

Abstract
Activation of microglia/macrophages is important in neonatal hypoxic–ischemic (HI) brain injury. Based on experimental studies, we identified macrophage/microglia-derived mediators with potential neurotoxic effects after neonatal HI and examined them in cerebrospinal fluid (CSF) from newborn infants after birth asphyxia. Galectin-3 is a novel inflammatory mediator produced by microglia/macrophages. Galectin-3 is chemotactic for inflammatory cells and activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase resulting in production and release of reactive oxygen species (ROS). Matrix metalloproteinase-9 (MMP-9) is a tissue-degrading protease expressed by activated microglia in the immature brain after HI. Both galectin-3 and MMP-9 contribute to brain injury in animal models for neonatal HI. Quinolinic acid (QUIN) is a neurotoxic N-methyl-d-aspartate (NMDA) receptor agonist also produced by activated microglia/macrophages. Galectin-3 and MMP-9 were measured by ELISA and QUIN by mass spectrometry. Asphyxiated infants (n = 20) had higher levels of galectin-3 (mean (SEM) 2.64 (0.43) ng/mL) and QUIN (335.42 (58.9) nM) than controls (n = 15) (1.36 (0.46) ng/mL and 116.56 (16.46) nM, respectively), p < 0.05 and p < 0.01. Infants with septic infections (n = 10) did not differ from controls. Asphyxiated infants with abnormal outcome had higher levels of galectin-3 (3.96 (0.67) ng/mL) than those with normal outcome (1.76 (0.32) ng/mL), p = 0.02, and the difference remained significant in the clinically relevant group of infants with moderate encephalopathy. MMP-9 was detected in few infants with no difference between groups. The potentially neurotoxic macrophage/microglia-derived mediators galectin-3 and QUIN are increased in CSF after birth asphyxia and could serve as markers and may contribute to injury.
Keywords: Hypoxic–ischemic brain injury, Microglia, Neonate, Cerebrospinal fluid galectin-3, Quinolinic acid
Introduction
Birth asphyxia with hypoxic–ischemic (HI) brain injury is an important cause of permanent neurological and cognitive impairment in newborn infants [1]. The immunoinflammatory system is activated in the secondary neurotoxic cascade after HI [2, 3] and microglial cells may contribute to perinatal brain injury [4–6]. Exposure to intrauterine inflammation also increases the risk for cerebral palsy in term infants and may contribute to encephalopathy and to susceptibility to asphyxiating factors [6–8]. It has been suggested that activated microglial cells cause brain injury through the production of proinflammatory cytokines [3, 9, 10], excitotoxins [11, 12], and reactive oxygen species (ROS) [13], but the direct mechanisms by which inflammatory cell activation contributes to injury after neonatal HI are not fully elucidated.
Galectin-3 is a β-galactoside-binding lectin, important in cell adhesion, cell proliferation, and apoptotic regulation. In recent years, it has also been demonstrated that galectin-3 is involved in the inflammatory process. Galectin-3 is produced by activated tissue macrophages [14] including microglial cells [15]. The mRNA expression is increased 10–20-fold after experimental neonatal HI [16] and there is a massive staining for galectin-3 in activated microglia adjacent to injury after HI in newborn mice [17]. Galectin-3 is involved in phagocyte recruitment and activation after inflammation [18, 19], has strong chemotactic properties for monocytes and macrophages [20], and activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in primed inflammatory cells leading to the production and release of ROS [14, 21]. Galectin-3 is also important in the regulation of macrophage phenotypes by driving cell populations towards an alternative activation associated with a more chronic, reparative inflammation [22]. The dual role of galectin-3 is reflected by conflicting results in adult and neonatal brain injury models. In the adult stroke model, galectin-3 is protective [23] while our previous studies show that galectin-3 contributes to brain injury after neonatal HI [17]. Galectin-3 activation and release has not been studied in association with brain injury in the clinical setting.
The immature brain is highly susceptible to excitotoxins acting on the N-methyl-d-aspartate (NMDA) receptor [24]. Quinolinic acid (QUIN) is a neurotoxic metabolite of l-tryptophan that is used to produce experimental neuronal injury [25]. QUIN acts mainly as a NMDA receptor agonist [25] but can also induce ROS formation and oxidative injury [26]. QUIN is produced by activated microglia/macrophages [11, 12, 27] and the synthesis is promoted by proinflammatory cytokines [28]. QUIN levels in injured brain regions increase 2–4 days after insult in models of transient ischemia [27, 29] and QUIN inhibition attenuates ischemic injury in adult brain [30]. Cerebrospinal fluid (CSF) QUIN concentrations are elevated in various inflammatory and noninflammatory cerebral diseases also in the pediatric population [31] but have not previously been studied in neonatal HI injury.
