 - glutathione S-transferase T1. The power of interaction is show by the distance between the two genes on the figure A in horizontal direction. The redundancy/synergy power is show in the figure B by the color intensity.
In the study, different clinical markers were analyzed and the association has found with BS clinical score. The BS clinical score is a computed tomography, which measures pulmonary involvement, therapeutic effects and selection of patients for transplantation, which detects anatomical changes of the lung parenchyma [29,38,39]. The BS has low variation between examiners, good reproducibility, high sensitivity and specificity, and high correlation with pulmonary function test [29]. Values obtained in the BS can predict severity associated with deterioration of the structure of the lung parenchyma, which later in clinical evolution can be observed by other variables such as BMI and lung function.
The most important cause of mortality and morbidity is the increase of oxidizing reactions; in this way, understand the mechanisms associated with this process is important to the clinical dynamics of the disease and to comprehend the mechanisms by which the disease shows a variability in its severity among patients with similar CFTR genotype. The association of clinical, laboratory, and genetic markers, such as modifier genes, with CF’s severity can provide better understanding of clinical variability finding among these patients being important in the clinical prognosis. In addition, further studies should be developed to investigate the possibility to use products related to GSH pathway as a possible target of new drugs to CF and also to understand the oxidative process which takes place in the lungs of these patients.
Conclusion
The described results with the support of the MDR analysis tool demonstrated a genetic interaction of CF severity with the BS, and gene polymorphisms related to GSH pathway, showing that the BS is an important marker of CF severity, since it indicates early pulmonary disease, and contribute to determine patients of a risk group without the search of other clinical and laboratory markers. In this context, polymorphisms in genes associated with metabolism of GSH act on the CF’s severity.
Abbreviations
ACE: Angiotensin-converting enzyme; ADRB2: Beta-2-adrenergic receptor; BMI: Body mass index; BS: Bhalla score; CD14: Cluster of differentiation 14; CF: Cystic fibrosis; CFTR: Cystic fibrosis transmembrane regulator; CYP1A1: Cytochrome P450, family 1, subfamily A, polypeptide 1; DNA: Deoxyribonucleic acid; FEF25-75%: Forced expiratory flow between 25% and 75% of FVC; FEV1: Forced expiratory volume; FVC: Forced vital capacity; GCLC: Glutathione cysteine ligase, subunit catalytic; GSH: Glutathione; GSTM1: Glutathione S-transferase Mu 1; GSTP1: Glutathione S-transferase Pi 1; GSTT1: Glutathione S-transferase Theta 1; HWE: Hardy-Weinberg equilibrium; MAF: Minor allele frequency; MBL2: Mannose-binding Lectin-2; MDR: Multifactor dimensionality reduction; MLPA: Multiplex ligation-dependent probe amplification; OEGE: Online encyclopedia for genetic epidemiology; PCR: Polymerase chain reaction; RFLP: Restriction fragment length polymorphisms; TGF-β1: Transforming growth factor Beta-1; WHO: World Health Organization.
Competing interests
Authors declare that they have no competing interests.
Authors’ contributions
FALM: made substantial contributions to conception and design, acquisition of data, analysis and interpretation data included in this study; involved in drafting the manuscript and revising it for critically important intellectual content. CSB: carried out the molecular genetic studies and drafted the manuscript. AFR: has been involved in drafting the manuscript and revising it critically for important intellectual content. RS: carried out analyses and data interpretation, as well as has been involved in manuscript revising. JDR: made substantial contributions to conception and design, acquisition of data, and data interpretation included in this study; involved in drafting the manuscript and revising it for critically important intellectual content. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
GCLC-129C>T polymorphism in GCLC gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
GCLC-3506A>G polymorphism in GCLC gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTM1 gene deletion polymorphism in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTT1 gene deletion polymorphism in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTP1+313A>G polymorphism in GSTP1 gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
Contributor Information
Fernando Augusto de Lima Marson, Email: fernandolimamarson@hotmail.com.
Carmen Sílvia Bertuzzo, Email: bertuzzo@fcm.unicamp.br.
Rodrigo Secolin, Email: rsecolin@gmail.com.
Antônio Fernando Ribeiro, Email: anferi@uol.com.br.
José Dirceu Ribeiro, Email: jdirceuribeiro@gmail.com.
Acknowledgments
Authors are grateful to Taís Daiene Russo Hortencio, Luciana Cardoso Bonadia, Kátia Cristina Alberto Aguiar, Aline Gonçalves and Simoni Avansini for the assistance in data collection and organization of ideas, and to Maria Angela Ribeiro to spirometry analysis. We thank Professor Dr. Frauke Stanke and Professor Dr. Marilda de Souza Gonçalves to all corrections and contributions to this manuscript. We thank http://www.laboratoriomultiusuario.com.br and LAFIP/CIPED to help in the search. Funding Source: Fapesp.
References
- CFF – Cystic Fibrosis Foundation. 2012. http://www.cff.org. Access: 12.05.2012.
