

Abstract
Research has identified distinct neuronal circuits within the basolateral amygdala (BLA) that differentially mediate fear expression versus inhibition; however, molecular markers of these populations remain unknown. Here we examine whether optogenetic activation of a cellular subpopulation, which may correlate with the physiologically identified extinction neurons in the BLA, would differentially support fear conditioning versus fear inhibition/extinction. We first molecularly characterized Thy1-channelrhodopsin-2 (Thy1-ChR2-EYFP)-expressing neurons as a subpopulation of glutamatergic pyramidal neurons within the BLA. Optogenetic stimulation of these neurons inhibited a subpopulation of medial central amygdala neurons and shunted excitation from the lateral amygdala. Brief activation of these neurons during fear training disrupted later fear memory in male mice. Optogenetic activation during unreinforced stimulus exposure enhanced extinction retention, but had no effect on fear expression, locomotion, or open-field behavior. Together, these data suggest that the Thy1-expressing subpopulation of BLA pyramidal neurons provide an important molecular and pharmacological target for inhibiting fear and enhancing extinction and for furthering our understanding of the molecular mechanisms of fear processing.
Introduction
In humans, excessive associative fear is the hallmark of a number of neuropsychiatric disorders, including specific phobias, social phobias, panic disorder, and posttraumatic stress disorder (American Psychiatric Association, 2000; Kessler et al., 2005). Classical fear conditioning is perhaps the most powerful experimental model for studying the neural basis of associative learning and memory formation in mammals (Maren and Fanselow, 1996; Davis, 2000; LeDoux, 2000). In classical fear conditioning, subjects are presented with a neutral conditioned stimulus (CS), usually a tone, which is paired with an aversive unconditioned stimulus (US), usually a foot shock. After successive pairings, the tone acquires aversive properties and elicits fear responses when presented alone. In rodents, this fear is frequently quantified by measuring freezing behavior displayed during the presentation of the tone. Subsequently, when the tone is presented repeatedly in the absence of the shock, fear responses decline, a phenomenon called extinction. Extinction is not the forgetting of the original associative memory, but is rather thought to involve new learning, forming a new memory representation that the CS no longer predicts the US (Bouton et al., 2006; Myers and Davis, 2006). Considerable evidence using a variety of experimental models indicates that the amygdala is essential in the acquisition, consolidation, and extinction of conditioned fear (Davis, 2000; Myers and Davis, 2006; Herry et al., 2010).
A major challenge to understanding how the amygdala regulates emotional memories is identifying the neuronal circuits that differentially regulate fear acquisition, consolidation, and extinction. Recently, two electrophysiologically distinct populations of neurons within the basolateral amygdala (BLA) were identified to regulate fear responses in awake-behaving mice (Herry et al., 2008). Specifically, one population of neurons responded to the CS with increased firing frequency during and after fear conditioning. These same neurons reduced firing frequency during extinction training. A second population only responded to the CS with increased firing during extinction training. Importantly, the activity of these distinct neuronal populations predicts the behavioral output; increased firing of “extinction” neurons and decreased firing of “fear” neurons preceded a decrease in freezing behavior. These data suggest that there are distinct subpopulations of neurons within the BLA that are responsible for fear activation and fear inhibition. To date, however, the molecular identities of these neuronal populations have not been determined. The present study elucidates the role of a specific subpopulation of BLA neurons that may play a critical role in fear processing.
Here, we use a combination of optogenetic manipulation of neuronal activity in awake behaving mice, slice electrophysiology, immunocytochemistry, and in situ hybridization to identify specific neurons within the amygdala regulating long-term fear memories. We used two strains of mice with transgene expression driven by the Thy1-promoter (Thy1-YFP; Thy1-ChR2-EYFP), which target a specific subpopulation of pyramidal neurons within amygdala and other forebrain areas (Sugino et al., 2006). This enabled us to examine and manipulate neuronal activity within the same subpopulation of Thy1-labeled glutamatergic neurons within the BLA during and following fear conditioning and fear extinction learning.
Materials and Methods
Animals
Adult (6–10 weeks) B6.Cg-Tg(Thy1-COP4/EYFP)18Gfng/J (Thy1-ChR2-EYFP), and B6.Cg-Tg(Thy1-YFP)HJrs/J (Thy1-YFP) mice were purchased from Jackson Laboratories. All experimental mice were group housed (4–5 mice per cage) on a 12:12 h light:dark cycle in a temperature-controlled colony room and had unrestricted access to food and water. All procedures were conducted in accordance with policy guidelines set by the National Institutes of Health and were approved by the Emory University Institutional Animal Care and Use Committee.
Surgical procedures
For guide cannula implantation, mice were anesthetized deeply with a ketamine/dormitor mixture and placed in a stereotaxic instrument. Stereotaxic coordinates were derived from Paxinos and Franklin (2004). The head was positioned in the stereotaxic instrument so that the skull was level between lambda and bregma before implantation of the guide cannulas. Mice were implanted with a unilateral cannula aimed just dorsal to the BLA. Coordinates were 1.5 mm posterior, 3.4 mm lateral, and 4.8 mm below bregma. The side of guide cannula implantation was varied across mice. Dummy stylets were placed in the guide cannulas to keep them patent. Following surgery, mice were handled daily and their dummy stylets removed and replaced to habituate the mice to being restrained and to the insertion of the fiber optic.
Behavior and optogenetic stimulation
Cue-dependent fear conditioning.
Mice were pre-exposed to the conditioning chambers (Med Associates) 2 d before training. During cued-fear training, mice received five paired CS tones (30 s, 6 kHz, 75–80 db) and US shock (1 s, 0.5 mA) trials with a 90 s intertrial interval (ITI).
Cued-fear expression.
The expression of fear memory was tested 24 h after fear conditioning in a novel context. Mice were presented with 15 30 s tones with a 60 s ITI. Freezing during tone presentation was measured with FreezeView software (Coulbourn Instruments).
Fear extinction, retention, renewal, and reinstatement.
Fear extinction was measured 24 h after fear conditioning in a novel context. We used a suboptimal extinction procedure in which mice were presented with 15 30 s tones with a 60 s ITI. Freezing during tone presentation was measured with FreezeView software (Coulbourn Instruments). Forty-eight hours after initial extinction training, mice were tested for extinction retention as above. To test renewal, mice were placed back in the training context and presented with five initial training CS tones (30 s, 6 kHz, 75–80 db). To test fear reinstatement, mice were placed in an entirely new context and presented with a single unsignaled footshock. Mice were tested 24 h later and presented with five initial training CS tones (30 s, 6 kHz, 75–80 db).
Open field.
Open-field tests were conducted in a dimly lit room. Mice were implanted with fiber optics, then placed in the open-field apparatus (Med Associates) and allowed to explore for 5 min. Following the initial exploration time, mice were allowed to explore for an addition 5 min with optogenetic stimulation.
Optogenetic stimulation.