Microglial cells are also the main source of matrix metalloproteinase (MMP)-9 in the immature brain [32]. MMP-9 contributes to injury and breakdown of the blood–brain barrier (BBB) after experimental neonatal HI [32].
Although several studies indicate that inflammation and activation of microglia/macrophages are detrimental after HI, there is conflicting data in models of perinatal injury [4, 33] and reports of protective microglial effects in adult models of ischemia–reperfusion [34]. A more detailed knowledge of inflammatory mechanisms that promote injury or repair is needed.
In this study, we focus on potentially neurotoxic microglia/macrophage-derived inflammatory mediators identified in experimental studies. We evaluate CSF concentrations of galectin-3, QUIN, and MMP-9 after birth asphyxia and correlate the levels to clinical symptoms, prognosis, and long-term outcome. We also evaluate if systemic inflammation in the absence of asphyxia could induce elevated inflammatory markers in CSF.
Methods
Patients
Forty-five newborn infants treated at The Queen Silvia Children's Hospital were included in the study. The group was divided into birth asphyxia (n = 20), systemic infection/inflammation (n = 10), and controls (n = 15). The asphyxiated infants fulfilled the following criteria: (1) intrapartum distress with cardiotochographic late decelerations >1 h, absent variability or bradycardia >30 min, early passage of thick meconium or scalp pH <7.2. A small number of infants delivered after severe obstetric complications (acute placental abruption or uterine rupture) did not meet the intrapartum criteria. (2) Need for neonatal resuscitation with positive pressure ventilation >3 min, APGAR score ≤6 at 5′ or umbilical/first postnatal pH <7.0, and/or base deficit >16. The samples were obtained before hypothermia was introduced as a routine method and none of the infants were thus cooled.
The asphyxia group was further divided into mild (hyperalert, irritable, normal, or increased tone), moderate (lethargic, decreased tone, seizures), or severe (comatose, flaccid, failure to maintain ventilation, seizures not mandatory) encephalopathy [35, 36]. Infants were followed up by a trained pediatrician for ≥18 months and referred to a child neurologist and/or neuropsychologist if neurological abnormalities or developmental delay was suspected. An abnormal outcome was defined as death, cerebral palsy, mental retardation determined by WISC-III, or Griffith's standardized tests and/or epilepsy.
To evaluate the contribution of systemic inflammation to elevated inflammatory mediators in CSF, a subgroup of infants with clinical signs of sepsis but no signs of asphyxia was used (n = 10). All infants had CRP >100 mg/L and 6/10 included infants also had positive blood cultures. The control infants consisted of infants with clinical suspicion of infection but with negative bacterial cultures in blood and CSF in addition to CRP <50 mg/L (n = 15). The highest CRP value within 24 h before or after the spinal tap was used.
Infants with intracranial hemorrhage, cerebral malformation, elevated white blood cell count, or positive viral/bacterial cultures in CSF were excluded from all groups. Samples were also excluded if containing macroscopic blood.
CSF Sampling
CSF was obtained by spinal taps for diagnostic purposes. After 1.5 mL for clinical use had been tapped, ≤1.5 mL of CSF was collected and spun at 4,000 rpm for 10 min. Cell counts and CSF protein were analyzed as clinical routine samples. Samples were stored at −80 °C until analysis. Due to limited sample volumes, all compounds were not analyzed in every patient. Galectin-3 and MMP-9 were analyzed in 15/20 asphyxiated infants, 11/15 controls, and 8/10 infections. QUIN was analyzed in 18/20 asphyxias, 12/15 controls, and 10/10 infections. Parental consent was obtained and the study was approved by the Research Ethics Committee, Sahlgrenska University Hospital.
Galectin-3 and MMP-9 Analyses
Concentrations of galectin-3 and MMP-9 were measured using quantitative enzyme-linked immunosorbent assay (ELISA) according to the manufacturers' instructions. The detection level for the galectin-3 ELISA (BMS279; Bender MedSystems) was 0.150 ng/mL, while the MMP-9 (total) ELISA (Quantikine® DMP900; R&D Systems) detected MMP-9 (both 92 kDa pro- and 82 kDa active forms) concentrations down to 0.156 ng/mL.
QUIN Analysis
CSF QUIN was measured as the dihexafluoroisopropanol ester by electron impact negative chemical ionization mass spectrometry and gas chromatography with [13C7]-QUIN as internal standard [37].
Statistical Analysis
Results are given as mean (SEM). Groups were compared using the Kruskal–Wallis or the Mann–Whitney U test. The Kruskal–Wallis test was used with Dunn's correction when appropriate. Correlations were analyzed with Spearman's rank test and a p value <0.05 was considered statistically significant.
Results
Inflammatory Mediators in Asphyxiated Infants and Infants with Systemic Infection
Background data are given in Table 1. Severe obstetric complications (abruptio placentae, uterine rupture, dystocia, or tight nuchal cord) was present in 9/20 deliveries, while only one mother had clinical signs of chorioamnionitis.