- Drumm ML, Ziady AG, Davis PB. Genetic variation and clinical heterogeneity in cystic fibrosis. Annu Rev Pathol. 2012;7:267–282. doi: 10.1146/annurev-pathol-011811-120900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem BS, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens JM, Iannuzzi MC, Kerem BS, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui LC, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Cystic Fibrosis Genetic Analysis Consortium - CFGAC. The Cystic fibrosis mutation database. 2012. http://www.genet.sickkids.on.ca/cftr. Access: 06/03/2012.
- Stanke F, Becker T, Kumar V, Hedtfeld S, Becker C, Cuppens H, Tamm S, Yarden J, Laabs U, Siebert B, Fernandez L, Macek M Jr, Radojkovic D, Ballmann M, Greipel J, Cassiman JJ, Wienker TF, Tümmler B. Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia. J Med Genet. 2011;48(1):24–31. doi: 10.1136/jmg.2010.080937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro AM, Carriero N, Bjornson R, Hartge P, Rothman N, Chatterjee N. Power of data mining methods to detect genetic associations and interactions. Hum Hered. 2011;72(2):85–97. doi: 10.1159/000330579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieker MG, Sanders EAM, Rijkers GT, Ruven HJT, Van Der Ent CK. Disease modifying genes in cystic fibrosis. J Cyst Fibros. 2005;4(2):7–13. doi: 10.1016/j.jcf.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Davies JC, Griesenbach U, Alton E. Modifier genes in cystic fibrosis. Pediatr Pulmonol. 2005;39(45):383–391. doi: 10.1002/ppul.20198. [DOI] [PubMed] [Google Scholar]
- Faria EJ, Faria IC, Ribeiro JD, Ribeiro AF, Hessel G, Bertuzzo CS. Association of MBL2, TGF-beta1 and CD14 gene polymorphisms with lung disease severity in cystic fibrosis . J Bras Pneumol. 2009;35(4):334–342. doi: 10.1590/S1806-37132009000400007. [DOI] [PubMed] [Google Scholar]
- Lima CS, Ortega MM, Marson FA, Zulli R, Ribeiro AF, Bertuzzo CS. ystic fibrosis transmembrane conductance regulator gene mutations and glutathione S-transferase null genotypes in cystic fibrosis patients in Brazil . J Bras Pneumol. 2012;38(1):50–56. doi: 10.1590/S1806-37132012000100008. [DOI] [PubMed] [Google Scholar]
- Marson FA, Bertuzzo CS, Hortencio TD, Ribeiro JD, Bonadia LC, Ribeiro AF. The ACE gene D/I polymorphism as a modulator of severity of cystic fibrosis. BMC Pulm Med. 2012;8:12–41. doi: 10.1186/1471-2466-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson FA, Bertuzzo CS, Ribeiro AF, Ribeiro JD. Polymorphisms in ADRB2 gene can modulate the response to bronchodilators and the severity of cystic fibrosis. BMC Pulm Med. 2012;5:50. doi: 10.1186/1471-2466-12-50. 12(1):50. Print up ahead. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Li B, Zhou Y, Zhong N, Ran P. Genetic analysis of CC16, OGG1 and GCLC polymorphisms and susceptibility to COPD. Respirology. 2007;12(1):29–33. doi: 10.1111/j.1440-1843.2006.00958.x. [DOI] [PubMed] [Google Scholar]
- Henrion-Caude A, Flamant C, Roussey M, Housset C, Flahault A, Fryer AA, Chadelat K, Strange RC, Clement A. Liver disease in pediatric patients with cystic fibrosis is associated with glutathione S-transferase P1 polymorphism. Hepatology. 2002;36(4 pt 1):913–917. doi: 10.1053/jhep.2002.35534. [DOI] [PubMed] [Google Scholar]
- McKone EF, Shao J, Frangolias DD, Keener CL, Shephard CA, Farin FM, Tonelli MR, Pare PD, Sandford AJ, Aitken ML, Kavanagh TJ. Variants in the Glutamate-Cysteine-Ligase gene are associated with cystic fibrosis lung disease. Am J Resp and Crit Care Med. 2006;174(4):415–429. doi: 10.1164/rccm.200508-1281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillet-Fieux MN, Nguyen-Khoa T, Loriot MA, Kelly M, de Villartay P, Sermet I, Verrier P, Bonnefont JP, Beaune P, Lenoir G, Lacour B. Glutathione S-transferases related to P. aeruginosa lung infection in cystic fibrosis children: preliminary study. Clin Biochem. 2009;42(1–2):57–63. doi: 10.1016/j.clinbiochem.2008.09.116. [DOI] [PubMed] [Google Scholar]
- Baranov VS, Ivaschenko T, Bakay B, Aseev M, Belotserkovskaya R, Baranova H, Malet P, Perriot J, Mouraire P, Baskakov VN, Savitskyi GA, Gorbushin S, Deyneka SI, Michnin E, Barchuck A, Vakharlovsky V, Pavlov G, Shilko VI, Guembitzkaya T, Kovaleva L. Proportion of the GSTM1 0/0 genotype in some Slavic populations and its correlation with cystic fibrosis and some multifactorial diseases. Hum Genet. 1996;97(4):516–520. doi: 10.1007/BF02267078. [DOI] [PubMed] [Google Scholar]
- Hull J, Thomson AH. Contribution of genetic factors other than CFTR to disease severity in cystic fibrosis. Thorax. 1998;53(12):1018–1021. doi: 10.1136/thx.53.12.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterpone S, Cornetta T, Angioni A, Fiscarelli E, Lucidi V, Testa A, Cozzi R. DNA damage and related modifier genes in Italian cystic fibrosis patients. Biol Res. 2009;42(4):477–486. [PubMed] [Google Scholar]
- Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, Darrah RJ, Dorfman R, Sandford AJ, Corey M, Zielenski J, Durie P, Goddard K, Yankaskas JR, Wright FA, Knowles MR. Gene Modifier Study Group. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353(14):1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- Korytina GF, Iaibaeva DG, Viktorova TV. Polymorphism of glutathione-S-transferase M1 and P1 genes in patients with cystic fibrosis and chronic respiratory tract diseases. Genetika. 2004;40(3):401–408. [PubMed] [Google Scholar]
- National Center for Biotechnology Information – NCBI. 2012. http://www.ncbi.nlm.nih.gov/. Access: 20/05/2012.