Optogenetic stimulation was performed using a 50 mW, 473 nm fiber-optic-coupled laser (IkeCool). The laser-coupled fiber was connected to the fiber optic that was inserted into the guide cannula with a fiber optic rotary joint (Doric Lenses). Before each experiment, mice were restrained and the fiber optic was inserted directly through the guide cannula and secured using a modified dust cap (Plastics One). Light stimulation parameters were 2 s stimulation at 20 Hz, 15 ms pulses. Light power density during stimulation was estimated at 14.32–15.91 mW/mm2. For stimulation during fear acquisition, the light stimulation coterminated with the tone and shock. For stimulation during fear extinction, the light stimulation coterminated with each tone. Stimulation parameters during the open-field experiments were the same as described for fear conditioning and fear extinction except that pulses were delivered once every minute for the 5 min exploration time.
Dual fluorescent in situ hybridization
Thy1-ChR2-EYFP mice were anesthetized and decapitated. Brains were rapidly removed, frozen on dry ice, and stored at −80°C until processing. Tissue was sectioned at 16 μm on a cryostat and mounted on Superfrost Plus slides (Fisher Scientific). cDNA clones containing the coding sequence of mouse Vglut1 and the coding sequence for ChR2 were linearized with appropriate restriction enzymes. Fluorescently labeled riboprobes were generated using T7 (Vglut1) and SP6 (ChR2) RNA polymerase (Maxiscript SP6/T7 kit). The Vglut1 riboprobe was labeled with fluorescein and the ChR2 riboprobe was labeled with digoxigenin. Following a prehybridization procedure, the sections were hybridized with both riboprobes at 55°C for 16 h and then subjected to a series of stringent washes. Sections were then incubated with anti-fluorescein-polymerized horse-radish peroxidase (POD) and Fab fragments, followed by fluorescent amplification and peroxidase quenching, and then with anti-digoxigenin-POD, Fab fragments (Roche). Signals were amplified with the TSA Plus Fluorescein Fluorescence System or TSA Plus Cy5 Fluorescence System (PerkinElmer) following each series of primary antibodies. Sections were then stained with Hoechst, washed, and coverslipped with Mowiol mounting medium.
Immunohistochemistry
Standard chromogenic immunohistochemistry was used to determine c-fos expression after optical stimulation. Briefly, free-floating sections containing the BLA were rinsed in PBS, then rinsed in 0.014% phenylhydrazine, permeabilized with 0.5% Triton-X in PBS, and incubated for 48 h at 4°C with primary antibody against c-fos (1:1000, rabbit anti-c-fos; Santa Cruz Biotechnology). Sections were then rinsed in PBS and incubated at room temperature with goat anti-rabbit biotinylated secondary antibody (1:500) followed by avidin-biotin-peroxidase complex reagent (Vector Laboratories). Sections were rinsed in PBS and staining was visualized using a nickel-enhanced DAB reaction. Slides were coverslipped with DPX and allowed to dry before imaging. C-fos immunostaining targeting the BLA was performed ∼1 h after laser light stimulation of Thy1-ChR2-EYFP mice following the behavioral studies described below, using similar stimulation paradigms. We found that without stimulation (no stim), there was minimal c-fos activation, whereas with stimulation (stim), there was increased c-fos staining in BLA and lateral central amygdala (CeL) (see Fig. 2C).
Figure 2.

Channel rhodopsin and EYFP expression within the basolateral amygdala of the Thy1-ChR2-EYFP mouse line. A, Autoradiography of an in situ hybridization with a 35-S-labeled riboprobe targeting the channelrhodopsin (ChR2) gene from anterior (left) to more posterior (right) coronal sections including BLA. B, EYFP labeling of Thy1-ChR2-EYFP cells and fibers from anterior (topr left) to posterior (bottem right) coronal sections spanning the amygdala, demonstrating Thy1-ChR2-EYFP labeling in BLA, but not LA or CeA nuclei of the amygdala. This pattern of ChR2 and EYFP expression recapitulates what has been observed in the Thy1-EYFP line. C, C-fos immunostaining was performed ∼1 h after laser light stimulation of Thy1-ChR2-EYFP mice targeting the BLA, following the behavioral studies described below, using similar stimulation paradigms. We found that without stimulation, there was minimal c-fos activation, whereas with stimulation, there was increased c-fos staining in BLA and CeL. LaDL, Dorsolateral amygdala.
Immunofluorescence was used to determine the relative colocalization of yellow fluorescent protein (YFP) with specific markers for the major subpopulations of BLA interneurons, namely parvalbumin and calbindin. To determine relative expression of YFP within the population of BLA principal neurons, we performed additional experiments using an antibody raised against calcium/calmodulin-dependent protein kinase II α (CaMKIIα). CaMKIIα is exclusively expressed in the BLA principal neurons and not in GABAergic interneurons (McDonald et al., 2002). Free-floating Thy1-YFP mouse brain sections containing BLA were rinsed in PBS, permeabilized with 0.5% Triton-X in PBS, and incubated for 48 h at 4°C with the following primary antibodies in 0.5% Triton-X solution: polyclonal rabbit anti-parvalbumin antibody (1:1000; Swant), mouse anti-calbindin antibody (1:1000; Sigma Aldrich), and mouse anti-CaMKIIα antibody (1:1000; Cell Signaling Solutions). Subsequently, sections were rinsed in PBS and incubated at room temperature for 2 h with either Alexa-Fluor 488 goat anti-mouse IgG or Alexa-Fluor 568 goat anti-rabbit IgG (1:500; Invitrogen), depending on the primary antibody host. Sections were then rinsed, mounted on gelatin-coated glass slides, and coverslipped using Mowiol mounting medium (Sigma Aldrich). Confocal laser scanning microscopy was used to obtain high-resolution photomicrographs using an Orca R2 cooled CCD camera (Hammamatsu) mounted on a Leica DM5500B microscope (Leica Mircosystems).
Slice electrophysiology
Tissue preparation.
Acute, coronal slices (300 μm) containing medial central amygdala (CeM), BLA, and lateral amygdala (LA) were collected from 2- to 4-month-old Thy1-ChR2-EYFP mice for ex vivo electrophysiological recordings. Briefly, animals were decapitated under isoflurane anesthesia (Abbott Laboratories) and the brains were removed and immersed in ice-cold, oxygenated cutting solution that contained the following (in mm): 130 NaCl, 3.5 KCl, 1.1 KH2PO4, 6.0 MgCl2, 1.0 CaCl2, 30 NaHCO3, 10 glucose, 0.8 thiourea, 2 sodium pyruvate, 0.4 ascorbic acid, and 2 kynurenic acid. A block of tissue containing the amygdala was then mounted in a Leica VTS-1000 vibrating microtome (Leica Microsystems) and coronal slices were cut and hemisected. Slices were transferred to a holding chamber containing oxygenated cutting solution at 32°C for 40 min before being placed in room-temperature, oxygenated, artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3.5 KCl, 1.1 KH2PO4, 1.3 MgCl2, 2.5 CaCl2, 30 NaHCO3, 10 glucose, 0.8 thiourea, 2 sodium pyruvate, and 0.4 ascorbic acid.