Table 1.
Background data
| Asphyxia (n = 20) | Systemic infection (n = 10) | Control (n = 15) | ||
|---|---|---|---|---|
| Gestational age (weeks) | 39.14 (0.66) | 39.60 (0.71) | 39.8 (0.48) | NS |
| Sex (male/female) | 8/12 | 8/2 | 12/3 | |
| APGAR score at 5 min | 3.85 (0.61) | 9.50 (0.40) | 9.20 (0.22) | p < 0.0001 |
| CRP (mg/L) | 9.65 (3.22) | 139.2 (9.84) | 15.27 (4.71) | p < 0.0001 |
| Acidosis | ||||
| —pH | 6.92 (0.05) | |||
| —BD | 19.53 (1.65) | |||
| Age at LP (h) | 37.70 (4.48) | 37.00 (10.61) | 64.13 (10.67) | NS |
| CSF protein (mg/L) | 891.5 (163.5) | 625.1 (123.0) | 542.0 (78.78) | NS |
Results are given as mean (SEM), and statistical analysis was performed with Kruskal–Wallis test. Acidosis was evaluated in umbilical cord blood or first postnatal sample. Early blood gases were not routinely analyzed in control or infected infants
Asphyxiated infants had significantly higher levels of galectin-3 (mean (SEM) 2.64 (0.43) ng/mL) (n = 15) compared to control infants (1.36 (0.46) ng/mL) (n = 11), p < 0.05, while the levels in infants with infections (1.36 (0.47) ng/mL) (n = 8) did not differ from controls (Fig. 1).
Fig. 1.
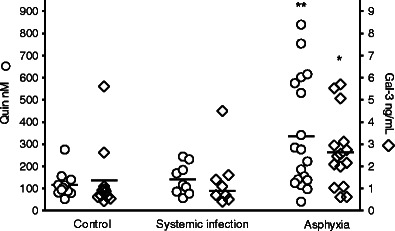
CSF concentrations of QUIN and galectin-3. The asphyxia group (n = 20) had significantly higher concentrations of QUIN (p < 0.01) and galectin-3 (p < 0.05) than the control group (n = 15), while the infection group (n = 10) did not differ from controls (Kruskal–Wallis test with Dunn's correction). Crossbars represent mean
The same pattern was seen for QUIN, with increased concentrations in asphyxiated infants (335.42 (58.9) nM) (n = 18) vs. controls (116.56 (16.46) nM) (n = 12), p < 0.01, while the concentrations in infants with systemic infection (140.92 (22.8) nM) (n = 10) were similar to controls (Fig. 1). In a subanalysis, infected infants with positive cultures (n = 6) were compared with control infants and no differences were found for either galectin-3 or QUIN. MMP-9 was detected in only one infant with severe asphyxia.
Inflammatory Mediators in Relation to Encephalopathy and Prognosis
The distribution of galectin-3 and QUIN concentrations according to encephalopathy is demonstrated in Fig. 2. Statistical comparisons were not performed due to the uneven number of patients in the different groups.
Fig. 2.
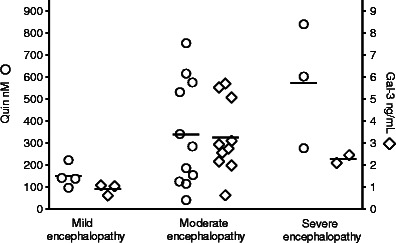
CSF concentrations of QUIN and galectin-3 in asphyxiated infants with mild (n = 4), moderate (n = 13), and severe (n = 3) encephalopathy. Crossbars represent mean
Infants with poor prognosis based on a severe clinical course (moderate and severe encephalopathy) had significantly higher concentrations of galectin-3 (3.08 (0.45) ng/mL) and QUIN (388.5 (69.4) nM) than infants with milder clinical symptoms and a predicted good prognosis (mild encephalopathy, controls, infections) (1.21 (0.25) ng/mL and 130.6 (12) nM), p < 0.0001 and p < 0.0001, respectively (Fig. 3).
Fig. 3.
