

Abstract
Objective:
Sepsis is a systemic inflammatory response associated with high mortality rates in the clinical setting. A multiplex endpoint polymerase chain reaction (PCR) based assay for rapid detection of enterobacteriaceae involved in septicemia, which included Internal Control (IC) and 16S rDNA, is presented here. To develop a panel of primers for DNA fragments of 16S rDNA, enterobacteriaceae, IC, and evaluate analytical sensitivity and specificity of the test
Materials and Methods:
Primers for amplification of enterobacteriaceae, IC, and16S rDNA were designed, and then PCR was performed. Minimal analytical sensitivity was determined by cloning and colony PCR, and specificity was tested on the basis of their respective standard strains. This study is a cross-sectional Model.
Results:
Our results showed the rpoB gene as the most promising target for detection of enterobacteriaceae by PCR amplification. Specificity and sensitivity of endpoint PCR were 100%, 100%, and 100%, and 10, 1, and 100 copies/reaction for enterobacteriaceae, IC, and 16S rDNA, respectively.
Conclusion:
The molecular panel presented offers the advantage of an easy, reliable, and cost-effective system when compared to other molecular detection methods. However, further evaluation is needed. Our assay holds promising for more rapid pathogens related in clinical sepsis.
Keywords: Blood culture, Intensive Care Unit, pathogenic bacteria, polymerase chain reaction, sepsis
INTRODUCTION
Throughout the world, blood stream infections (BSIs) show high rates of morbidity and mortality, with a mortality rate ranging from 20 to 70%.[1–5] BSIs account for 30–40% of all cases of severs sepsis and septic shock.[6] The true incidence of nosocomial BSI is unknown, but it is anticipated that about 250,000 cases occur annually in the USA.[7] Affirmation of the septicemia diagnosis depends on microbiological exams based on blood culture procedure, which usually takes 24–72 h.[8] Thus, in many cases, antibiotic therapy is initiated on the basis of clinical criteria. Inappropriate empirical antimicrobial therapy is an important predictor of death, especially among patients in critical condition, and is associated with the emergence of drug-resistant strains, increasing the treatment costs.[9] Nevertheless, it is suggested that adequate antimicrobial therapy should be initiated as early as possible because a delay by each hour results in a 7.6% increase in death rate.[10]
Faster diagnosis of BSIs by molecular assay using polymerase chain reaction (PCR) is an important tool for the detection of microorganisms with high sensitivity, even when targets are present at extremely low number [10–100 deoxyribo nucleic acid (DNA) copies].[11] Earlier implementation of appropriate antimicrobial treatment reduces morbidity and mortality.[12] Enterobacteriaceae is the most common gram-negative bacteria causing septicemia and there is approximately the same remediation protocol for them according to clinical and laboratory standards institute (CLSI).[13] Microbial investigation by PCR has been explored with a detection system known as multiplex PCR. This technique is based on simultaneous amplification of two or more primer pairs in a single reaction vessel, which, in turn, usually means reduced cost and time requirement. The multiplex PCR technique requires a complex balance among some parameters such as level of salts, primers, DNA polymerase, and compatible primer melting temperatures, resulting in a laborious experimental design.[14,15] One of the most important factors in PCR assay is checking the reaction by negative and positive controls.[16]
The aim of present study was to develop and standardize a set of primers and to evaluate the analytical sensitivity and specificity of specific DNA fragments of bacteria and internal control, which is controlled the whole of the process is needed for assay such as DNA extraction and PCR reaction, with a simple, fast, and cost-effective method. There isn't any stydy, which used rpoB gene and internal control (IC) in the same reaction to identification of enterobacteriaceae in bacteremia by PCR.
MATERIALS AND METHODS
This study is a cross-sectional kind that was performed in Isfahan University of Medical Science, Isfahan, Iran, in 2011–2012 as part of a research project number 390011. Reference strains of organisms were bought from reference laboratory of Iran and Pasture Institute of Iran and brought to the microbiology lab of bacterial and virology group, Medical School, Isfahan University of Medical Science.
