

Abstract
Matrix metalloproteinase inhibitors (MMPi) utilize zinc-binding groups (ZBGs) to chelate the catalytic Zn(II) ion resulting in enzyme inhibition. Adapting findings from the literature of Zn(II) ion sensors, we previously reported chelating sulfonamide inhibitors of MMP-2, some of which showed excellent selectivity over other gelatinases (MMP-9). Herein, we greatly expand our investigation of chelating sulfonamides as MMP inhibitors (MMPi) with the synthesis and screening of several new libraries consisting of 2-phenyl-7-sulfonamidobenzimidazole, 2-phenyl-7-sulfonamidobenzoxazole, 7-sulfonamidobenzimidazole, 7-sulfonamidobenzoxazole, and 2-(2-sulfonamidophenyl)-quinoline ZBG derivatives. A novel microwave irradiation synthetic procedure was utilized to rapidly and efficiently prepare these molecules. To better understand the coordination chemistry underlying these ZBGs, crystal structures of representative molecules with several first row transition metals were determined and differences in coordination preferences were considered. Surprisingly, only compounds with the 2-phenyl-7-sulfonamidobenzimidazole ZBG showed inhibition of MMP-2, suggesting that the specific structure of the ZBG can have a pronounced effect of inhibitory activity.
Introduction
Matrix metalloproteinases (MMPs) comprise a family of highly homologous Zn(II)-dependent endopeptidases involved in many important physiological processes, specifically the cleavage of extracellular proteins (1–3). MMPs are able to degrade proteins from the extracellular matrix (ECM) and the overexpression and misregulation of MMPs have been associated with a variety of pathologies including cardiovascular disease, arthritis, and inflammation (4–7). The role of MMPs in these diseases has made them therapeutic targets and for over three decades, various research groups have developed small molecules that show selective MMP inhibition (8, 9). Most MMPi employ a zinc-binding group (ZBG) to bind the active site metal ion (10, 11). The vast majority of MMPi use a hydroxamic acid as the ZBG, which is a strong chelator, but is limited by poor pharmacokinetics, low oral bioavailability, and inadequate selectivity for zinc (12).
In an effort to identify new scaffolds that could act as strong and potentially more selective ZBGs, we reported that chelators found in Zn(II) sensors, such as 8-sulfonamidoquinoline (13, 14) and 2-sulfonamidophenyl-benzimidazole (Figure 1) (15–17), could be used effectively as ZBGs for MMPi development (18). Of particular interest from this study was the observation that some sulfonamides displayed selectivity for MMP-2 over MMP-9, an unusual finding because these MMPs both belong to the gelatinase subclass of MMPs. Moreover, when comparing 8-sulfonamidoquinoline and 2-sulfonamidophenyl-benzimidazole derivatives (Figure 1), the 2-sulfonamidophenyl-benzimidazole compounds were more selective, which again clearly demonstrates the role of the ZBG in the selectivity of the inhibitors.
Figure 1.

Structure and IC50 values of select chelating 2-sulfonamidophenylbenzimidazole (BIS1, left) and 8-sulfonamidoquinoline (QS1, middle) inhibitors. The BIS1 inhibitor displays better overall selectivity for MMP-2 versus other MMPs. The proposed mode of binding for these chelating sulfonamide inhibitors is shown on the right.
Herein, we further explore the effect of the ZBG on the activity and selectivity of chelating sulfonamide-based MMPi. Five related sulfonamide libraries based on 7-sulfonamidobenzoxazole (ZBG1), 2-phenyl-7-sulfonamidobenzoxazole (ZBG2), 7-sulfonamidobenzimidazole (ZBG3), 2-phenyl-7-sulfonamidobenzimidazole (ZBG4), and 2-(2-sulfonamidophenyl)-quinoline (ZBG5) were synthesized. The coordination chemistry of these ligands was explored and the libraries were screened against MMP-2 and MMP-9. Overall, we find that even these relatively small changes to the ZBG have a pronounced effect on the inhibitory ability of these compounds, with the majority of compounds being far less effective then the chelating sulfonamides we previously identified. We have examined the coordination chemistry of these new ligands to gain added insight into their metal-binding behavior to facilitate further studies on their use in metalloenzyme inhibitors.
Experimental Methods
General
Starting materials and solvents were purchased from commercial suppliers (Sigma-Aldrich, Alfa Aesar, Fisher, etc.) and used as received. Microwave synthesis reactions were performed in 10 mL or 35 mL microwave vials using a CEM Discover S reactor. UV-visible spectra were collected on a Perkin Elmer Lambda 25 spectrophotometer in DMSO with 0.1% triethylamine. Column chromatography was performed using a Teledyne ISCO Combiflash system with prepacked silica cartridges. 1H/13C NMR spectra were recorded at ambient temperature on a 400 Varian FT-NMR instrument located in the Department of Chemistry and Biochemistry at the University of California, San Diego. Mass spectra were obtained at the Molecular Mass Spectrometry Facility in the Department of Chemistry and Biochemistry at the University of California, San Diego.
7-Nitrobenzoxazole (2)
To a toluene (25 mL) suspension of 3-nitro-2-aminophenol (1) (1 g, 6.5 mmol) in a 35 mL microwave tube was added triethylorthoformate (3.2 mL, 19.4 mmol) and a catalytic amount of p-toluenesulfonic acid monohydrate (62 mg, 0.3 mmol). The red suspension was stirred at 130 °C for 2 min in a microwave reactor, after which the reaction was complete by TLC. The orange solution was then cooled to −20 °C and the precipitate was isolated by filtration as a light pink solid (991 mg, 93%). 1H NMR (CD3OD, 400 MHz) δ 7.67 (t, J = 8.4 Hz, 1H), 8.12 (dd, J1 = 8.0 Hz, J2 = 0.8 Hz, 1H), 8.26 (dd, J1 = 8.4 Hz, J2 = 0.8 Hz, 1H), 8.79 (s, 1H). ESI-MS m/z 165.06 (M+H)+, 186.91 (M+Na)+.
