

Abstract
Locomotion and cue-dependent behaviors are modified through corticostriatal signaling whereby short-term increases in dopamine availability can provoke persistent changes in glutamate release that contribute to neuropsychiatric disorders, including Parkinson's disease and drug dependence. We found that withdrawal of mice from repeated amphetamine treatment caused a chronic presynaptic depression (CPD) in glutamate release that was most pronounced in corticostriatal terminals with a low probability of release and lasted >50 d in treated mice. An amphetamine challenge reversed CPD via a dopamine D1-receptor-dependent paradoxical presynaptic potentiation (PPP) that increased corticostriatal activity in direct pathway medium spiny neurons. This PPP was correlated with locomotor responses after a drug challenge, suggesting that it may underlie the sensitization process. Experiments in brain slices and in vivo indicated that dopamine regulation of acetylcholine release from tonically active interneurons contributes to CPD, PPP, locomotor sensitization, and cognitive ability. Therefore, a chronic decrease in corticostriatal activity during withdrawal is regulated around a new physiological range by tonically active interneurons and returns to normal upon reexposure to amphetamine, suggesting that this paradoxical return of striatal activity to a more stable, normalized state may represent an additional source of drug motivation during abstinence.
Introduction
The neocortex refines volitional movements and goal-directed behaviors through the corticostriatal–basal ganglia–thalamocortical feedback loop (Albin et al., 1989; Jog et al., 1999). The input of this neural network consists of glutamatergic cortical afferents that excite D1-class (D1R) and D2-class dopamine receptor (D2R)-expressing striatal medium-sized spiny neurons (MSNs), which form distinct direct and indirect pathways that promote and suppress competing motor movements, respectively (Pennartz et al., 1994; Nicola et al., 2000). Modulation of these excitatory corticostriatal synapses is determined by the availability of dopamine and acetylcholine, which are necessary for the establishment of reward, attention, and motor learning (Kalivas and Volkow, 2005; Cepeda et al., 2010). Emerging evidence suggests that abnormalities in the availability of these neuromodulators may promote an imbalance between direct and indirect striatal pathways (Beutler et al., 2011; Kozorovitskiy et al., 2012; Wang et al., 2012) to produce the motor and neuropsychological symptoms of Parkinsonism and drug dependence (Kalivas et al., 2003; Bamford and Cepeda, 2009).
Addiction is considered a chronic, allostatic condition (Ahmed and Koob, 2005) characterized by drug seeking behaviors and relapse after withdrawal (Kalivas and Volkow, 2005). Psychostimulants have a high potential for abuse because they acutely increase brain dopamine levels (Sulzer, 2011) and their repeated use can trigger long-lasting changes in striatal glutamate (Pierce et al., 1996; Cornish et al., 1999; Kalivas et al., 2003; McFarland et al., 2003) and acetylcholine (Abercrombie and DeBoer, 1997; Bamford et al., 2008) availability that are reversible upon drug challenge.
The striatum is partitioned into distinct regions based on connectivity and function. Although psychostimulant-induced synaptic plasticity has been described in the nucleus accumbens (Kalivas and Volkow, 2005), several other neuronal loci are also likely involved, including the dorsal striatum (Everitt and Robbins, 2005; Porrino et al., 2007). Specifically, the dorsal striatum participates in habit formation (Yin et al., 2004; Everitt and Robbins, 2005), motoric responses (Bamford et al., 2008; Beutler et al., 2011; Ferguson et al., 2011), cognitive flexibility (Stalnaker et al., 2009; Darvas and Palmiter, 2011), and cue-dependent behaviors (Darvas and Palmiter, 2011; Quintana et al., 2012) that are characteristic of drug seeking behaviors. However, it remains unclear how repeated dopamine release can promote long-lasting plasticity in striatal synapses that might account for these behaviors.
Here, we assessed behaviorally relevant changes in corticostriatal activity in the dorsal striatum after acute and repeated amphetamine by combining electrophysiological recordings in MSNs and acetylcholine-releasing tonically active striatal interneurons (TANs) with optical recordings from individual synaptic terminals. These measurements were complemented with behavioral, biochemical, and electrophysiological recordings in TANs and MSNs from choline acetyltransferase knock-out (KO) mice in which acetylcholine synthesis was selectively reduced after injection of AAV1-Cre-GFP into the dorsal striatum to determine how acetylcholine availability can produce cognitive inflexibility, locomotor sensitization, and long-lasting, yet reversible dopamine-dependent changes in corticostriatal function.