Primer design
A panel of six primers was developed for detection of enterobacteriaceae organisms related to sepsis, 16S rDNA, and IC. A series of nucleotide sequence of each organism was selected from the GenBank database returned from the rpoB gene and 16S rDNA for all of the microorganisms and X chromosome of Drosophila melanogaster for IC. Each respective set of consensual sequence was generated by Mega4 clustalW software.[17] The sequences were submitted to Allel ID6 software to design a primer for template. Then, the designed primer was checked in Mega4, Oligo6, Oligo Explorer software for finding the sequence and annual temperature, and simulating a set of primers for simultaneous amplification, respectively. The primer sets and genes target sequences are listed in Table 1.
Table 1.
Sequences of primers used

Multiple oligonucleotides were then tested in silicon regarding specificity of the BLAST tool (BLAST N and Primer BLAST) against all nucleotide sequences available in GenBank.
Microorganisms
Reference strains of organisms such as Escherichia coli (ATCC 25922), Enterobacter aerogenes (ATCC 13044), Proteus mirabilis (ATCC 7002), Klebsiella pneumonia (ATCC 13882), Salmonella enterica (ATCC 35640), and Shigella boyidii (ATCC 9207) were bought from reference laboratory of Iran and Pasture Institute of Iran.
DNA preparation
We used six different bacterial strains such as: Enterococcus faecalis ATCC 29212 and Staphylococcus aureus ATCC 29213 as Gram positive and Acinetobacter baumannii ATCC 19606, E. coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and K. pneumonia ATCC 13882 as Gram negative using three different kits: a-Qiamp DNA Mini Kit (Qiagen, Germany), b-Fermentas (Fermentas, Lithuania), and c-Cinagen DNA extraction kit (dNTP, Iran) to evaluate efficiency was applied for DNA extraction of strains maintained in solid media as follows: 200 μl of bacterial suspension (0.5 McFarland) was subjected to individual extraction protocols as indicated by each manufacturer. Qualitative and quantitative assessment of DNA preparation was performed through agarose gel and spectrophotometric measurements.
Bacteria and D. melanogaster (IC) DNA quantitation
The linearized standard PTZ75 plasmid was used for generation of a standard. The standards of 16S rDNA, K. pneumonia, and IC were made and measured according to the Applied Biosystems’ protocol. Then, sensitivity tests were performed by TA cloning assay (Fermentas, Lithuania) according to manufacturer's instructions, and colony PCR of mentioned microbial strains and IC was carried out. The obtained band of each dilution is plotted against the log10 of the corresponding initial template concentration. It was done by testing eight different dilutions, each one in triplicate, for 3 days by PCR. The concentration was 1 × 108 to 1 plasmid DNA target sequences copy number/reaction and repeat for the others.
Polymerase chain reaction
The reaction was performed at a defined volume of 25 μl and included 50-500 ng DNA, 12.5 μl Master Mix (Takar a, Japan), and 0.5 μl of 10 pM primer (Faza Biotech, Iran). To achieve a common optimal annealing temperature for all primer pairs, individual reactions were performed at 60°C. The initial denaturation was performed at 94°C for 5 min, followed by 35 cycles encompassing a 30-second step at 94°C, a 30-second step at 60°C, and a 1-min step at 72°C. An extension step at 72°C for 10 min ended the procedure. The amplicons obtained were submitted to electrophoresis in 1% agarose gel and stained by ethidium bromide (0.5 μg/ml) for UV light analysis and digitized (UV doc).
Analytical sensitivity and specificity of the test
Sensitivity was tested with serial dilutions of different plasmids containing the target sequences of the 16S rDNA, K. pneumonia, and IC, ranging from 1 to 108 copies/reaction. Specificity tests were performed employing pure cultures DNA of microorganisms included or not included in the panel such as: A. baumannii (ATCC 19606), K. pneumonia (ATCC 13882), P. aeruginosa (ATCC 27853), E. faecalis (ATCC 29212), S. aureus (ATCC 29213), and human DNA.
Statistical analysis
The one-way analysis of variance (ANOVA) test was used to compare the three different extraction kits. P values below 0.05 were considered significant.
RESULTS
Analysis of multiple oligonucleotide sequences suggested by Oligo Analyzer software resulted in a panel of three pairs of compatible primers for amplification of respective microorganisms in the same protocol. The 16S rDNA primers were designed by degenerative technology for amplification of all the bacteria and generated amplification of 1505 bp, the IC generated amplification of 684 bp, and K. pneumonia generated amplification of 512 bp [Table 1]. Analysis of all primers was carried out at an annealing temperature of 60°C. Multiplex PCR was performed with K. pneumonia, IC, and 16S rDNA [Figure 1].