7-Aminobenzoxazole (4)
To a solution of 4-nitrobenzoxazole (2) (1.91 g, 11.6 mmol) in 160 mL of methanol was added 10% Pd/C (1.24 g, 1.16 mmol) portionwise. The suspension was placed under H2(g) atmosphere for 1 h (P = 40 psi) and then filtered through a pad of celite. The transparent solution was evaporated to yield a black solid (1.42 g, 91%). 1H NMR (CD3OD, 400 MHz) δ 6.63 (dd, J1 = 8.0 Hz, J2 = 0.8 Hz, 1H), 6.87 (dd, J1 = 8.4 Hz, J2 = 0.8 Hz, 1H), 7.12 (t, J = 8.4 Hz, 1H), 8.25 (s, 1H). ESI-MS m/z 135.22 (M+H)+.
7-Sulfonamidobenzoxazole (ZBG1)
To a solution of 4-aminobenzoxazole (50 mg, 0.37 mmol) in pyridine (1 mL) was added 1.5 eq (0.56 mmol) of sulfonyl chloride. The transparent solution was heated in the microwave at 130 °C for 3 min (Power = 300 W). The solution was then poured into 5 mL of water and the precipitate was filtered off, rinsed with water, and dried under vacuum. If no precipitate formed, the aqueous phase was extracted twice with chloroform, the combined organic layers were washed with a solution of HCl (1M), dried with MgSO4, filted, and evaporated under reduced pressure. The crude product was either purified by column of chromatography or recrystallized from an appropriate solvent.
2-Phenyl-7-nitrobenzoxazole (3)
To a solution of 2-amino-4-nitrophenol (1) (1.0 g, 6.48 mmol) in trimethylorthobenzoate (3.4 mL, 19.5 mmol) and a catalytic amount of p-toluenesulfonic acid (61 mg, 0.32 mmol). The solution was then heated in a microwave reactor at 130 °C for 15 min. Toluene (10 mL) was added to the reaction mixture and the precipitate was vacuum filtered and washed with toluene (1.35 g, 87%). 1H NMR (CDCl3, 500 MHz) δ 7.49 (dt, J1 = 8.0 Hz, J2 = 1.0 Hz, 1H), 7.57 (t, J = 7.0 Hz, 2H), 7.62 (dt, J1 = 7.0 Hz, J2 = 1.0 Hz, 1H), 7.92 (dd, J1 = 8.0 Hz, J2 = 0.5 Hz, 1H), 8.20 (dd, J1 = 8.0 Hz, J2 = 0.5 Hz, 1H), 8.37 (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3, 100 MHz) 166.44, 152.46, 137.03, 132.86, 129.06, 128.52, 125.78, 124.31, 120.98, 116.55. ESI-MS m/z 241.21 (M+H)+.
2-Phenyl-7-aminobenzoxazole (5)
To a solution of 2-phenyl-7-nitrobenzoxazole (3) (1.2 g, 5.0 mmol) in 400 mL methanol was added 10% Pd/C (527 mg, 0.5 mmol) portionwise. The suspension was placed under H2(g) atmosphere for 30 min (P = 40 psi) and then filtered through a pad of celite. The transparent solution is evaporated to yield a black solid that was recrystallized from ethanol (850 mg, 71%). 1H NMR (CD3OD, 500 MHz) δ 6.65 (dd, J1 = 8.0 Hz, J2 = 1.0 Hz, 1H), 6.91 (dd, J1 = 8.5 Hz, J2 = 1.0 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 7.56 (m, 3H), 8.19 (m, 2H). 13C NMR (CD3OD, 100 MHz) δ 159.72, 151.70, 141.59, 131.66, 129.71, 129.23, 127.37, 127.18, 126.78, 108.53, 97.99. ESI-MS m/z 211.23 (M+H)+.
2-Phenyl-7-sulfonamidobenzoxazole (ZBG2)
To a solution of 2-phenyl-7-aminobenzoxazole (50 mg, 0.24 mmol) in pyridine (1 mL) was added 1.5 eq (0.36 mmol) of sulfonyl chloride. The transparent solution was heated in the microwave at 130°C for 3 min (Power = 300 W). The solution was then poured into 5 mL of water and the precipitate was filtered off, rinsed with water and dried under vaccum. If no precipitate was formed, the aqueous phase was extracted twice with chloroform. The combined organic layers were washed with a solution of HCl (1M), dried and evaporated under reduced pressure. The crude product was either purified by column of chromatography or recrystallized from an appropriate solvent.
7-Nitrobenzimidazole (7)
To a solution of 3-nitro-1,2-phenylenediamine (6) (1 g, 6.53 mmol) in toluene (25mL) was added triethylorthoformate (3.25 mL, 19.59 mmol) with a catalytic amount of p-toluenesulfonic acid (60 mg, 0.38 mmol). The solution was heated in the microwave at 120 °C for 1 min, followed by cooling to −20 °C to produce a precipitate. The precipitate was isolated by vacuum filtration (1.0 g, 94%). 1H NMR (CD3OD, 400 MHz) δ 7.46 (t, J = 8.0 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 8.23 (d, J = 7.6 Hz, 1H), 8.46 (s, 1H). ESI-MS m/z 164.22 (M+H)+.