- Koide S, Kugiyama K, Sugiyama S, Nakamura S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. JACC. 2003;41(4):539–545. doi: 10.1016/S0735-1097(02)02866-8. [DOI] [PubMed] [Google Scholar]
- Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-S-transferase family of enzymes. Mutat Res. 2001;482(1–2):21–26. doi: 10.1016/s0027-5107(01)00206-8. [DOI] [PubMed] [Google Scholar]
- Amorim LMF, Rossini A, Mendonça G, Lotsch P, Simão TA, Gallo CM, Pinto L. CYP1A1, GSTM1, and GSTT1 polymorphisms and breast cancer risk in Brazilian women. Cancer Lett. 2002;181(12):179–186. doi: 10.1016/s0304-3835(02)00058-7. [DOI] [PubMed] [Google Scholar]
- Harries LW, Stubbins MJ, Forman D, Howard GCW, Wolf CR. Identification of genetic polymorphisms at the glutathione S-transferase Pi locus and association with susceptibility to bladder, testicular and prostate cancer. Carcinogenesis. 1997;18(4):641–644. doi: 10.1093/carcin/18.4.641. [DOI] [PubMed] [Google Scholar]
- Santos CIS, Ribeiro JD, Ribeiro AF, Hessel G. Critical analysis of scoring systems used in the assessment of Cystic Fibrosis severity: state of the art. J Bras Pneumol. 2004;30(3):286–298. [Google Scholar]
- WHO. Antro [programa de computador]. Version 3.0.1. Geneva: WORLD HEALTH ORGANIZATION; 2006. [Google Scholar]
- WHO. AntroPLUS [programa de computador]. Version 1.0.2. Geneva: WORLD HEALTH ORGANIZATION; 2007. [Google Scholar]
- American Thoracic Society (ATS) 2012. http://www.thoracic.org/. Access: 03/04/2012.
- Bonadia LC. Correlação entre aspectos clínicos, moleculares e fisiológicos de pacientes adultos com hipótese diagnóstica de fibrose cística de um centro de referência no Brasil. Brazil, State University of Campinas, PhD Thesis: University of Campinas, Departement of Genetics; 2011. (Phd thesis). [Google Scholar]
- Abdel-Rahman SZ, El-Zein RA, Anwar WA, Au WW. A multiplex PCR procedure for polymorphic analysis of GSTM1 e GSTT1 genes in population studies. Cancer Lett. 1996;107(2):229–233. doi: 10.1016/0304-3835(96)04832-X. [DOI] [PubMed] [Google Scholar]
- Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics. 2003;19(3):376–382. doi: 10.1093/bioinformatics/btf869. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Moore JH. Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, mising data, phenocopy, and genetic heterogeneity. Genet Epidemiol. 2003;24(2):150–157. doi: 10.1002/gepi.10218. [DOI] [PubMed] [Google Scholar]
- Moore JH, Gilbert JC, Tsai CT, Chiang FT, Holden T, Barney N, White BC. A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J Theor Biol. 2006;241(2):252–261. doi: 10.1016/j.jtbi.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Albi G, Rayón-Aledo JC, Caballero P, Rosado P, García-Esparza E. Cystic fibrosis in images: the Bhalla scoring system for computed tomography in paediatric patients. Radiology. 2012;54(3):260–268. doi: 10.1016/j.rx.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Bhalla M, Turcios N, Aponte V, Jenkins M, Leitman BS, McCauley DI, Naidich DP. Cystic fibrosis: scoring system with thin-section CT. Radiology. 1991;179(3):783–788. doi: 10.1148/radiology.179.3.2027992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GCLC-129C>T polymorphism in GCLC gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
GCLC-3506A>G polymorphism in GCLC gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTM1 gene deletion polymorphism in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTT1 gene deletion polymorphism in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.
The GSTP1+313A>G polymorphism in GSTP1 gene in association with clinical variables in cystic fibrosis patients distributed by CFTR mutation.