Figure 1.
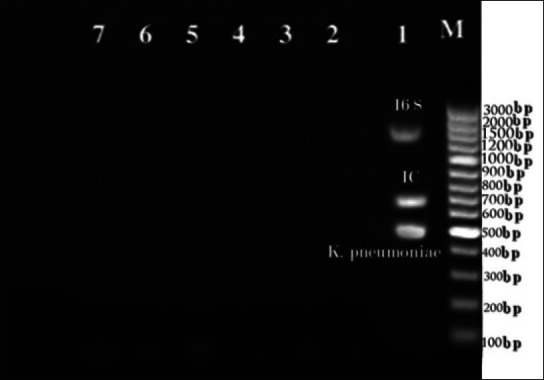
Simultaneous amplification in one vessel of Klebsiella pneumonia, Internal control (spiked IC) and 16S rDNA with optimized common annealing temperature (60° C) and specificity test of the PCR system designed for: (1) K. pneumonia (2) S. aureus (3) E. facium (4) P. aeruginosa (5) A. baumani (6) Human DNA (7) H2O. The bacterial concentration is (0.5 McFarland) of each strain. (M) Molecular Standard is indicated on the right
The quality of extracted bacterial DNA with the three kits was compared for each one of the six different bacterial species, by using five replicate samples per germ. The mean OD values obtained with Qiagen, Fermentas, and Cinagen kits were 1.9 ± 0.07, 1.6 ± 0.09, and 1.4 ± 0.11, respectively. The concentration of bacterial DNA (ng/μl) obtained with Qiagen, Fermentas, and Cinagen kits are reported in Table 2. P values were calculated by one-way ANOVA. Post-hoc test (Bonferroni) showed statistical significance between kits two by two (P < 0.05). The best extraction was obtained by Qiamp DNA Mini Kit and this kit was used for DNA extraction of bacterial strains.
Table 2.
Comparison the bacterial DNA with 3 different extraction kits

Sensitivity analysis showed a lower threshold for detection of microbial DNA at 10 copy number (K. pneumonia), 100 copy numbers (16S rDNA), and 1 copy/reaction, (IC). In the multiplex PCR, the analytical sensitivity was 10-fold less than the separate reaction [Figure 2]. Optimal specificity results were obtained with all primer pairs when compared to divergent microbial and human DNA [Figure 1].
Figure 2.
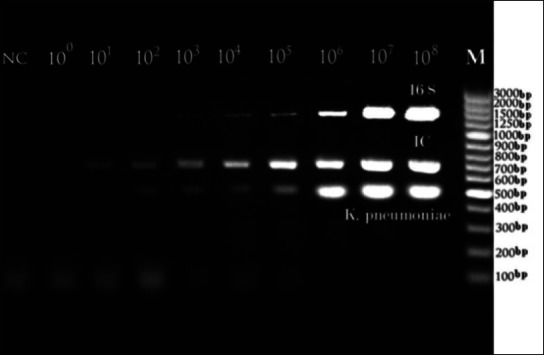
Analytical Sensitivity test of the plasmid DNA target sequences of the PCR system designed for 16S rDNA, IC and K. pneumonia in multiplex PCR reaction. (1) 108 CN, (2) 107 CN, (3) 106 CN, (4) 105 CN, (5) 104 CN, (6) 103 CN, (7) 102 CN, (8) 101 CN, 100 and negative control in order from right to left. (M) The position of the Molecular Standard is indicated on the right. CN: Copy number
DISCUSSION
Diagnostic procedures for sepsis require a 24–72 h period for confirmation of an infectious etiology and identification of the pathogen, and antibiotic therapy is usually started on an empirical basis.[8] However, the survey of comparing blood cultures and another technique for detection of microorganisms related to sepsis involves more rapid and accurate reporting when performed using PCR-based methods.[18–20] DNA isolation and amplification is very important for almost all laboratories involved in molecular biology research of microorganisms, especially those dedicated to difficult samples. Purity and quality of this DNA has to be verified before doing any amplification reactions; therefore, there is an increasing need for new, fast, and reliable method.[21]
The time required for specific enterobacteriaceae diagnosis using the multiplex PCR developed in the present study is about 4 h. It must be considered that this panel includes the detection of IC for confirming DNA extraction and PCR reaction in view of the difficulty of samples. Considering the high sensitivity of PCR-based methods, specific measures must be followed to prevent false-positive results; such measures include sequencing using specific primers for confirming the obtained amplicons.[22] Our results showed a detection limit variable between 1 and 100 copies of the target sequences when they were used separately, but in multiplex PCR, the detection limit is 10 times lower than this. Similar detection limits were observed for different molecular techniques when applied to different samples.[23,24] On the other hand, we used IC to prevent false-negative results. Other studies have proposed variety methods of PCR-based diagnostic procedures.