7-Aminobenzimidazole (9)
To a solution of 7-nitrobenzimidazole (7) (495 mg, 3.03 mmol) in 50 mL of methanol was added 10% Pd/C (322 mg, 0.3 mmol) portionwise. The suspension was placed under H2(g) atmosphere for 3.5 h (P = 40 psi) and then filtered through a pad of celite. The transparent solution was evaporated to yield a black solid (403 mg, 99%). 1H NMR (CD3OD, 400 MHz) δ 6.55 (dd, J1 = 7.6 Hz, J2 = 0.8 Hz, 1H), 6.89 (dd, J1 = 8.0 Hz, J2 = 0.8 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 8.03 (s, 1H). ESI-MS m/z 134.21 (M+H)+.
7-Sulfonamidobenzimidazole (ZBG3)
To a solution of 7-aminobenzimidazole (50 mg, 0.37 mmol) in pyridine (2 mL) was added 1.5 eq (0.37 mmol) of sulfonyl chloride. The transparent solution was heated at 100 °C for ~4 h (until complete by TLC). The pyridine was then evaporated off and 15 mL of water was added to the remaining solution and stirred for 15 min. The precipitate was filtered off and rinsed with water and recrystallized from an appropriate solvent. If no precipitate was formed, the aqueous phase was extracted three times with chloroform. The organic layer was then washed with a solution of 1M HCl, dried with MgSO4 and evaporated under vacuum to obtain an oil that was either purified by column of chromatography or recrystallized from an appropriate solvent.
2-Phenyl-7-nitrobenzimidazole (8)
To a solution of 3-nitro-1,2-phenylenediamine (6) (100 mg, 0.65 mmol) in toluene (25 mL) was added trimethylorthobenzoate (336 µL, 1.9 mmol) and a catalytic amounts of p-toluenesulfonic acid (6.2 mg, 0.03 mmol). The solution was heated in the microwave at 130°C for 1 min. The solution was then cooled down to −20°C. The precipitate was vacuum filtered, rinsed with ether, and then collected (78 mg, 50%). 1H NMR (DMSO, 400 MHz) δ 8.12–8.15 (m, 6H), 8.16 (s, 1H), 8.18 (d, J = 0.8 Hz, 1H), 8.19 (d, J = 0.8 Hz, 1H). ESI-MS m/z 240.25 (M+H)+.
2-Phenyl-7-aminobenzimidazole (10)
To a solution of 2-phenyl-7-nitrobenzimidazole (8) (300 mg, 1.25 mmol) in 140 mL methanol was added 10% Pd/C (133 mg, 0.12 mmol) portionwise. The suspension was placed under H2(g) atmosphere for 30 min (P = 40 psi) and then filtered through a pad of celite. The transparent solution is evaporated to yield a black solid (258 mg, 81%). 1H NMR (DMSO, 500 MHz) δ 5.24 (s, NH, 2H), 6.37 (d, J = 7.5, Hz 1H), 6.75 (brs, 1H), 6.90 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.0 Hz, 1H), 7.53 (t, J = 7.5 Hz, 2H), 8.12 (d, J = 8.0 Hz, 2H). ESI-MS m/z 210.33 (M+H)+.
2-Phenyl-7-sulfonamidobenzimidazole (ZBG4)
To a solution of 2-phenyl-7-aminobenzimidazole (50 mg, 0.24 mmol) in pyridine (1 mL) was added 2 eq (0.48 mmol) of sulfonyl chloride. The transparent solution was heated in the microwave at 130 °C for 3 min (Power = 300W). The solution was then poured into 5 mL of water and the precipitate was filtered off, rinsed with water, and dried under vacuum. If no precipitate was formed, the aqueous phase was extracted twice with chloroform. The combined organic layers were washed with a solution of 1M HCl, dried with MgSO4, filtered, and evaporated under reduced pressure. The crude product was either purified by column of chromatography or recrystallized from an appropriate solvent.
tert-Butyl-(2-(quinolin-2-yl)phenyl)carbamate (12)
To a solution of 2-bromoquinoline (500 mg, 2.40 mmol) in 9 mL of toluene and 9 mL of 2M K2CO3 was added 2-(N-Bocamino) phenylboronic acid (11) (1.15 g, 3.6 mmol), and a catalytic amount of triphenylphosphine (277 mg, 0.24 mmol). The solution was refluxed at 100 °C overnight. Then water was added and the solution was extracted three times with dichloromethane. The organic layer was dried with anhydrous magnesium sulfate and was purified by column chromatography (Hex/EtOAc, 100/0 to 96/4) to isolate the desired product (761 mg, 99%). 1H NMR (CHCl3, 400 MHz) δ 7.13 (td, J1 = 6.0 Hz, J2 = 0.8 Hz, 1H), 7.42 (td, J1 = 6.4 Hz, J2 = 0.8 Hz, 1H), 7.58 (t, J = 6.0 Hz, 1H), 7.77 (td, J1 = 6.8 Hz, J2 = 0.8 Hz, 1H), 7.82 (dd, J1 = 6.4 Hz, J2 = 0.8 Hz, 1H), 7.85 (d, J = 6.4 Hz, 1H), 7.89 (d, J = 6.4 Hz, 1H), 8.08 (d, J = 6.4 Hz, 1H), 8.27 (d, J = 6.8 Hz, 1H), 8.42 (d, J = 6.4 Hz, 1H). ESI-MS m/z 320.98 (M+H)+, 320.98 (M+Na)+.