Patch clamp.
Slices were placed in a Warner Series 20 recording chamber (Warner Instruments) mounted on the fixed stage of a Leica DM-LFS microscope (Leica Microsystems). Slices were fully submerged and continuously perfused at a rate of 1–2 ml/min with heated (32°C) and oxygenated ACSF. Before patch clamping, a concentric bipolar stimulating electrode (FHC) was placed in the LA, and a 200 μm fiber-optic cable (Thorlabs) connected to a 488 nm laser (Shanghai Lasers) was placed at the external capsule, lateral to and aimed at the BLA. The BLA, LA, and CeM were identified using a mouse brain atlas (Paxinos and Franklin, 2004), and neurons were selected for recording under IR-DIC illumination with a 40× water-immersion objective. For all experiments, whole-cell patch-clamp configuration was established and cell responses were recorded in current-clamp mode. For measuring responses to optical stimulation, pulses of 0.6–1.2 mW, 488 nm light were delivered at 0.2 or 20 Hz. For electrical stimulation, 150 μs pulses were delivered to the LA. Images were captured with a Hamamatsu Orca ER CCD camera controlled by SimplePCI software (Compix). Whole-cell patch-clamp recordings were conducted using thin-walled borosilicate glass-patch electrodes (WPI), which were pulled on a P-97 Flaming/Brown micropipette puller (Sutter Instruments). Patch electrodes had resistances ranging from 4 to 7 MΩ when filled with standard patch solution that contained the following (in mm unless otherwise noted): 138 K-gluconate, 2 KCl, 3 MgCl2, 5 phosphocreatine, 2 K-ATP, 0.2 NaGTP, 10 HEPES, and 3 mg/ml biocytin. The patch solution was adjusted to a pH of 7.3 with KOH and had a final osmolarity of ∼280 mOsm. Junction potentials were offset before patching neurons. Series resistances and input resistance were monitored online with a 50 pA hyperpolarizing step (200 ms) given between stimulation sweeps, and neurons with more than a 15% change in series resistance were discarded. Recordings were obtained using a MultiClamp 700A amplifier (Molecular Devices), a Digidata 1322A A/D interface, and pClamp 10 software (Molecular Devices). Data were filtered at 5 kHz and sampled at 10 kHz. Neurons were excluded from analysis if their resting membrane potential (Vm) was more positive than −55 mV or if their action potentials did not surpass +5 mV. Neurons were also excluded if the recording location was incorrect based on post hoc visualization of recorded neurons using a Leica DM5500B confocal, spinning disk laser microscope (Leica Microsystems) equipped with an Orca R2 cooled CCD camera (Hamamatsu) after fixation with 10% buffered formalin (Fisher Scientific) and staining with Alexa Fluor 568 streptavidin (Invitrogen). In some recordings, SR 95531 (Tocris Bioscience) was applied by gravity perfusion at 5 μm in the circulating ACSF after being stored frozen as concentrated stock in dH2O.
Statistical analysis
Statistical analyses were performed using SPSS 19 and Prism 5. For all behavioral experiments, freezing behavior was analyzed using a repeated-measures ANOVA with optogenetic stimulation as the between-subjects factor and tone as the within-subjects factor. Averaged freezing values were also analyzed using a Student's t test between treatment groups. Activity measurements in the open field (time in surround, time in center, distance traveled) were also analyzed using a Student's t test between treatment groups. In electrophysiological experiments, spike number was analyzed using a repeated-measure ANOVA.
Results
Thy1-ChR2-EYFP neurons are a subpopulation of glutamatergic neurons within the BLA
To characterize this population of neurons, we used Thy1-YFP transgenic mice that have been described previously (Feng et al., 2000) and a novel transgenic mouse strain, Thy1-ChR2-EYFP (B6.Cg-Tg(Thy1-COP4/EYFP)18Gfng/J), that expresses channelrhodopsin-2 (ChR2) fused to enhanced YFP also under the control of the Thy1 promoter (Arenkiel et al., 2007). The expression patterns of YFP and ChR2 driven by the Thy1 promoter in both strains are extremely similar within the BLA (Fig. 1A,B). YFP and channelrhodopsin-2 expression indicate strong transgene expression specifically within the BLA, and not within the lateral or central amygdala (Fig. 1A,B). To further characterize this Thy-1-ChR2-expressing subpopulation, we performed in situ hybridization of the Thy1-ChR2-YFP mice, finding that, whereas all Thy1-ChR2-expressing neurons also express vGlut1, a marker of excitatory neurons, only a subset of the vGlut1-expressing neurons express ChR2 (Fig. 1C–H). Further, we used immunocytochemistry to analyze the colocalization of another excitatory neuronal marker, CaMKIIα, as well as two inhibitory markers, parvalbumin and calbindin, with the Thy1-expressing cells. We found that, similar to vGlut1, Thy1-YFP-expressing neurons represent a subset of CaMKIIα cells, but do not overlap with calbindin or parvalbumin populations (Fig. 1I–Q). Overall, these immunofluorescence studies revealed significant overlap in the localization of Thy1-YFP with a marker for BLA principal neurons, CaMKIIα, in the somatodendritic compartment of a large number of neurons scattered throughout the BLA. Significantly, there was no overlap with either of the markers for BLA interneurons, parvalbumin nor calbindin. These data suggest that the Thy1-YFP and Thy1-ChR2-EYFP neurons represent a distinct subpopulation of amygdala excitatory, principal neurons. Finally, radiolabeled in situ hybridization of brains from Thy1-ChR2-EYFP mice supported previously reported expression patterns (Feng et al., 2000; Porrero et al., 2010), with particularly strong expression of ChR2 in the BLA (Fig. 2A). Endogenous EYFP expression in Thy1-ChR2-EYFP mice indicates strong transgene expression specifically within the BLA, but not LA or central amygdala (CeA) (Fig. 2B).
Figure 1.