[25–27] For example, a molecular method could detect 62 pathogens via PCR followed by hybridization assay;[28] however, this assay does not include microorganisms’ differentiation. In the present study, a set of primers was reported for detecting the most common gram-negative bacteria included in enterobacteriaceae which involve 30–40% of microorganisms related to sepsis. It is a faster method and showed higher sensitivity than blood culture. In the other hand, there was an IC for detecting the whole of DNA extraction and PCR reaction process, which made ensure of the result especially about negative ones. Furthermore, fast and sensitive molecular panel-based method for detecting enterobacteriaceae causing sepsis directly from blood specimen appears promising for a rapid fine tuning of empirical antibiotic therapy.[20]
CONCLUSION
In comparison to the multiple conventional methods where all primers are mixed in a single vessel, there is no delay in diagnostic time. However, there is a 16S primer accompanying an IC, which confirmed the existence of bacteria and accuracy of all steps of PCR process. We conclude that the molecular method developed is a reliable and fast method for detecting sepsis related to enterobacteriaceae; however, the role of this method in standard strains was assessed and its sensitivity and specificity in analytical step was measured, but further studies must be undertaken on clinical samples to confirm its application in diagnostic laboratory.
ACKNOWLEDGMENT
The authors are grateful to Vice Chancellor for Research of the Isfahan University of Medical Sciences for providing financial support for this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Almyroudis NG, Segal BH. Prevention and treatment of invasive fungal diseases in neutropenic patients. Curr Opin Infect Dis. 2009;22:385–93. doi: 10.1097/QCO.0b013e32832e074d. [DOI] [PubMed] [Google Scholar]
- 3.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Occurrence and outcomes of sepsis: Influence of race. Crit Care Med. 2007;35:763–8. doi: 10.1097/01.CCM.0000256726.80998.BF. [DOI] [PubMed] [Google Scholar]
- 4.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–50. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 5.Melamed A, Sorvillo FJ. The burden of sepsis-associated mortality in the United States from 1999 to 2005: An analysis of multiple-cause-of-death data. Crit Care. 2009;13:R28. doi: 10.1186/cc7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bochud PY, Glauser MP, Calandra T International Sepsis Forum. Antibiotics in sepsis. Intensive Care Med. 2001;27:S33–48. doi: 10.1007/pl00003796. [DOI] [PubMed] [Google Scholar]
- 7.Pittet D, Li N, Woolson RF, Wenzel RP. Microbiological factors influencing the outcome of nosocomial bloodstream infections: A 6-year validated, population-based model. Clin Infect Dis. 1997;24:1068–78. doi: 10.1086/513640. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AC, West TE, Peacock SJ. Surviving sepsis in developing countries. Crit Care Med. 2008;36:2487–8. doi: 10.1097/CCM.0b013e318177762d. [DOI] [PubMed] [Google Scholar]
- 9.Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. 1999;115:462–74. doi: 10.1378/chest.115.2.462. [DOI] [PubMed] [Google Scholar]
- 10.Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34:1589–96. doi: 10.1097/01.CCM.0000217961.75225.E9. [DOI] [PubMed] [Google Scholar]
- 11.Cuchacovich R. Clinical applications of the polymerase chain reaction: An update. Infect Dis Clin North Am. 2006;20:735–58. doi: 10.1016/j.idc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Zaragoza R, Artero A, Camarena JJ, Sancho S, González R, Nogueira JM. The influence of inadequate empirical antimicrobial treatment on patients with bloodstream infections in an intensive care unit. Clin Microbiol Infect. 2003;9:412–8. doi: 10.1046/j.1469-0691.2003.00656.x. [DOI] [PubMed] [Google Scholar]
- 13.Clinical and Laboratory Standards Institute. CLSI document M100-S21. West Valley Road, Suite 1400, Wayne, Pennsylvania 19087 USA: Clinical and Laboratory Standards Institute; 2011. Performance Standards for AntimicrobialSusceptibility Testing; Twenty-First Informational Supplement. [Google Scholar]