2-(2-Aminophenyl)quinolone (13)
In a dry round bottom flask was placed tert-butyl (2-(quinolin-2-yl)phenyl)carbamate (12) (300 mg, 0.93 mmol) and dry dichloromethane was added under nitrogen atmosphere. Trifluoroacetic acid was then added and the solution was stirred at room temperature for 3.5 h. The solution was evaporated to dryness and the precipitate was dissolved in dichloromethane and washed with 1M NaOH. The product was then purified by column chromatography (Hex/EtOAc, 100/0 to 98/2). 1H NMR (CHCl3, 400 MHz) δ 6.20 (brs, 2H, NH2), 6.80–6.84 (m, 2H), 7.21 (td, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.51 (t, J = 7.2 Hz, 1H), 7.68–7.72 (m, 2H), 7.80–7.85 (m, 2H), 8.04 (d, J = 8.8 Hz, 1H), 8.20 (d, J = 8.8 Hz, 1H). ESI-MS m/z 221.34 (M+H)+.
2-(2-Sulfonamidophenyl)quinoline (ZBG5)
To a solution of 2-(2-aminophenyl)quinoline (50 mg, 0.23 mmol) in pyridine (2 mL) is added 2 eq (0.45 mmol) of sulfonyl chloride. The transparent solution was heated in the microwave at 130°C for 3 min (Power = 300W). The solution was then poured into 5 mL of water and the precipitate was filtered off, rinsed with water and dried under vacuum. If no precipitate was formed, the aqueous phase was extracted twice with chloroform. The combined organic layers were washed with a solution of 1M HCl, dried with MgSO4, filtered, and evaporated under reduced pressure. The crude product was either purified by column of chromatography or recrystallized from an appropriate solvent.
Inhibition Assays
MMP activity assays were carried out in white NUNC 96-well plates as previously described (19–21). Each well contained a total volume of 100 µL: 60 µL of buffer (50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, pH 7.5), 20 µL of human recombinant MMP-2, or −9 (1.16 U or 0.9 U, respectively per well, BIOMOL International), and 10 µL of the inhibitor solution (50 µM final concentration). After a 30 min incubation period at 37 °C, the reaction was initiated by the addition of 10 µL fluorogenic MMP substrate (4 µM final concentration, Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2·AcOH, BIOMOL International). Kinetic measurements were recorded using a Bio-Tek Flx 800 fluorescence plate reader every min for 20 min with excitation and emission wavelengths at 320 and 400 nm, respectively. IC50 values were calculated using GraphPad Prism 5 software. All assays were run in duplicate.
Results
Synthesis
The synthesis of the 7-sulfonamidobenzoxazole (ZBG1, Scheme 1) and 7-sulfonamidobenzimidazole derivatives (ZBG3) began with an acid-catalyzed condensation of triethylorthoformate with either 2-amino-3-nitrophenol (1) or 3-nitro-1,2-phenylenediamine (6), respectively. The reported procedures for this type of reaction usually involve refluxing conditions in toluene for >1.5 h (2, 4, 7). By exploring a variety of microwave irradiation reaction conditions, it was determined that the transformation could be completed in 2 min at 130 °C. In addition, the products (2, 7) were easily isolated by filtration in >90% yield and did not require further purification. Reduction of the nitro group by hydrogenation gave the primary amines (4, 9) in >90% yield. For the final step, amine 4 was combined with a sulfonyl chloride in neat pyridine for 3 min using a previously reported microwave procedure to generate the different chelating sulfonamide fragments (18). In the case of 9, the microwave-mediated sulfonamide coupling procedure generated the disubstituted sulfonamides on the 3- and 7-position even when only one equivalent of sulfonyl chloride was utilized under microwave conditions. In order to obtain the monosubstituted ZBG3 compounds on the 7-position, a previously reported conventional heating procedure was utilized (5).
Scheme 1.

Synthesis of new inhibitors using a three-step, microwave procedure.
The synthesis of the 2-phenyl-7-sulfonamidobenzoxazole (ZBG2) and 2-phenyl-7-sulfonamidobenzimidazole derivatives (ZBG4) was performed using the same general synthetic route (Scheme 1); however, the reactivity of 2-amino-3-nitrophenol (1) and 3-nitro-1,2-phenylenediamine (6) with trimethylorthobenzoate were slightly different. Using the aforementioned microwave condensation, the conversion of the 3-nitro-1,2-phenylenediamine (6) into the 2-phenyl-7-nitrobenzimidazole (8) was complete within 1 min at 130 °C with isolated yields of ~50%. In contrast, 2-amino-3-nitrophenol (1) required longer reaction times (15 min) in order to achieve full conversion to 2-phenyl-7-nitrobenzoxazole (3) in good yields (87%). Subsequent reduction of the nitro groups of compounds 3 and 8 gave the free amines (5, 10) in >81% yield. The sulfonamide couplings were performed using a microwave procedure generating the desired sulfonamide inhibitors (18).
Finally, 2-(2-sulfonamidophenyl)-quinoline derivatives (ZBG5, Scheme 1) were synthesized in three steps via a Suzuki coupling between the 2-bromoquinoline and N-Boc-phenylboronic acid (11). After the Suzuki coupling (12), removal of the Boc group with TFA and microwave-mediated coupling with sulfonyl chlorides yielded the desired compounds. For all of the aforementioned chelating sulfonamide ZBGs, eight derivatives were synthesized in order to produce small fragment libraries suitable for in vitro screening against MMPs (Scheme 1).
Coordination Chemistry of ZBGs
In order to gain a better fundamental understanding into the coordination chemistry of these ZBGs, which is directly relevant to understanding their ability or inability to inhibit metalloproteinases (22–24), metal complexes with these ligands were prepared and their structures determined by single-crystal X-ray diffraction methods. Complexes were generally formed between the tosylated ligand and transition metal salts with the addition of triethylamine as a base. Most of the complexes could be crystallized by diffusing ether into a solution of the complex dissolved in a mixture of CH2Cl2 and MeOH (see Supporting Information and Table S1 for details). Surprisingly, not all of the chelating sulfonamides formed the expected, simple bidentate chelates, and others even showed unexpected reactivity in the presence of metal ions.