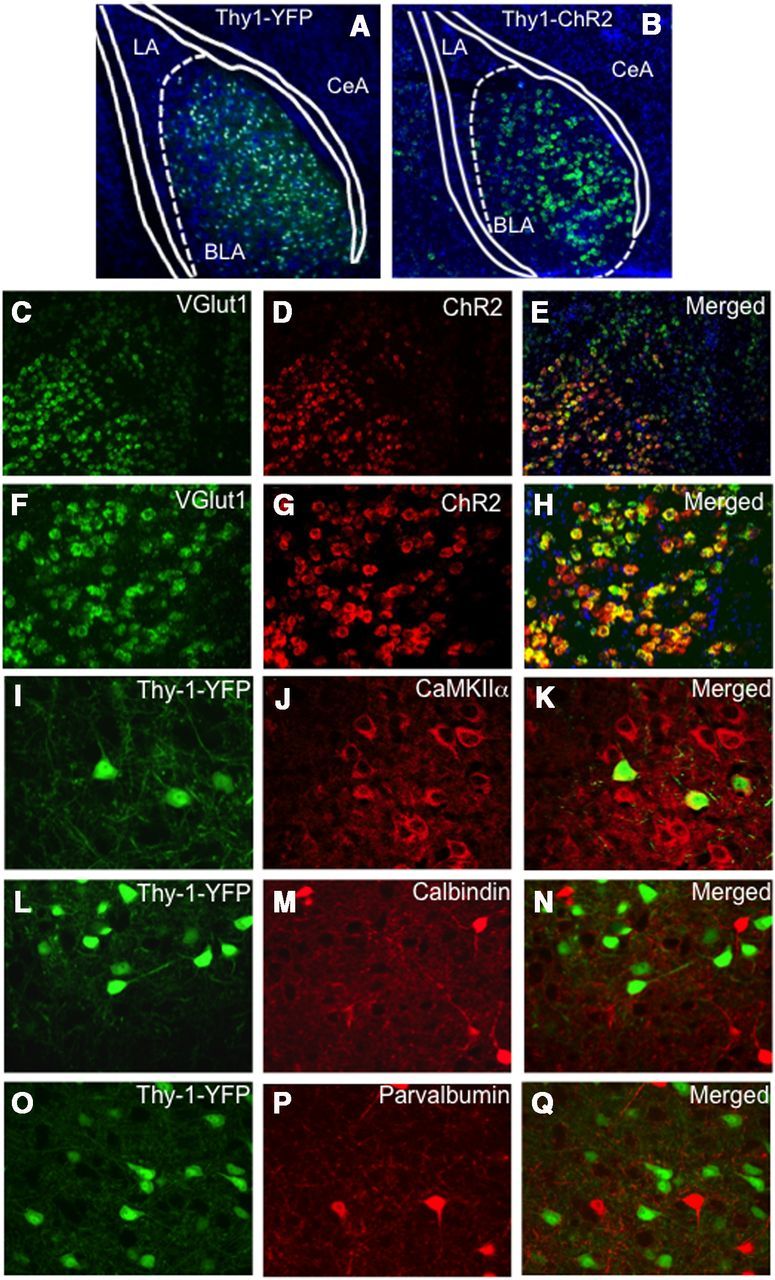
Molecular characterization of Thy1-expressing pyramidal neurons within basolateral amygdala transgene expression driven by the Thy1 promoter in two lines of mice (Thy1-YFP, Thy1-ChR2-EYFP). Extremely similar expression patterns of the two transgenes driven by the Thy1 promoter are observed in the BLA between the two lines. Although the expression pattern is the same, more cells appear labeled in A because of the thickness of the section (45 μm in A vs 16 μm in B). A, A coronal section of a Thy1-YFP mouse, double-labeled with DAPI (blue) staining all cells and YFP labeling the Thy1 subpopulation, in which the Thy1-YFP pyramidal neurons are only located in the BLA, and not the LA or CeA regions. B, A coronal section of a Thy1-ChR2-EYFP mouse identifying ChR2 transgene expression (green) using fluorescent in situ hybridization in which the Thy1-ChR2 neurons are only located in the BLA, and not the LA or CeA regions. Dual in situ hybridization for ChR2 and Vglut1 demonstrating that ChR2 neurons in the BLA are glutamatergic. C–E, Coronal sections (10×) of a Thy1-ChR2-EYFP mouse labeled with VGlut1 mRNA (green, C), ChR2 mRNA (red, D), and merged (E). F–H, Coronal sections (20×) of a Thy1-ChR2-EYFP mouse labeled with VGlut1 mRNA (green, F), ChR2 mRNA (red, G), and merged (H). All Vglut1 neurons do not express ChR2; however, virtually all ChR2 neurons express Vglut1 within the BLA. I–Q, Dual immunocytochemistry characterization of the Thy1-EYFP neuronal population in the Thy1-EYFP mouse line (green) and its colocalization with a subset of CaMKIIα neurons (I–K), but lack of colocalization with the inhibitory calbindin (L–N) or parvalbumin (O–Q) populations.
Electrophysiological correlates of optogenetic activation of Thy1-expressing subpopulation of BLA neurons
Activity of CeM neurons has been linked to increased conditioned freezing (Haubensak et al., 2010); thus, we first examined whether Thy1-ChR2-YFP-expressing neurons within the BLA could inhibit CeM neurons.
Current-clamp recordings from BLA Thy1-ChR2-EYFP neurons demonstrated high-fidelity spiking in response to 20 Hz laser light pulses (488 nm, 15 ms, 0.6–1.2 mW), a frequency commonly used for optogenetic behavioral activation (Fig. 3A; Johansen et al., 2010; Tye et al., 2011; Yizhar et al., 2011). Next, neurons in the CeM were patch-clamped to examine postsynaptic responses to photoactivation of Thy1-ChR2-EYFP-expressing neurons in the BLA (15 ms, 0.2 Hz), as well as to bipolar electrical stimulation of the LA (150 μs, 0.2 Hz; Fig. 3B). Photoactivation of BLA Thy1-ChR2-EYFP-expressing neurons evoked a postsynaptic potential (PSP) in every recorded neuron in the CeM (n = 12). Seven of 12 (58%) CeM neurons exhibited a biphasic response in which an EPSP was shunted by an IPSP (Fig. 3C). The remaining CeM neurons responded with a monophasic EPSP. Notably, in those CeM neurons showing a biphasic response, the IPSP was completely abolished in all neurons tested by bath application of the GABAA receptor antagonist, SR95531 (5 μm; Fig. 3D), and repeated (20 Hz) photoactivation of the BLA elicited a sustained inhibitory response that abolished spiking in CeM neurons depolarized to action potential threshold (F(2,4) = 9.89, p < 0.05; Fig. 3E).
Figure 3.

Electrophysiological effects within the BLA and CeM of optogenetic activation of BLA-Thy1 neurons. A, A representative current-clamp recording depicts typical, high-fidelity spiking of a BLA-Thy1 pyramidal neuron in response to laser light activation (blue bars) at 20 Hz with a pulse width of 15 ms. B, Slice electrophysiology experiments were performed on Thy1-ChR2-EYFP mice with neurons patch clamped in the CeM, a fiber-optic light source targeting the BLA, and a bipolar stimulating electrode placed in the LA. A diagram of the experimental setup is overlaid on a photomicrograph depicting YFP expression (green) in Thy1 neurons of the BLA and a recorded, biocytin-filled neuron in the CeM (red). C, A representative current-clamp recording from a CeM neuron depicts a biphasic response (left) to optogenetic BLA-Thy1 activation (blue bar) and a shunt of an action potential elicited by LA stimulation (stim). Responses without action potentials were averaged for five consecutive sweeps (n = 12). D, A representative, biphasic response in a CeM neuron, held at a membrane potential of −55 mV with direct current injection, to light activation of BLA-Thy1 neurons (left). The response became purely depolarizing following bath application of 5 μm SR95531 and was able to elicit an action potential (right) (n = 3). E, A representative current-clamp recording of a free-firing CeM neuron, depolarized to action potential threshold with direct current, depicts blockade of ongoing spiking by a 20 Hz, 1 s train of optogenetic BLA-Thy1 activation (blue bars) (n = 12). F, Current-clamp recordings of a CeM neuron, depolarized near threshold, depict 20 Hz optogenetic activation (blue bars) of BLA-Thy1 neurons inhibiting the recorded cell (top) and shunting LA-stimulation-induced excitation (bottom), which itself evoked action potentials (middle). Stimulation artifacts were cropped at −25 and −50 mV for clarity.