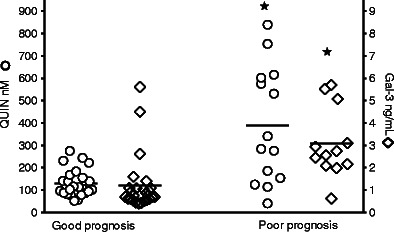
CSF concentrations of QUIN and galectin-3 in asphyxiated infants with good or poor prognosis. Infants with poor prognosis (n = 12/14) based on a severe clinical course (moderate and severe encephalopathy) had significantly higher concentrations of galectin-3 and QUIN (p < 0.0001 and p < 0.0001, Mann–Whitney U test) than infants with milder clinical symptoms (n = 25) and a predicted good prognosis (mild encephalopathy, controls, infections). Crossbars represent mean
Inflammatory Mediators and Outcome
An abnormal outcome was found in 8/20 asphyxiated infants, including one infant who died in the neonatal period. Cerebral palsy of the bilateral spastic or dyskinetic type was found in 4/7, mental retardation in 3/7, and severe epilepsy in 2/7 surviving infants. Asphyxiated infants with abnormal outcome had higher levels of galectin-3 (3.96 (0.67) ng/mL) (n = 6) than those with normal outcome (1.76 (0.32) ng/mL) (n = 9), p = 0.02. QUIN levels were higher (435.7 (88.9) nM) (n = 8) in infants with an abnormal vs. those with a normal (255.2 (72.7) nM) (n = 10) outcome, but the difference did not reach statistical significance (Fig. 4). Infants with septic infections and control infants had all normal outcome.
Fig. 4.
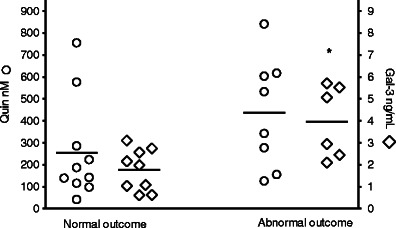
CSF concentrations of QUIN and galectin-3 in asphyxiated infants with normal (n = 12) or abnormal (n = 8) outcome. Galectin-3 levels were significantly higher in infants with abnormal outcome (p = 0.02, Mann–Whitney U test). Crossbars represent mean
Within the group of infants with moderate encephalopathy (n = 12), where prognosis is hard to predict based on clinical signs, infants with abnormal outcome had higher levels of galectin-3 (4.81 (0.64) ng/mL) (n = 4) than those with normal outcome (2.19 (0.36) ng/mL) (n = 6), p = 0.02. No difference in QUIN concentrations was seen (Fig. 5).
Fig. 5.
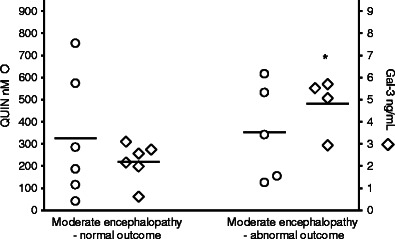
CSF concentrations of QUIN and galectin-3 in asphyxiated infants with moderate encephalopathy and normal (n = 8) or abnormal (n = 5) outcome. Galectin-3 levels were significantly higher in infants with abnormal outcome (p = 0.02, Mann–Whitney U test). Crossbars represent mean
Clinical Correlations
There was a significant correlation between galectin-3 and QUIN levels, p = 0.01. Concentrations of QUIN and galectin-3 did not correlate with CRP, CSF protein, or age at lumbar puncture in the whole group or in the subgroups.
Discussion
In this study, we found increased concentrations of QUIN and galectin-3 in CSF from asphyxiated infants. These increases were more pronounced in infants with severe clinical course and poor prognosis. Galectin-3 was also elevated in asphyxiated infants with abnormal outcome and the elevation remained significant within the clinically important group with moderate encephalopathy. Systemic infection/inflammation without signs of asphyxia did not result in elevated CSF concentrations.
The pathogenesis of birth asphyxia is complex and several other factors including chorioamnionitis may contribute to neonatal encephalopathy as well as perinatal brain injury [7, 8]. In order to define a group where perinatal asphyxia was indeed the probable cause of encephalopathy, we used well-established criteria including signs of fetal distress, neonatal need for resuscitation, and most importantly, metabolic acidosis. Previous studies show that a large proportion of encephalopathic infants fulfilling very similar criteria for asphyxia have acutely evolving injuries on magnetic resonance imaging (MRI) [38], suggesting that the insult took place at or near the time of birth. In addition, nearly half of the asphyxiated infants in our study were exposed to severe obstetric complications that may directly impair oxygen delivery and they presented with a considerable metabolic acidosis (mean pH 6.9 and base deficit 20). In clinical intervention studies using hypothermia, eligible infants with moderate to severe encephalopathy have to meet inclusion criteria for significant birth asphyxia (pH <7, base deficit >16, low APGAR score at 10′ or prolonged need for resuscitation) [39, 40]. Seventeen out of 20 asphyxiated infants in our study fulfilled these criteria even if our study also included infants with mild encephalopathy. The infants with adverse outcome also met the internationally accepted clinical criteria for cerebral palsy caused by an acute intrapartum event [41].
Blood gases were not routinely obtained at birth in the then apparently healthy infection and control infants. Since isolated metabolic acidosis is a common clinical finding not associated with neurological sequels in infants without encephalopathy, the control and infected infants are likely to be a true low-risk group even if early blood gases for comparisons with the asphyxiated infants were not available.