Upon reaction with ZnCl2, ZBG1a undergoes a chemical rearrangement to 3-tosyl-7-hydroxybenzimidazole (THBI). The structure of the THBI product was confirmed by X-ray crystallography (Figure 3). The C–O bond length is 1.358(2) Å, consistent with an aromatic alcohol. The hetrocycle C–N bond lengths are 1.304(3) and 1.388(3) Å for the imine and amine nitrogen atoms, respectively. It is important to note that under similar crystallization conditions but in the absence of Zn(II), ZBG1a readily crystallizes without undergoing any rearrangement (Figure 3). However, ZBG1a does transform to THBI in the presence of Zn(NO3)2, further confirming the role of Zn(II) in this rearrangement.
Figure 3.

ZBG1a (left) undergoes a rearrangement to THBI (right) in the presence of Zn(II). Thermal ellipsoids are shown at 50% probability and most hydrogen atoms have been omitted for clarity.
The rearranged THBI ligand binds to Zn(II) ions by forming a neutral Zn4L6Cl2 cluster (Figure 4). The cluster consists of two pairs of symmetry-related Zn(II) ions. The first type of Zn(II) ion is coordinated by four different THBI ligands, adopting a distorted octahedral geometry. The Zn(II) ion is chelated by two THBI ligands, as well as being bridged to its symmetric equivalent by the hydroxy groups of two other symmetry-equivalent THBI molecules. The second type of zinc ions are 4-coordinate, bound by the imide nitrogen of two THBI and two deprotonated hydroxy groups of two more THBI ligands. The fourth coordination site on each tetrahedral Zn(II) ion is occupied by a chloride ion.
Figure 4.

Asymmetric unit of the Zn4THBI6Cl2 cluster (left). Hydrogen atoms and a diethyl ether molecule have been excluded for clarity. Thermal ellipsoids are shown at 50% probability and hydrogen atoms have been removed for clarity. The full cluster (middle) is shown with sulfonamide substituents removed for clarity. The complex connectivity (right) is highlighted by showing each, unique ligand in a different color with Zn(II) ions in white and chloride ions removed for clarity.
The coordination chemistry of ZBG1a with Co(acac)2Ni(acac)2 (acac = acetylacetonate), and Cu2OAc4 were also explored. Interestingly, with these metal ions, no rearrangement of the ZBG1a ligand was observed. In all of the complexes, the sulfonamide group was not deprotonated and the ligand was found to bind only through imine nitrogen atom (Figure 5). The Co(II) and Ni(II) complexes form mononuclear complexes where the metal ions adopt a distorted octahedral geometry with the equatorial plane comprised of two chelating acac ligands and the axial positions occupied by two monodentate ZBG1a molecules. The M–N bond lengths are 2.223(2) and 2.158(2) Å for the Co(II) and Ni(II) complexes, respectively. The Cu(II) complex is similar to the Co(II) and Ni(II) compounds, in that the ZBG1a binds in the same manner (Cu–N = 2.203 Å); however, the overall structure of the complex is dinuclear, maintaining the Cu2OAc4 paddlewheel structure (Figure 5).
Figure 5.

Complexes of ZBG1a (left to right) with Co(II), Ni(II), and Cu(II). Thermal ellipsoids are shown at 50% probability and most hydrogen atoms have been removed for clarity. In the case of the Cu(II) complex the asymmetric unit contains two independent molecules, but only one is shown.
The reaction between ZBG2a and ZnCl2 yielded a mononuclear complex with a slightly distorted tetrahedral Zn(II) ion coordinated by two chloride ligands and one ZBG2a molecule through its imine and deprotonated sulfonamide nitrogen atoms (Figure 6). The Zn–N bond distances are 2.116(3) and 2.038(3) Å for the imine and sulfonamide donors, respectively. The charge of the anionic complex is balanced by a protonated triethylamine molecule in the crystal lattice. Attempts to crystallize free ZBG2a or other metal complexes with this ligand, including Co(II), Ni(II), and Cu(II) were unsuccessful.
Figure 6.

Structure of Zn(ZBG2a)Cl2. The asymmetric unit contains two anionic complexes, two protonated triethylamine molecules, and a methanol molecule. Thermal ellipsoids are shown at 50% probability and hydrogen atoms have been omitted for clarity.