Electrical stimulation of the LA evoked a monophasic EPSP in eight of 12 (67%) CeM neurons (Fig. 3C). Notably, in the seven CeM neurons that showed a biphasic EPSP/IPSP response to BLA photoactivation, electrical stimulation of the LA evoked either a monophasic EPSP (4 neurons) or no response (3 neurons). In the four CeM neurons that received convergent synaptic input from the LA and BLA, photoactivation of the BLA shunted action potentials driven by electrical stimulation of the LA at both low (0.05 Hz; Fig. 3C) and high (20 Hz) stimulation frequencies (Fig. 3F). Together, these data suggest that this subpopulation of BLA pyramidal neurons inhibits a population of CeM neurons and can shunt direct excitatory drive from the LA.
Activation of Thy1-expressing neurons inhibits fear consolidation
To examine the behavioral effects of BLA Thy1-ChR2-EYFP neuronal activation, mice were implanted with guide cannulae just dorsal to the BLA to enable light activation of the Thy1-ChR2-EYFP neurons. After recovery and habituation to handling, mice underwent fear conditioning. Fear conditioning involved five tone–shock pairings that coterminated with a discrete 2 s optical activation of Thy1-ChR2-EYFP neurons within the BLA (Fig. 4A). We used the same 20 Hz activation frequency as used for the above slice studies and which had been previously reported to be successful in other brain regions when combined with behavior (Arenkiel et al., 2007; Carter et al., 2009; Ciocchi et al., 2010; Gunaydin et al., 2010; Johansen et al., 2010; Zhang et al., 2010). Notably, BLA optical activation was only present during these brief 2 s pulses during CS–US training period. There was no effect of optical activation on fear expression during training (Fig. 4B,C).
Figure 4.
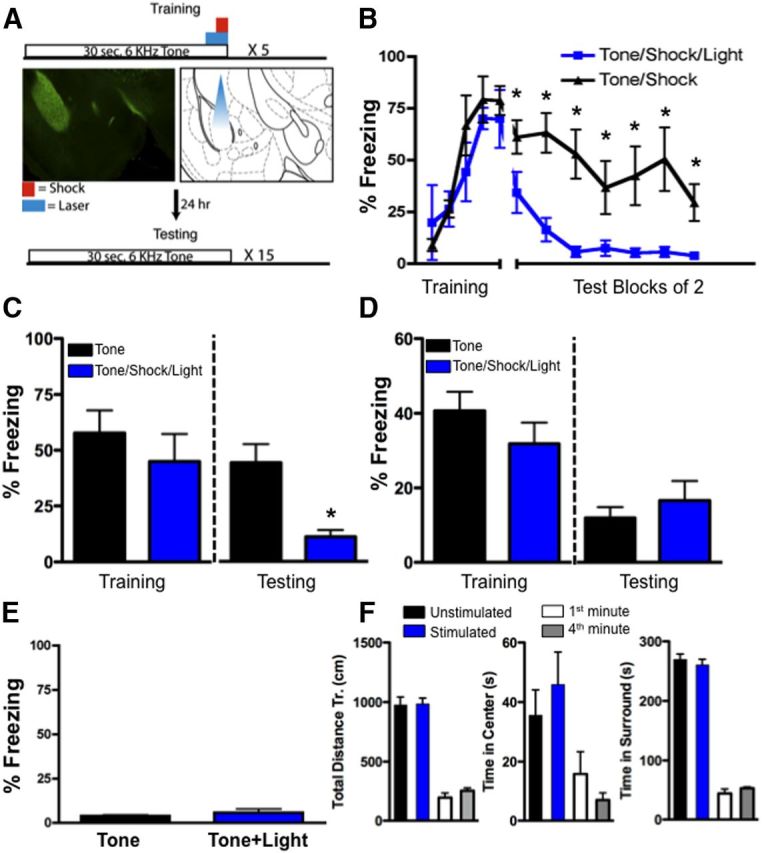
Discrete optogenetic activation of BLA Thy1 neurons inhibits fear memory formation without locomotor or anxiety-like effects. A, Training paradigm. Mice were trained with five trials of tones (30 s, 6 kHz) which coterminated with a 1 s footshock (0.5 mA). Thy1-ChR2-EYFP mice received either sham stimulation or the 30 s tone also coterminated with a 2 s (20 Hz, 15 ms) blue light targeting BLA. Left, EYFP expression in BLA. Right, Anticipated light cone with fiber optic cable. B, Freezing during training and 24 h later when mice were tested (averaged blocks of 2 trials) in the absence of optogenetic activation and in the presence of the conditioned tone cues. Mice that had been trained in conjunction with optogenetic activation (n = 8) show minimal expression of fear (n = 7 controls), C, Representation of data from A as a bar graph. Left, Average freezing responses to the last three tones during training and in the presence or absence of optogenetic stimulation. Right, Average freezing responses from A during testing in the absence of optogenetic stimulation. Mice that had been trained in conjunction with optogenetic activation show minimal expression of fear. D, Optogenetic stimulation in Thy1-EYFP mice lacking ChR2 expression has no effect on conditioned fear. Left, Average freezing responses to the last three tones during training and in the presence (n = 8) and absence (n = 9) of optogenetic stimulation. Right, Average freezing responses during testing in the absence of optogenetic stimulation. E, Optogenetic stimulation during training in Thy1-ChR2-EYFP mice in the absence of a US does not support fear learning (n = 7), thus optogenetic stimulation of Thy1-expressing neurons of the BLA alone cannot serve as a US. Bar graph represents average freezing responses to 10 tone presentations. F, Mice received the same stimulation procedure (2 s, 20 Hz, 15 ms blue light) during a standard open-field testing procedure. Left, Locomotion in the open field [total distance traveled (Tr.)] in control (n = 6) versus stimulated groups (n = 6) of Thy1-ChR2-EYFP mice. Center, Anxiety-like behavior (time in center of open-field) in control versus stimulated groups. Right, Time in surround within open field in control versus stimulated groups. Additionally, there were no time-dependent changes in neophobia during the behavioral test that would mask a lack of effect of optogenetic stimulation during the second 5 min period. White and gray bars represent the same measures as above during the first minute and fourth minute, respectively, within the initial unstimulated testing period. Error bars represent mean ± SEM. *p ≤ 0.05.