- 14.Settanni L, Corsetti A. The use of multiplex PCR to detect and differentiate food-and beverage-associated microorganisms: a review. J Microbiol Methods. 2007;69:1–22. doi: 10.1016/j.mimet.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Singh A, Goering RV, Simjee S, Foley SL, Zervos MJ. Application of molecular techniques to the study of hospital infection. Clin Microbiol Rev. 2006;19:512–30. doi: 10.1128/CMR.00025-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radstrom P, Lofstrom C, Lovenklev M, Knutsson R, Wolffs P. Strategies for overcoming PCR inhibition. In: Diefenbach CW, Dveksler GS, editors. PCR primer: A laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2003. pp. 149–61. [Google Scholar]
- 17.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity ofprogressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J, Millar BC, Moore JE, Murphy K, Webb H, Fox AJ, et al. Employment of broad-range 16S rRNA PCR to detect aetiological agents of infection from clinical specimens in patients with acute meningitis--rapid separation of 16S rRNA PCR amplicons without the need for cloning. J Appl Microbiol. 2003;94:197–206. doi: 10.1046/j.1365-2672.2003.01839.x. [DOI] [PubMed] [Google Scholar]
- 19.Mancini N, Carletti S, Ghidoli N, Cichero P, Burioni R, Clementi M. The era of molecular and other non-culture-based methods in diagnosis of sepsis. Clin Microbiol Rev. 2010;23:235–51. doi: 10.1128/CMR.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reier-Nilsen T, Farstad T, Nakstad B, Lauvrak V, Steinbakk M. Comparison of broad range 16S rDNA PCR and conventional blood culture for diagnosis of sepsis in the newborn: A case control study. BMC Pediatr. 2009;9:5. doi: 10.1186/1471-2431-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barken KB, Haagensen JA, Tolker-Nielsen T. Advances in nucleic acid-baseddiagnostics of bacterial infections. Clin Chim Acta. 2007;384:1–11. doi: 10.1016/j.cca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Jordan JA, Durso MB. Comparison of 16S rRNA gene PCR and BACTEC 9240 fordetection of neonatal bacteremia. J Clin Microbiol. 2000;38:2574–8. doi: 10.1128/jcm.38.7.2574-2578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zucol F, Ammann RA, Berger C, Aebi C, Altwegg M, Niggli FK, et al. Real-time quantitative broad-range PCR assay for detection of the 16S rRNA gene followed by sequencing for species identification. J Clin Microbiol. 2006;44:2750–9. doi: 10.1128/JCM.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothman RE, Majmudar MD, Kelen GD, Madico G, Gaydos CA, Walker T, et al. Detection of bacteremia in emergency department patients at risk for infective endocarditis using universal 16S rRNA primers in a decontaminated polymerase chain reaction assay. J Infect Dis. 2002;186:1677–81. doi: 10.1086/345367. [DOI] [PubMed] [Google Scholar]
- 25.Cursons RT, Jeyerajah E, Sleigh JW. The use of polymerase chain reaction to detect septicemia in critically ill patients. Crit Care Med. 1999;27:937–40. doi: 10.1097/00003246-199905000-00029. [DOI] [PubMed] [Google Scholar]
- 26.Rantakokko-Jalava K, Nikkari S, Jalava J, Eerola E, Skurnik M, Meurman O, et al. Direct amplification of rRNA genes in diagnosis of bacterial infections. J Clin Microbiol. 2000;38:32–9. doi: 10.1128/jcm.38.1.32-39.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto Y. PCR in diagnosis of infection: A Detection of bacteria in cerebrospinal fluids. Clin Diagn Lab Immunol. 2002;9:508–14. doi: 10.1128/CDLI.9.3.508-514.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klausegger A, Hell M, Berger A, Zinober K, Baier S, Jones N, et al. Gram type-specific broad-range PCR amplification for rapid detection of 62 pathogenic bacteria. J Clin Microbiol. 1999;37:464–6. doi: 10.1128/jcm.37.2.464-466.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]