Even if the study group fulfilled strict criteria for intrapartum asphyxia, we cannot exclude that other factors including chorioamnionitis contributed to the susceptibility to asphyxia. Only one mother had clinical chorioamnionitis, but placentas were not examined and subclinical infections cannot be ruled out. However, the lack of microglia response and CSF inflammatory reaction in infants with systemic inflammation/infection makes it less likely that chorioamnionitis in itself, or the systemic inflammation seen in the asphyxiated infants, could explain the CSF changes found in infants with encephalopathy.
QUIN in CSF has previously been studied under normal as well as various pathological conditions in the pediatric population [42, 43], but there are no studies in newborns or after HI brain injury. The normal QUIN levels are inversely related to age with a mean concentration of 31 nM (range 20–64) in infants below 1 year of age [42]. We report significantly higher levels in newborn control infants with a mean concentration of 116 nM. This increase may be due to the trauma of normal labor or be part of the mild inflammatory activation associated with normal birth. The latter seems less likely since infants with a pronounced systemic inflammatory response included in this study did not differ from controls. In asphyxiated infants, QUIN concentrations were elevated approximately threefold, and the concentrations were similar to those previously found in infants with intraventricular hemorrhage [43], a clinical condition that is associated with an intense and prolonged inflammatory response in CSF [44].
QUIN concentrations were also associated with a severe clinical course and poor prognosis, but the differences between asphyxiated infants with various outcomes did not reach statistical significance. QUIN concentrations may, however, be difficult to evaluate since timing is important for the absolute value. In experimental ischemia, QUIN is significantly elevated after 2 days and increases further thereafter [29]. Serial CSF samples after trauma show an increase in QUIN concentrations up to 48 h [45] and increased QUIN levels were associated with mortality, when corrected for time of sampling [45]. In our study, sampling was performed at a mean of 38 h after birth in the asphyxiated group, suggesting that many infants had not reached their peak levels and that the values may not fully reflect the extent of injury. It is, however, not likely that the timing could explain the difference between groups since there was no difference in age at sampling between groups and no correlation between QUIN levels and age at sampling. In addition, there was a tendency towards later sampling in the control group suggesting that the control levels might even be overestimated in relation to the asphyxiated group.
To our knowledge, galectin-3 has not previously been measured in CSF from newborns or in association with brain injury. We report detectable levels in all samples and a significant increase in asphyxiated infants and infants with a severe clinical course, poor prognosis, and abnormal outcome. In a separate analysis of the clinically important group of infants with moderate encephalopathy, the difference in galectin-3 levels between infants with normal or abnormal outcome remained significant although the number of infants was small. Markers of abnormal outcome are of specific importance in this group since only 30–50 % of these infants develop a permanent brain injury despite similar clinical courses [1]. We have previously shown that in the experimental setting of neonatal HI, the effect, but not the expression, of galectin-3 is gender-dependent [17]. In this study, the groups were too small to allow for subgroup analyses, and we cannot exclude that the uneven distribution of boys and girls in the different study groups may have influenced the results.
The pattern of activation and CSF increases were similar for QUIN and galectin-3 and the concentrations in individual infants significantly correlated. This suggests that galectin-3 and QUIN are part of the same inflammatory response. The galectin-3 pattern of increase and correlation to severity of clinical course and outcome is also similar to that seen in previous studies of proinflammatory cytokines [2].
MMP-9 was found in only one infant with severe asphyxia. This may be due to the detection method. Normal CSF values are below the detection level of the ELISA used [46] and even in severe CNS inflammatory disease, MMP-9 is detected in only a minority of patients [47].
An important question is whether galectin-3 and QUIN are produced within the brain or if systemic cells of the monocyte/macrophage lineage are responsible for the increase found in CSF. Galectin-3 as well as QUIN are produced by activated microglia–macrophages as part of the inflammatory response after focal cerebral insults [11, 15, 27]. QUIN is produced by local de novo synthesis after focal ischemia [48] but an increased production of QUIN within the brain is also seen in systemic inflammation [27, 49]. In experimental studies, systemic inflammation induced by the bacterial toxin lipopolysaccharide (LPS) results in increased CSF concentrations of QUIN [27, 49], but in patients with HIV, elevated serum concentrations do not correlate with CSF levels, supporting an intracerebral source of QUIN [50]. Galectin-3 activation in focal vs. systemic inflammatory response is less well studied and it is not known whether HI or sepsis result in a systemic galectin-3 production.
Another possible mechanism by which systemically produced inflammatory mediators can contribute to elevated CSF concentration is through leakage over an injured BBB. Serum QUIN concentrations normally exceed those found in CSF [50], while serum levels of galectin-3 under normal or inflammatory conditions are not known. In our study, CSF protein content did not differ between asphyxiated infants and controls suggesting that there were no major differences in BBB damage. The absence of correlation between CSF protein and QUIN or galectin-3 concentrations also suggests that BBB breakdown is not responsible for the increased levels seen in CSF. CSF protein level was, however, not analyzed in all samples (7/45 missing) and in the absence of simultaneous blood samples, we cannot exclude a systemic contribution. It is, however, likely that brain microglia/macrophages are an important source of CSF galectin-3 and QUIN after asphyxia. Increased expression of galectin-3 at mRNA [16] as well as protein [17] is seen after experimental perinatal HI and intrathecal production of QUIN with associated increases in CSF is seen in experimental brain ischemia and inflammation [29, 48], as well as clinical traumatic brain injury [31, 51].