Twenty-four hours after fear conditioning, mice were tested in a novel context for freezing responses to the CS. Mice that had previously received CS–US training coterminating with laser photoactivation displayed significantly less freezing in response to CS presentation compared with mice receiving sham stimulation (F(1,12) = 17.84, p = 0.001; t12 = 4.224, p = 0.001; Fig. 4B,C). These data suggest that photoactivation of BLA Thy1-ChR2-EYFP neurons reduced long-term fear memory consolidation. These results are consistent with the hypotheses that optogenetic activation of the Thy1 subpopulation may decrease general fear/anxiety responses, interfere with amygdala functioning, or specifically inhibit conditioned fear. We sought to separate these possibilities with subsequent experiments. To determine whether tissue heating/damage was the cause of the blockade of fear consolidation, we replicated the above experiments using Thy1-YFP mice, which lack ChR2 expression. Here we show that optogenetic stimulation in mice lacking ChR2 does not inhibit conditioned fear (Fig. 4D). Thus, the above effect was unlikely due to tissue heating or damage. Previous data suggested that optogenetic stimulation of the lateral amygdala could serve as an unconditioned stimulus (Johansen et al., 2010). Thus, to determine whether optogenetic stimulation of Thy1-ChR2-expressing neurons within the BLA could serve as an unconditioned stimulus, we paired tone CS presentations with optogenetic stimulation of the BLA and tested the mice for freezing 24 h later. Freezing responses during testing were negligible, suggesting that optogenetic stimulation of Thy1-ChR2-expressing neurons within the BLA is unable to support fear learning in the absence of true unconditioned stimulus (Fig. 4E).
Next, to determine whether the inhibition of freezing was due to a general effect on anxiety-like or unconditioned fear responses, we observed behavior and locomotion in an open-field paradigm during optogenetic activation of Thy1-ChR2-EYFP cells in the BLA. Mice were tested in an open field in the absence or presence of the same 2 s optical stimulation parameters used during fear conditioning (Fig. 4F). Optogenetic stimulation of the BLA did not cause any significant alteration in locomotion or anxiety-like behavior, as measured by total distance traveled (t5 = 0.19, p > 0.05), time spent in the center of the open field (t5 = 0.69, p > 0.05), or time spent in the surround (t5 = 0.62, p > 0.05; Fig. 4F). In addition, there were no time-dependent differences in these open-field measures over the first 5 min period in the absence of optogenetic stimulation (ps > 0.05; Fig. 4F), suggesting that decreased neophobia did not mask an apparent effect of optogenetic stimulation on anxiety. Combined, these data suggest that our discrete optical stimulation parameters, when coterminated with tone and shock, likely have a specific inhibitory effect on the consolidation of long-term fear memory. Moreover, these data suggest that Thy1-ChR2-EYFP-expressing neurons may be part of a specific neural circuit inhibiting fear.
Activation of Thy1-expressing neurons enhances consolidation of fear extinction
If Thy1-ChR2-expressing neurons are part of a specific neural circuit inhibiting fear, then they should also enhance extinction learning. In contrast, if this activation were leading to a general disruption of amygdala processing, we would expect to see a disruption of extinction, as amygdala plasticity is required for extinction learning. To test this hypothesis, mice were trained in a cued fear conditioning paradigm, then 24 h later were extinction trained, followed by testing for extinction retention. Mice were fear conditioned with tone–shock pairings alone and then received optical stimulation during extinction training. Fifteen presentations of the tone CS coterminated with a 2 s optical stimulation of the BLA during extinction training (Fig. 5A), using the same optical stimulation parameters described above. During extinction training, optical activation of Thy1-ChR2 neurons in the BLA had no effect on freezing (F(1,8) = 0.019, p > 0.05; t8 = 0.1821, p > 0.05; Fig. 5B,C), again indicating that our discrete activation parameters had no effect on fear expression or freezing behavior alone.
Figure 5.
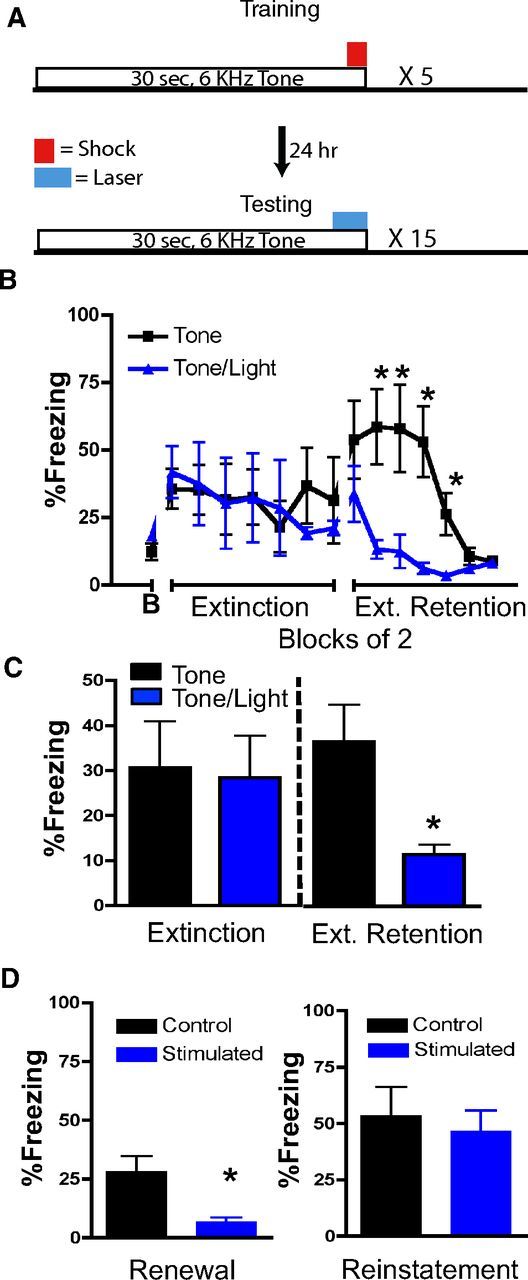
Discrete optogenetic activation of Thy1 neurons within BLA enhances extinction learning. A, Top, Fear training paradigm. All cannulated Thy1-ChR2-EYFP mice were trained with five trials of tones (30 s, 6 kHz), which coterminated with a 0.5 s footshock (0.5 mA). Bottom, Twenty-four hours later, in a separate context, all mice received 15 trials of tones (30 s, 6 kHz) coterminating with either sham stimulation or with a 2 s, BLA-targeted, blue light (20 Hz, 15 ms). B, No difference between sham versus optogenetically activated groups was seen during extinction training (B, Baseline freezing during extinction before CS presentation). However, 24 h later, mice that received the blue light during extinction (n = 5) showed much lower freezing to the cues compared with controls (n = 5), consistent with dense extinction (Ext.) retention. C, Representation of data from B as a bar graph. Left, Average freezing responses during extinction to 15 tone presentations and in the presence or absence of optogenetic stimulation. Right, Average freezing responses from B during extinction retention testing in the absence of optogenetic stimulation. Mice that had been extinction trained in conjunction with optogenetic activation showed enhanced extinction retention. D, Twenty-four hours later, mice were fear tested in the initial conditioned context. Those without optogenetic stimulation (n = 5) showed renewal of fear, whereas those with optogenetic stimulation (n = 5) had minimal freezing to the tone. Following footshock reinstatement in a new context, all mice showed similar levels of tone-activated freezing behavior. Error bars represent mean ± SEM. *p ≤ 0.05.