In contrast to the other ZBGs studied here, ZBG3a readily forms complexes and crystallizes with late first-row transition metals. Co(II) and Zn(II) yielded isostructural complexes where the metal ion adopts a distorted trigonal bipyramidal geometry. In both complexes, two chelating ZBG3a ligands occupy both equatorial and axial sites, with the third equatorial position occupied by an monodentate ZBG3a ligand coordinated via a single imine nitrogen atom (Figure 7). The axial Zn–N bond lengths are 2.261(3) and 2.319(3) Å while the corresponding Co–N distances are slightly shorter at 2.239(3) and 2.258(3) Å. The equatorial Zn–N bond lengths are 2.005(3) and 2.008(3) Å while the Co(II) complex displays slightly longer distances of 2.021(3) and 2.026(3) Å. The ZBG3a molecule binds with a M– N distance of 2.023 and 2.047 Å for Zn(II) and Co(II), respectively. ZBG3a also readily forms a complex with Cu(II), with three ZBG3a ligands bound in a similar fashion as found with Co(II) and Zn(II), but the Cu(II) center has a distorted square pyramidal geometry (Figure 7). The axial ligand is the imine nitrogen atom from one of the chelating ZBG3a ligands. The Cu–N bond distances for equatorial imine nitrogen atoms are 1.993(4) and 1.978(4) Å, while the axial Cu–N distance is much longer at 2.249(4) Å due to Jahn-Teller distortion. The two sulfonamide nitrogen atoms bind to copper at distances of 2.075(4) and 2.131(4) Å. Finally, the Ni(II) complex with ZBG3a displayed the most distinct structure, with the Ni(II) ion coordinated in a distorted octahedral geometry by two chelating ZBG3a ligands and two water molecules (Figure 7). The two sulfonamide nitrogen donor atoms are positioned trans to each other on the coordination sphere. While the water ligands are held in a cis arrangement. The nickel ion binds to the sulfonamide nitrogen atoms at distances of 2.177(5) and 2.239(5) Å, while the Ni–N imine bond distances are 2.031(5) and 2.069(5) Å. The bite angle for the chelating ZBG3a ligands in all of these complexes are very similar at ~81–83°.
Figure 7.

Transition metal complexes of ZBG3a. Asymmetric units of Zn(II), Co(II), Cu(II), and Ni(II) complexes (top left to bottom right). Thermal ellipsoids are shown at 50% probability and hydrogen atoms and solvent molecules have been removed for clarity.
To date, attempts to isolate and crystallize complexes with ZBG4 or ZBG5 under a variety of reaction conditions have not been successful. However, the free ZBG4a ligand was crystallized as a protonated cation, which served to confirm the connectivity of this compound (Figure S1).
In Vitro Activity Against MMPs
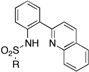
The five new libraries (i.e. 40 compounds, each based on one of the five ZBGs, Table 1) were screened for inhibitory activity against MMP-2 and MMP-9. A standard in vitro fluorescent substrate assay was utilized, and each compound was screened at a fixed concentration of 50 µM. In the case of MMP-2, the derivatives of ZBG1, ZBG2, and ZBG3 showed little or no inhibition at 50 µM. All of the ZBG5 compounds showed some activity against both MMP-2 and MMP-9, and three inhibitors (ZBG5d, ZBG5f, ZBG5i) gave nearly 30% inhibition against both MMPs at 50 µM (Table 1). However, the 2-phenyl-7-sulfonamidobenzimidazole (ZBG4) derivatives showed the best overall activity at 50 µM, with five of eight compounds showing >40% inhibition. Because all of the ZBG derivatives in this study utilize the same sulfonamide ‘backbones’ (R groups, Table 1), the assay results indicate that modest changes to the ZBG are playing a significant role in generating effective MMP inhibition. This is rather remarkable considering the structural similarity of all the ZBGs presented here, in addition to the related sulfonamides that were already reported (18). Among the ZBG4 derivatives, the best hit was the 4-trifluoromethylphenyl derivative (ZBG4b) that was determined to have an IC50 value of 40 µM against MMP-2 (Table 1). This is consistent with our earlier studies (see compounds BIS1 and QS1, Figure 1) (18), where the 4-trifluoromethylphenyl derivatives were also found to be the most active compounds against MMP-2 (out of ~40 different sulfonamide substituents). Clearly, the inhibitory activity of these compounds is surprisingly sensitive to the nature of the ZBG. This is likely due to a combination of affects, such as sterics, but may also reflect differences in the coordination chemistry of these ligands as well (vide supra).
Table 1.
Summary of MMP inhibition results. The compound name is listed above the percent inhibition obtain at a ligand concentration of 50 µM against MMP-2 (top) and MMP-9 (bottom). Note that fewer ZBG1 derivatives were prepared because of observed ligand instability (see text).
| R = | ||||||||
|---|---|---|---|---|---|---|---|---|
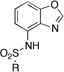 |
ZBG1b 0 0 |
n.d. |
ZBG1d 0 0 |
n.d. | n.d. |
ZBG1g 0 0 |
n.d. |
ZBG1i 0 0 |
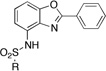 |
ZBG2b 0 0 |
ZBG2c 0 0 |
ZBG2d 0 0 |
ZBG2e 0 0 |
ZBG2f 0 0 |
ZBG2g 0 0 |
ZBG2h 7 0 |
ZBG2i 0 0 |
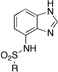 |
ZBG3b 0 0 |
ZBG3c 5 0 |
ZBG3d 0 0 |
ZBG3e 0 0 |
ZBG3f 0 0 |
ZBG3g 10 0 |
ZBG3h 0 0 |
ZBG3i 0 1 |
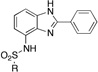 |
ZBG4b 69a 26 |
ZBG4c 0 11 |
ZBG4d 50 14 |
ZBG4e 51 26 |
ZBG4f 10 20 |
ZBG4g 50 23 |
ZBG4h 41 17 |
ZBG4i 0 0 |
 |
ZBG5b 10 5 |
ZBG5c 5 18 |
ZBG5d 27 26 |
ZBG5e 18 18 |
ZBG5f 27 28 |
ZBG5g 9 9 |
ZBG5h 10 3 |
ZBG5i 27 23 |
An IC50 value of 40 µM was determined against MMP-2
Discussion
Previous studies have shown that sulfonamide compounds such as 1 form stable chelates upon binding Zn(II) and other metal ions (13, 25). Our earlier studies show that such molecules can bind to the active site Zn(II) ions in MMPs eliciting good inhibition of these enzymes (18). In this study, we sought to develop a better structure-activity relationship with respect to the nature of these chelating sulfonamide ZBGs. ZBG1 and ZBG3 were designed to examine relatively conservative structural and electronic changes, looking at the impact of the quinoline ring on the activity of these compounds. ZBG1 and ZBG3 change the fused, 6-membered ring quinoline to a fused 5-membered benzoxazole and benzimidazole ligand, respectively. However, when exploring the coordination chemistry of ZBG1, it was found that the benzoxazole ring was unstable in the presence of Zn(II) and rearranges to yield 7-hydroxylbenzimidazole derivatives (Figure 3). A similar rearrangement of this heterocycle has been reported in the literature upon treatment with a reducing agent such as NaBH4 (26). Therefore, we attribute the poor inhibition activity of ZBG1 derivatives to this chemical rearrangement, which destroys the desired chelator shortly after binding to Zn(II). In contrast, the benzoimidazole analog (ZBG3) did not rearrange upon binding Zn(II) and readily formed stable complexes with a variety of transition metal ions. Nonetheless, these compounds did not show activity against MMP-2. The coordinative ability of ZBG3 and compound 1 are very similar (18), but they do provide slightly different bite angles (79° for ZBG3 compared to 83° for compound 1) upon binding Zn(II), which may produce different binding orientations in the MMP active that results in the observed loss of activity. Regardless, by examining the simple coordination chemistry of these ligands, one can a priori rule out the use of ZBG1 in metalloprotein inhibitors, but conclude that ZBG3 is a feasible candidate for inhibitor development.