Irrespective of their possible origins, both QUIN and galectin-3 have possible neurotoxic effects. QUIN is a well-known neurotoxin, acting as an NMDA receptor agonist [25] as well as promoting oxidative injury [26]. Inhibition of QUIN production reduces injury in adult ischemia–reperfusion injury [30], but the immature brain has not been studied. Interestingly, studies in rats suggest that the immature brain has a specific sensitivity to QUIN with a maximal vulnerability at postnatal day 7 when the brain maturity closely resembles that seen in newborn infants [52]. In addition, QUIN-induced ROS formation is dependent on the presence of free ferrous iron [26]. Free iron is released after experimental HI in newborn pigs [53] and is present in CSF from asphyxiated infants [54]. QUIN is neurotoxic in vitro in concentrations as low as 100 nM [55] and studies in patients with encephalitis and intracerebral production of QUIN show that QUIN concentrations in the brain is tenfold higher than that found in CSF [50]. It is, thus, likely that brain QUIN levels in the asphyxiated patient could reach toxic levels.
Galectin-3 contributes to injury after experimental neonatal HI possibly by modulating the microglia response and MMP-9 activation [17]. This is in stark contrast to findings in adult stroke models where galectin-3 is expressed in a microglia population with protective properties [56] and transgenic mice lacking functional galectin-3 have aggravated injury [23]. Galectin-3 may, thus, serve as a useful marker of severity of injury in the newborn asphyxiated infant, but further studies are needed to determine whether galectin-3 contributes to injury also in the clinical setting.
In conclusion, our study demonstrates elevated levels of macrophage/microglia-derived potentially neurotoxic inflammatory mediators in CSF from asphyxiated infants with severe clinical course and adverse outcome. These mediators have been identified in experimental studies and our study confirms their clinical relevance. In addition, the role of microglia in HI injury is under current debate and a more detailed knowledge of inflammatory mechanisms that contribute to injury or repair can, in the future, lead to targeted therapeutic interventions.
Acknowledgments
This work was supported by the Frimurar-Barnhus Foundation, the Wilhelm and Martina Lundgren Foundation, Linnéa and Josef Carlsson Foundation, The Jerring Foundation, Göteborg Medical Society, and the Gustaf the Vth 80-Year Foundation.
Conflict of Interest
None.
Footnotes
This work was conducted at Perinatal Center, University of Gothenburg.
References
- 1.Volpe J. Hypoxic-ischemic encephalopathy; clinical aspects. Neurology of the Newborn. 5th Edition ed.; 2008. p. 440–84.
- 2.Savman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. 1998;43(6):746–751. doi: 10.1203/00006450-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, et al. Chemokine and inflammatory cell response to hypoxia–ischemia in immature rats. Pediatr Res. 1999;45(4 Pt 1):500–509. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- 4.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic–ischemic injury. Ann Neurol. 2002;52(1):54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 5.Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121(3):619–628. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- 6.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71(4):444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 7.Nelson KB, Grether JK. Potentially asphyxiating conditions and spastic cerebral palsy in infants of normal birth weight. Am J Obstet Gynecol. 1998;179(2):507–513. doi: 10.1016/s0002-9378(98)70387-4. [DOI] [PubMed] [Google Scholar]
- 8.Badawi N, Kurinczuk JJ, Keogh JM, Alessandri LM, O'Sullivan F, Burton PR, et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case–control study. BMJ. 1998;317(7172):1554–1558. doi: 10.1136/bmj.317.7172.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin D, Chinookoswong N, Miller G. The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxia–ischemia. Exp Neurol. 1994;130(2):362–367. doi: 10.1006/exnr.1994.1215. [DOI] [PubMed] [Google Scholar]
- 10.Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22(14):5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyes MP, Saito K, Major EO, Milstien S, Markey SP, Vickers JH. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from l-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain. 1993;116(Pt 6):1425–1450. doi: 10.1093/brain/116.6.1425. [DOI] [PubMed] [Google Scholar]
- 12.Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005;49(1):15–23. doi: 10.1002/glia.20090. [DOI] [PubMed] [Google Scholar]