However, when the mice were retested for extinction retention in the absence of optical stimulation, mice previously extinction trained with optical stimulation displayed significantly less fear compared with mice receiving sham stimulation. This suggests that optical stimulation of BLA Thy1-ChR2-EYFP neurons enhanced extinction consolidation (F(1,8) = 9.0, p = 0.017; t8 = 2.972, p = 0.017; Fig. 5B,C). Thus, optical stimulation within the BLA did not result in a general impairment of amygdala functioning due to damage or enhanced feedback inhibition onto interneurons producing reduced learning or plasticity, two potential explanations for the deficit in fear learning we observed previously. In both cases, we would also expect to see an inhibition of extinction of fear rather than the enhanced fear extinction we observed (note that a number of other manipulations such as pharmacological blockade of amygdala functioning, blockade of NMDA functioning, or amygdala lesions all inhibit both fear learning and extinction learning; Myers and Davis, 2006; Johansen et al., 2011). Therefore, these data are consistent with a relatively specific effect of BLA Thy1-ChR2-EYFP neuronal activation on enhancing inhibitory/extinction learning.
We next tested whether the enhanced extinction consolidation would transfer to a new context. It is generally accepted that extinction is a form of context-dependent learning, not an erasure of the original fear memory (Myers et al., 2006). Placing mice back into the original training context typically results in a return of freezing, termed “fear renewal”. Here, we demonstrate that optogenetic-induced enhanced extinction consolidation also inhibits renewal of fear (F(1,8) = 9.4, p = 0.01; Fig. 5D). These data suggest that the enhanced extinction learning is potentially due to a strengthening of the CS–no-US association, rendering context to be less relevant and enabling the transfer of extinction across contexts.
One could argue that the blockade of spontaneous recovery (enhanced extinction retention) and renewal of fear with optogenetic stimulation of BLA Thy1-ChR2-EYFP neurons represents erasure or unlearning of the fear memory. To test this possibility, we examined both groups of mice in a reinstatement test. Mice that were previously extinction trained were subjected to a single unsignaled footshock in an entirely new context. We then assessed tone-evoked freezing 24 h later (reinstatement test). Here, we demonstrate that the fear memory was fully reinstated following a single unsignaled footshock (F(1,8) = 0.19, p > 0.05; Fig. 5D), consistent with a savings of the prior memory trace, indicating that the fear memory was not erased. Rather, optogenetic stimulation of Thy1-ChR2-EYFP-expressing BLA neurons appears to directly enhance fear extinction consolidation and may also render the extinction memory independent of context modulation, which is critical to the process of fear renewal. In addition, mice can express fear normally and can be retrained to an additional cue following optogenetic-enhanced extinction, suggesting that activation of Thy1-ChR2-EYFP-expressing neurons does not permanently alter amygdala function.
Discussion
Our data demonstrate that pairing activation of a specific neuronal population only during the CS–US presentation inhibited the memory for the association between the CS and US, and thus inhibited fear consolidation. Conversely, during extinction training, pairing activation of Thy1-ChR2-expressing neurons during a very discrete time window during presentation of the CS alone enhanced the CS–no-US association, and thus enhanced extinction retention. It is important to note the associative nature of the pairing of the optogenetic stimulation with the CS–US or CS–no-US as compared to previous reports using prolonged stimulation parameters examining fear and anxiety-like behavior (Goshen et al., 2011; Tye et al., 2011). The present data suggest that the BLA contains a subpopulation of glutamatergic neurons that may be specialized for inhibiting fear responses.
We demonstrate that a Thy1-ChR2-EYFP-expressing subpopulation of BLA neurons can inhibit CeM neuronal activity. In contrast, electrical stimulation of the LA evoked a purely excitatory response in CeM neurons. Significantly, concurrent optogenetic activation of Thy1-ChR2-EYFP neurons within the BLA shunted the excitatory LA driven response and prevented CeM neuron activation. Discrete optogenetic activation of Thy1-ChR2-EYFP-expressing neurons in the BLA during cued fear conditioning inhibits fear consolidation, whereas activation during suboptimal fear extinction training enhances the consolidation of the extinguished fear response. The observed enhancement of extinction learning was sufficient to allow transfer of the extinguished response to the training context in a fear renewal test. This did not, however, disrupt the initial memory trace, because the fear response was fully reinstated with a single unsignaled footshock. It is unlikely that the observed extinction enhancement was due to a blockade of reconsolidation because of the duration of exposure to the CS in the present study. Several recent studies demonstrate that one of the key factors in distinguishing reconsolidation processes from extinction processes is the duration of CS exposure. In the present study, mice were exposed to 15 CS tones over a period of 22 min, which is beyond what has been reported to initiate reconsolidation, but does initiate extinction (Pedreira and Maldonado, 2003; Suzuki et al., 2004; Bredy and Barad, 2008; Pérez-Cuesta and Maldonado, 2009; Kirtley and Thomas, 2010; Inda et al., 2011; de la Fuente et al., 2011). Thus, despite the long-held role of excitatory neurotransmission within the BLA in fear, the current study demonstrates that activation of a population of glutamatergic neurons within the BLA specifically inhibits fear, apparently without disrupting the original memory trace.
Importantly, activation of Thy1-ChR2-EYFP-expressing neurons during the open-field test did not reduce anxiety-like behavior or alter locomotion. Although possible, this lack of effect on anxiety was unlikely due to a lack of neophobia during the second 5 min period of testing, as anxiety-like behavior did not vary as a function of time during the unstimulated time of the open-field test (Fig. 4F). In addition, optogenetic stimulation was unable to support fear learning in the absence of an unconditioned stimulus (Fig. 4E), indicating behavioral specificity of our stimulation parameters. In another study, optical activation of the lateral amygdala paired with presentation of a conditioned stimulus during training resulted in an increase in freezing during testing 24 h later (Johansen et al., 2010), suggesting that activation of LA pyramidal neurons is sufficient to produce long-term fear memory when associated with a CS in the absence of a US. These data further support our electrophysiological results of electrical stimulation of the LA activating CeM neurons, which could be shunted by optogenetic stimulation of BLA Thy1-ChR2-expressing neurons (Fig. 2). As activity of CeM neurons has been linked to increased conditioned freezing (Haubensak et al., 2010), our electrophysiological and behavioral data fit nicely with the role of this nucleus regulating fear responses.