While ZBG1 and ZBG3 were synthesized as analogues to compound QS1, ZBG2 and ZBG4 were synthesized to gain more insight around the phenylbenzimidazole sulfonamide ZBG found in compound BIS1 (27). ZBG2 and ZBG4 essentially preserve the steric and electronic features of compound BIS1, but place the sulfonamide substituent in a position on the chelator that is more similar to that found in ZBG1, ZBG3, and compound QS1. The result is that while the sulfonamide moiety of compound BIS1 generates a 6-membered chelate ring when bound to a metal ion, ZBG2 and ZBG4 will form a 5-membered chelate similar to that for ZBG3 or QS1. Thus, ZBG2 and ZBG4 are structurally related to compound BIS1 but certain features similar to QS1, making these ZBGs a hybrid our two earlier scaffolds (18, 27). Although the ZBG2 derivatives did not show any activity against MMP-2 or MMP-9 at 50 µM, most of the ZBG4 derivatives were active against both MMP-2 and MMP-9, with one compound (ZBG4b) giving an IC50 of 40 µM against MMP-2. ZBG4b possesses a 4-trifluoromethylphenyl sulfonamide substituent, which is consistent with the best hits from our earlier study (18). Interestingly, the only difference between ZBG3 and ZBG4 is the added phenyl ring on the 2-position of the ZBG. The significant difference in the activity between the 4-trifluoromethylphenyl derivatives of these ZBGs highlights the subtle nature of identifying effective scaffolds for developing metalloprotein inhibitors.
Conclusions
In summary, we have developed an efficient microwave procedure to synthesize benzimidazole and benzoxazole compounds and their sulfonamide derivatives. The resulting compounds, inspired from our previous findings (18, 27) and work on biocompatible Zn(II) sensors (15–17, 25, 28), were screened against the Zn(II)-dependent MMP-2 and MMP-9 metalloenzymes to identify compounds with good inhibitory activity. Much to our surprise, none of the new ZBGs were more potent than our original hits (BIS1 and QS1, Figure 1) and indeed most displayed very poor activity, despite the high similarity of the ZBGs. By examining the coordination chemistry and reactivity of these ligands with various transition metal ions, we identified unexpected reactivity that in some cases explains the poor inhibition activity of the compounds. Specifically, ZBG1 was found to be reactive in the presence of Zn(II), and several other ZBGs did not readily form metal complexes under standard conditions. The work here highlights how even subtle changes to the ZBG of a metalloprotein inhibitor can have a pronounced effect on activity, which suggests to us that more studies are required to better define, understand, and exploit the coordination chemistry of these ligands for use against important medicinal targets.
Supplementary Material
Acknowledgement
We thank Dr. Y. Su for performing mass spectrometry experiments. This work was supported by a grant from the National Institutes of Health (R21 HL094571). D.P.M. is supported by a SMART scholarship from the Office of Secretary Defense - Test and Evaluation (Grant No. N00244-09-1-0081).