- 13.Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys. 1998;353(2):312–321. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- 14.Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR., Jr Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol. 1995;147(4):1016–1028. [PMC free article] [PubMed] [Google Scholar]
- 15.Walther M, Kuklinski S, Pesheva P, Guntinas-Lichius O, Angelov DN, Neiss WF, et al. Galectin-3 is upregulated in microglial cells in response to ischemic brain lesions, but not to facial nerve axotomy. J Neurosci Res. 2000;61(4):430–435. doi: 10.1002/1097-4547(20000815)61:4<430::AID-JNR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.Hedtjarn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia–ischemia. J Cereb Blood Flow Metab. 2004;24(12):1333–1351. doi: 10.1097/01.WCB.0000141559.17620.36. [DOI] [PubMed] [Google Scholar]
- 17.Doverhag C, Hedtjarn M, Poirier F, Mallard C, Hagberg H, Karlsson A, et al. Galectin-3 contributes to neonatal hypoxic–ischemic brain injury. Neurobiol Dis. 2010;38(1):36–46. doi: 10.1016/j.nbd.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Colnot C, Ripoche MA, Milon G, Montagutelli X, Crocker PR, Poirier F. Maintenance of granulocyte numbers during acute peritonitis is defective in galectin-3-null mutant mice. Immunology. 1998;94(3):290–296. doi: 10.1046/j.1365-2567.1998.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu DK, Yang RY, Pan Z, Yu L, Salomon DR, Fung-Leung WP, et al. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol. 2000;156(3):1073–1083. doi: 10.1016/S0002-9440(10)64975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165(4):2156–2164. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson A, Follin P, Leffler H, Dahlgren C. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood. 1998;91(9):3430–3438. [PubMed] [Google Scholar]
- 22.MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180(4):2650–2658. doi: 10.4049/jimmunol.180.4.2650. [DOI] [PubMed] [Google Scholar]
- 23.Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32(30):10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston MV. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005;15(3):234–240. doi: 10.1111/j.1750-3639.2005.tb00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45(3):309–379. [PubMed] [Google Scholar]
- 26.Stipek S, Stastny F, Platenik J, Crkovska J, Zima T. The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem Int. 1997;30(2):233–237. [PubMed] [Google Scholar]
- 27.Heyes MP, Saito K, Chen CY, Proescholdt MG, Nowak TS, Jr, Li J, et al. Species heterogeneity between gerbils and rats: quinolinate production by microglia and astrocytes and accumulations in response to ischemic brain injury and systemic immune activation. J Neurochem. 1997;69(4):1519–1529. doi: 10.1046/j.1471-4159.1997.69041519.x. [DOI] [PubMed] [Google Scholar]
- 28.Saito K, Seishima M, Noma A, Markey SP, Heyes MP. Cytokine and drug modulation of kynurenine pathway metabolism by blood mononuclear cells. Adv Exp Med Biol. 1996;398:161–165. doi: 10.1007/978-1-4613-0381-7_26. [DOI] [PubMed] [Google Scholar]
- 29.Heyes MP, Nowak TS., Jr Delayed increases in regional brain quinolinic acid follow transient ischemia in the gerbil. J Cereb Blood Flow Metab. 1990;10(5):660–667. doi: 10.1038/jcbfm.1990.119. [DOI] [PubMed] [Google Scholar]
- 30.Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[−N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab. 1999;19(7):771–777. doi: 10.1097/00004647-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Heyes MP, Saito K, Crowley JS, Davis LE, Demitrack MA, Der M, et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain. 1992;115(Pt 5):1249–1273. doi: 10.1093/brain/115.5.1249. [DOI] [PubMed] [Google Scholar]
- 32.Svedin P, Hagberg H, Savman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia–ischemia. J Neurosci. 2007;27(7):1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189(1):58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, et al. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27(3):488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- 35.Levene MI, Sands C, Grindulis H, Moore JR. Comparison of two methods of predicting outcome in perinatal asphyxia. Lancet. 1986;1(8472):67–69. doi: 10.1016/s0140-6736(86)90718-x. [DOI] [PubMed] [Google Scholar]
- 36.Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress. A clinical and electroencephalographic study. Arch Neurol. 1976;33(10):696–705. doi: 10.1001/archneur.1976.00500100030012. [DOI] [PubMed] [Google Scholar]
- 37.Heyes MP, Markey SP. (18O)Quinolinic acid: its esterification without back exchange for use as internal standard in the quantification of brain and CSF quinolinic acid. Biomed Environ Mass Spectrom. 1988;15(5):291–293. doi: 10.1002/bms.1200150509. [DOI] [PubMed] [Google Scholar]