Our findings are consistent with an earlier report (Herry et al., 2008) suggesting that the previously identified extinction neurons represent a subset of a larger population of neurons within the BLA that are more generally involved in inhibiting fear memory. Interestingly, the slice physiology data suggest that some neurons in the CeM are inhibited while others are excited by optogenetic photoactivation of BLA Thy1-ChR2-EYFP neurons. In these studies of auditory fear, Thy1-ChR2-EYFP neuronal activation within the BLA appears to selectively support inhibition of fear learning and enhancement of fear extinction. One possible mechanism by which Thy1-ChR2-EYFP neurons inhibit fear may be via indirect inhibition of neurons in the CeM, as we showed for a number of CeM neurons upon optogenetic activation of these BLA neurons. Indeed, in a previous report, optical activation of BLA fibers within the lateral CeA inhibited anxiety-like behavior, whereas inhibition of the same fibers enhanced anxiety-like behavior (Tye et al., 2011). Our current data support a similar mechanism for fear inhibition. Here, we observed feedforward inhibition of CeM neurons via optical stimulation of Thy1-ChR2-expressing BLA neurons (Fig. 3). It is likely that that the Thy1-ChR2-EYFP neurons examined in the present study represent a similar population of neurons driving CeA responses involved in anxiety-like behavior, although this was not definitively determined. It is also possible that Thy1-ChR2-EYFP-expressing neurons represent a microcircuit regulating direct local responses within the BLA, but are synaptically connected to neurons that project to the CeA.
In the Herry and colleagues' (2008) study, extinction neurons represented 14% of total recorded units, which is a much smaller population than those activated here. In addition, extinction neurons are preferentially synaptically connected to the mPFC, placing them in an ideal position to regulate fear extinction. Thy1-ChR2-EYFP-expressing neurons make up a sizable portion of large spiny glutamatergic neurons in the BLA, as identified with in situ hybridization and immunohistochemistry in Thy1-YFP transgenic mice. Currently, it is unknown whether Thy1-YFP-expressing neurons of the BLA receive afferent inputs from the mPFC, or how they may be different from other glutamatergic neurons within the BLA. However, Thy1-YFP-expressing BLA neurons also send efferent fibers along the stria terminalis, terminating primarily in the nucleus accumbens (NAc) and caudate nucleus, as well as the bed nucleus of the stria terminalis (Porrero et al., 2010). The nucleus accumbens has previously been implicated in fear extinction (Pezze and Feldon, 2004; Holtzman-Assif et al., 2010; Muschamp et al., 2011), suggesting an essential role of this BLA–NAc pathway in fear inhibition/extinction. Overall, the circuit connectivity between mPFC, NAc, and BLA may act in concert with the CS-responding Thy1-YFP neurons to drive extinction learning in contrast to supporting continued fear expression.
Notably, there is a wealth of prior data showing that simply activating or inactivating the amygdala as a whole does not produce the patterns of functional responses that we have observed. Prior data demonstrate that general inhibition of the BLA through GABA agonists or NMDA antagonists block the acquisition/consolidation of fear learning (Wilensky et al., 1999) as well as block the extinction of fear (Falls et al., 1992). There is also evidence that general pharmacological enhancement of NMDA function in the BLA enhances fear acquisition (Maren et al., 1996) as well as fear extinction (Walker et al., 2002). Thus, our results are most parsimoniously interpreted as the specific activation of a functional subpopulation of pyramidal neurons.
In conclusion, these data suggest that activation of specific, genetically marked subpopulations of principal neurons within the BLA may selectively inhibit fear. It should be noted, however, that the use of transgenics in the current study do not allow us to rule out the possibility that stimulation of fibers passing through the BLA resulted in some of the observed electrophysiological or behavioral effects. Further work remains to be done to more fully characterize the circuit within which Thy1-ChR2-EYFP neurons inhibit fear and to understand whether this property is shared by all of the Thy1-YFP subpopulation or just those whose activation leads to CeM inhibition. Identification of molecular markers of neurons that selectively inhibit fear or enhance extinction would be particularly useful for enhancing our understanding of fear processes. Additionally, cell-type-specific amplification of mRNA may allow the identification of specific receptor targets that are preferentially expressed on this subset of cells, potentially leading to novel targets for disorders of fear regulation.
Footnotes
This work was supported by The Center for Behavioral Neuroscience of the National Science Foundation under Agreement No. IBN-9876754, NIH Grant DA019624, and the Yerkes National Primate Research Center Base Grant 2P51RR000165-51.
References
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, Feng G. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4 ed. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60:352–360. doi: 10.1016/j.biopsych.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, de Lecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci. 2009;29:10939–10949. doi: 10.1523/JNEUROSCI.1205-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Müller C, Lüthi A. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton J 2nd, editor. The amygdala. Oxford: Oxford UP; 2000. pp. 213–287. [Google Scholar]
- Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Goshen I, Brodsky M, Prakash R, Wallace J, Gradinaru V, Ramakrishnan C, Deisseroth K. Dynamics of retrieval strategies for remote memories. Cell. 2011;147:678–689. doi: 10.1016/j.cell.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387–392. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Lüthi A, Anderson DJ. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Lüthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Holtzman-Assif O, Laurent V, Westbrook RF. Blockade of dopamine activity in the nucleus accumbens impairs learning extinction of conditioned fear. Learn Mem. 2010;17:71–75. doi: 10.1101/lm.1668310. [DOI] [PubMed] [Google Scholar]
- Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. J Neurosci. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, LeDoux JE. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A. 2010;107:12692–12697. doi: 10.1073/pnas.1002418107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirtley A, Thomas KL. The exclusive induction of extinction is gated by BDNF. Learn Mem. 2010;17:612–619. doi: 10.1101/lm.1877010. [DOI] [PubMed] [Google Scholar]
- de la Fuente V, Freudenthal R, Romano A. Reconsolidation or extinction: transcription factor switch in the determination of memory course after retrieval. J Neurosci. 2011;31:5562–5573. doi: 10.1523/JNEUROSCI.6066-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Maren S, Fanselow MS. The amygdala and fear conditioning: has the nut been cracked? Neuron. 1996;16:237–240. doi: 10.1016/S0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037/0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- McDonald A, Muller J, Mascagni F. GABAergic innervation of alpha type II calcium/calmodulin-dependent protein kinase immunoreactive pyramidal neurons in the rat basolateral amygdala. J Comp Neurol. 2002;446:199–218. doi: 10.1002/cne.10204. [DOI] [PubMed] [Google Scholar]
- Muschamp JW, Van't Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, Meloni EG, Carlezon WA., Jr Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31:3095–3103. doi: 10.1523/JNEUROSCI.5973-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2006;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2004. [Google Scholar]
- Pedreira ME, Maldonado H. Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron. 2003;38:863–869. doi: 10.1016/S0896-6273(03)00352-0. [DOI] [PubMed] [Google Scholar]
- Pérez-Cuesta LM, Maldonado H. Memory reconsolidation and extinction in the crab: mutual exclusion or coexistence? Learn Mem. 2009;16:714–721. doi: 10.1101/lm.1544609. [DOI] [PubMed] [Google Scholar]
- Pezze MA, Feldon J. Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol. 2004;74:301–320. doi: 10.1016/j.pneurobio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Porrero C, Rubio-Garrido P, Avendaño C, Clascá F. Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res. 2010;1345:59–72. doi: 10.1016/j.brainres.2010.05.061. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci. 1999;19:48RC. doi: 10.1523/JNEUROSCI.19-24-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]