References
- 1.Coussens LM, Fingleton B, Matrisian LM. Matrix Metalloproteinase Inhibitors and Cancer: Trials and Tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 2.Beckman J, et al. Human DNA Polymerase I Uses a Combination of Positive and Negative Selectivity To Polymerize Purine dNTPs with High Fidelity. Biochemistry. 2006;46(2):448. doi: 10.1021/bi061243s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whittaker M, Floyd CD, Brown P, Gearing AJH. Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.Bréhu L, Fernandes A-C, Lavergne O. Regioselective preparation of 5-amino- and 6-amino*1,3-benzoxazole-4,7-diones from symmetrical diaminophenol and aminoresorcinol. Tet. Lett. 2005;46(9):1437. [Google Scholar]
- 5.Elkin V, et al. Hydrogenation on palladium-containing granulated catalysts 3. Synthesis of aminobenzimidazoles by catalytic hydrogenation of dinitroanilines. Russ. Chem. Bull. 2007;56(6):1216. [Google Scholar]
- 6.Lindsey ML. MMP Induction and Inhibition in Myocardial Infarction. Heart Failure Rev. 2004;9:7–19. doi: 10.1023/B:HREV.0000011390.44039.b7. [DOI] [PubMed] [Google Scholar]
- 7.Katritzky AR, Musgrave RP, Rachwal B, Zaklika C. Synthesis of 4-Hydroxy-1-methylbenzimidazole and 7-Hydroxy-1-methylbenzimidazole. Heterocycles. 1995;41(2):345–352. [Google Scholar]
- 8.Nuti E, Tuccinardi T, Rossello A. Matrix Metalloproteinase Inhibitors: New Challenges in the Era of Post Broad-Spectrum Inhibitors. Curr. Pharma. Design. 2007;13:2087. doi: 10.2174/138161207781039706. [DOI] [PubMed] [Google Scholar]
- 9.Pirard B. Insight into the structural determinants for selective inhibition of matrix metalloproteinases. Drug Discov. Today. 2007;12:640–646. doi: 10.1016/j.drudis.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Rao BG. Recent Developments in the Design of Specific Matrix Metalloproteinase Inhibitors aided by Structural and Computational Studies. Curr. Pharma. Design. 2005;11:295–322. doi: 10.2174/1381612053382115. [DOI] [PubMed] [Google Scholar]
- 11.Yiotakis A, Dive V. Synthetic active site-directed inhibitors of metzincins: Achievement and perspectives. Mol. Asp. Med. 2008;29(5):329. doi: 10.1016/j.mam.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem. 2007;2(2):152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 13.Hendrickson KM, Geue JP, Wyness O, Lincoln SF, Ward AD. Coordination and Fluorescence of the Intracellular Zn2+Probe [2-methyl-8-(4-Toluenesulfonamido)-6-quinolyloxy]acetic Acid (Zinquin A) in Ternary Zn2+Complexes. J. Am. Chem. Soc. 2003;125:3889–3895. doi: 10.1021/ja020685y. [DOI] [PubMed] [Google Scholar]
- 14.McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. In Situ Imaging of Metals in Cells and Tissues. Chem. Rev. 2009;109(10):4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henary MM, Fahrni CJ. Excited state intramolecular proton transfer and metal ion complexation of 2-(2 '-hydroxyphenyl)benzazoles in aqueous solution. J. Phys. Chem. A. 2002;106(21):5210–5220. [Google Scholar]
- 16.Henary MM, et al. Excited-state intramolecular proton transfer in 2-(2'-arylsulfonamidophenyl)benzimidazole derivatives: The effect of donor and acceptor substituents. J. Org. Chem. 2007;72(13):4784–4797. doi: 10.1021/jo070433l. [DOI] [PubMed] [Google Scholar]
- 17.Henary MM, Wu YG, Fahrni CJ. Zinc(II)-selective ratiometric fluorescent sensors based on inhibition of excited-state intramolecular proton transfer. Chem. Eur. J. 2004;10(12):3015–3025. doi: 10.1002/chem.200305299. [DOI] [PubMed] [Google Scholar]
- 18.Macías B, et al. Synthesis and Structural Characterization of Zinc Complexes with Sulfonamides containing 8-Aminoquinoleine. Z. Anorg. Allg. Chem. 2003;629(2):255–260. [Google Scholar]
- 19.Puerta DT, et al. Heterocyclic Zinc-Binding Groups for Use in Next-Generation Matrix Metalloproteinase Inhibitors: Potency, Toxicity, and Reactivity. J. Biol. Inorg. Chem. 2006;11:131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 20.Puerta DT, Lewis JA, Cohen SM. New beginnings for matrix metalloproteinase inhibitors: identification of high-affinity zinc-binding groups. J. Am. Chem. Soc. 2004;126:8388–8389. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 21.Puerta DT, Mongan J, Tran BL, McCammon JA, Cohen SM. Potent, selective pyrone-based inhibitors of stromelysin-1. J. Am. Chem. Soc. 2005;127:14148–14149. doi: 10.1021/ja054558o. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen FE, Cohen SM. Using Model Complexes To Augment and Advance Metalloproteinase Inhibitor Design. Inorg. Chem. 2004;43:3038–3047. doi: 10.1021/ic035388o. [DOI] [PubMed] [Google Scholar]
- 23.Puerta DT, Cohen SM. Elucidating Drug-Metalloprotein Interactions with Tris(pyrazolyl)borate Model Complexes. Inorg. Chem. 2002;41:5075–5082. doi: 10.1021/ic0204272. [DOI] [PubMed] [Google Scholar]
- 24.Puerta DT, Cohen SM. Examination of Novel Zinc-Binding Groups for Use in Matrix Metalloproteinase Inhibitors. Inorg. Chem. 2003;42:3423–3430. doi: 10.1021/ic026029g. [DOI] [PubMed] [Google Scholar]
- 25.Fahrni CJ, O'Halloran TV. Aqueous Coordination Chemistry of Quinoline-Based Fluorescence Probes for the Biological Chemistry of Zinc. J. Am. Chem. Soc. 1999;121:11448–11458. [Google Scholar]
- 26.Katritzky Alan R, Musgrave Richard A, Rachwal Bogumila, Zaklika Chris. Synthesis of 4-Hydroxy-1-methylbenzimidazole and 7-Hydroxy-1-methylbenzimidazole. Heterocycles. 1995;41(2):345–352. [Google Scholar]
- 27.Martin D, Rouffet M, Cohen SM. Illuminating Metal-Ion Sensors: Benzimidazolesulfonamide Metal Complexes. Inorg. Chem. 2010;49(22):10226–10228. doi: 10.1021/ic101700t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahrni CJ, Henary MM, VanDerveer DG. Excited-state intramolecular proton transfer in 2-(2 '-tosylaminophenyl)benzimidazole. J. Phys. Chem. A. 2002;106(34):7655–7663. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.