- 38.Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361(9359):736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- 39.Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663–670. doi: 10.1016/S0140-6736(05)17946-X. [DOI] [PubMed] [Google Scholar]
- 40.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. NEJM. 2005;353(15):1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- 41.MacLennan A. A template for defining a causal relation between acute intrapartum events and cerebral palsy: international consensus statement. BMJ. 1999;319(7216):1054–1059. doi: 10.1136/bmj.319.7216.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobbie MS, Surtees RA. Concentrations of quinolinic acid in cerebrospinal fluid measured by gas chromatography and electron-impact ionisation mass spectrometry. Age-related changes in a paediatric reference population. J Chromatogr B: Biomed Sci Appl. 1997;696(1):53–58. doi: 10.1016/s0378-4347(97)00221-1. [DOI] [PubMed] [Google Scholar]
- 43.Heyes MP, Saito K, Milstien S, Schiff SJ. Quinolinic acid in tumors, hemorrhage and bacterial infections of the central nervous system in children. J Neurol Sci. 1995;133(1–2):112–118. doi: 10.1016/0022-510x(95)00164-w. [DOI] [PubMed] [Google Scholar]
- 44.Savman K, Blennow M, Hagberg H, Tarkowski E, Thoresen M, Whitelaw A. Cytokine response in cerebrospinal fluid from preterm infants with posthaemorrhagic ventricular dilatation. Acta Paediatr. 2002;91(12):1357–1363. doi: 10.1111/j.1651-2227.2002.tb02834.x. [DOI] [PubMed] [Google Scholar]
- 45.Sinz EH, Kochanek PM, Heyes MP, Wisniewski SR, Bell MJ, Clark RS, et al. Quinolinic acid is increased in CSF and associated with mortality after traumatic brain injury in humans. J Cereb Blood Flow Metab. 1998;18(6):610–615. doi: 10.1097/00004647-199806000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Maliszewska M, Mader M, Scholl U, Azeh I, Hardeland R, Felgenhauer K, et al. Development of an ultrasensitive enzyme immunoassay for the determination of matrix metalloproteinase-9 (MMP-9) levels in normal human cerebrospinal fluid. J Neuroimmunol. 2001;116(2):233–237. doi: 10.1016/s0165-5728(01)00304-6. [DOI] [PubMed] [Google Scholar]
- 47.Fainardi E, Castellazzi M, Bellini T, Manfrinato MC, Baldi E, Casetta I, et al. Cerebrospinal fluid and serum levels and intrathecal production of active matrix metalloproteinase-9 (MMP-9) as markers of disease activity in patients with multiple sclerosis. Mult Scler. 2006;12(3):294–301. doi: 10.1191/135248506ms1274oa. [DOI] [PubMed] [Google Scholar]
- 48.Heyes MP, Morrison PF. Quantification of local de novo synthesis versus blood contributions to quinolinic acid concentrations in brain and systemic tissues. J Neurochem. 1997;68(1):280–288. doi: 10.1046/j.1471-4159.1997.68010280.x. [DOI] [PubMed] [Google Scholar]
- 49.Heyes MP, Quearry BJ, Markey SP. Systemic endotoxin increases l-tryptophan, 5-hydroxyindoleacetic acid, 3-hydroxykynurenine and quinolinic acid content of mouse cerebral cortex. Brain Res. 1989;491(1):173–179. doi: 10.1016/0006-8993(89)90101-7. [DOI] [PubMed] [Google Scholar]
- 50.Heyes MP, Saito K, Lackner A, Wiley CA, Achim CL, Markey SP. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB. 1998;12(10):881–896. doi: 10.1096/fasebj.12.10.881. [DOI] [PubMed] [Google Scholar]
- 51.Bell MJ, Kochanek PM, Heyes MP, Wisniewski SR, Sinz EH, Clark RS, et al. Quinolinic acid in the cerebrospinal fluid of children after traumatic brain injury. Crit Care Med. 1999;27(3):493–497. doi: 10.1097/00003246-199903000-00023. [DOI] [PubMed] [Google Scholar]
- 52.Trescher WH, McDonald JW, Johnston MV. Quinolinate-induced injury is enhanced in developing rat brain. Brain Res Dev Brain Res. 1994;83(2):224–232. doi: 10.1016/0165-3806(94)00141-3. [DOI] [PubMed] [Google Scholar]
- 53.Savman K, Nilsson UA, Thoresen M, Kjellmer I. Non-protein-bound iron in brain interstitium of newborn pigs after hypoxia. Dev Neurosci. 2005;27(2–4):176–184. doi: 10.1159/000085990. [DOI] [PubMed] [Google Scholar]
- 54.Ogihara T, Hirano K, Ogihara H, Misaki K, Hiroi M, Morinobu T, et al. Non-protein-bound transition metals and hydroxyl radical generation in cerebrospinal fluid of newborn infants with hypoxic ischemic encephalopathy. Pediatr Res. 2003;53(4):594–599. doi: 10.1203/01.PDR.0000054685.87405.59. [DOI] [PubMed] [Google Scholar]
- 55.Whetsell WO, Jr, Schwarcz R. Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci Lett. 1989;97(3):271–275. doi: 10.1016/0304-3940(89)90609-5. [DOI] [PubMed] [Google Scholar]
- 56.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]