Materials and Methods
Animals
All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council of the National Academies, Washington, DC) and were approved by the institutional animal care and use committee at the University of Washington. Mice 4–9 weeks of age were housed 5 to a cage in a modified specific pathogen free vivarium with a 12 h light/dark cycle and ad libitum access to food and water, except during locomotor recording. C57BL/6 male mice (n = 203), were obtained from The Jackson Laboratory. Hemizygous Drd1- and Drd2-EGFP BAC transgenic mice (n = 22) were generated as described previously and bred onto a C57BL/6 background (Cepeda et al., 2008; generously provided by Dr. Michael Levine, University of California, Los Angeles). For terminal procedures, mice were anesthetized with Nembutal (200 mg/kg i.p.) or with ketamine (650 mg/kg i.p.) and xylazine (44 mg/kg i.p.) before killing. For some experiments, mice were treated with Reserpine (5 mg/kg i.p.) and killed 12 h later at the onset of behavioral arrest. Conditional choline acetyltransferase (Chatlox/lox) mice (n = 48), which have exons 3 and 4 flanked by loxP sites, were generated as described previously (Misgeld et al., 2002; generously provided by Dr. Joshua Sanes, Harvard University, Cambridge, MA) and back-crossed for 12 generations onto a C57BL/6 genetic background. Acetylcholine synthesis was selectively reduced after injection of AAV1-Cre-GFP into the dorsal striatum (vChAT-KO). Sham control mice (sham-KO; n = 46) were wild-type littermates of homozygous conditional KO mice and were injected with the same AAV1-Cre-GFP.
Surgery
AAV1-Cre-GFP virus was generated and titered as described previously (Quintana et al., 2012). The virus was prepared in HEK293 cells with AAV1 coat serotype, purified by sucrose and CsCI gradient centrifugation steps, and then resuspended in HBSS at a titer of 2 × 109 viral genomes/ml. AAV1-Cre-GFP was injected into two sites of the dorsal striatum, in one hemisphere only (unilateral injection), or into four sites of the dorsal striatum (bilateral injection). For unilateral injections, 0.5 μl of virus was injected 1.3 mm anterior of bregma, +1.5 mm lateral to midline, and 3 mm ventral from the skull surface and 0 mm anterior of bregma, +1.75 mm lateral to midline, and 3 mm ventral from the skull surface. For bilateral injections, 0.5 μl of virus was injected 1.3 mm anterior of bregma, ±1.5 mm lateral to midline, and 3 mm ventral from the skull surface, and 0 mm anterior of bregma, ±1.75 mm lateral to midline, and 3 mm ventral from the skull surface. Unilateral and bilateral injections were performed on 3- to 4-week-old mice that had either no (sham-KO) or both (vChAT-KO) choline acetyltransferase alleles targeted with loxP sites. Bilateral injections were also made in older, 3- to 4-month-old mice and these data were pooled with younger mice because there was no difference in animal behaviors (data not shown). All surgeries were performed on anesthetized (with isoflurane) animals. After the surgeries, the animals were allowed to recover for 2–3 weeks before they were submitted to further experimental procedures.
Behavior
Locomotor sensitization.
Locomotor responses were determined using animal activity monitor cages (San Diego Instruments). Four infrared beams separated by 8.8 cm that cross the width of each chamber were connected to an IBM computer, which recorded the number of times each beam was broken. Some locomotor responses were measured in static mouse cages (37.2 cm D × 23.4 cm W × 14 cm H) with 16 photo cells per side (Columbus Instruments). Locomotor activity was measured in ambulations (2 consecutive beam interruptions) summated over 5 min intervals. On each test day, animals were acclimated to individual activity chambers for 90 min to allow them to become accustomed to the behavioral cage before subsequent injections of either amphetamine or saline. After the injection, the animals were placed back into their respective activity chambers and ambulations were recorded for 90 min. To separate the effects of novelty from the pharmacological effects of the drug, animals were acclimated to the locomotor chambers and injected with saline on experiment days 1, 2, 26, and 27.
Novelty locomotion.
Locomotor activity was recorded in the same locomotion chambers as described in the amphetamine sensitization procedure. Ambulations recorded over the first 60 min spent by the animals in the novel environment, constituted by the location and arrangement of the locomotion chambers, were scored as novelty-induced activity.
Motor coordination.
Learning-independent motor coordination was measured using the balance beam procedure. Mice had to traverse 60 cm along a cylindrical rod (15 mm in diameter) that was elevated (30 cm) above a cushioned table. The mice were placed on one end of the beam and allowed to walk to the other side. Mice that fell were placed back on the beam at the position where they fell and allowed to continue. The test was performed only once and the number of slips was recorded.
Motor skill learning.
The latency to fall from a rotarod (Rotamex 4/8 system; Columbus Instruments) was recorded over 3 consecutive days with 4 trials per day and an intertrial interval of 10 min. For each trial, mice were placed on the rotarod, which began at 4 rpm and accelerated to 40 rpm over the course of 5 min.
Visuospatial function and spatial reference memory.
The Morris water maze was used to assess visuospatial learning and spatial reference memory. Mice were trained to locate a hidden platform over a period of 4 d with 4 90 s trials per day. On each trial, mice were released into the pool from a different location. All sessions were recorded with a camera and analyzed with Ethovision software (Noldus). The circular pool was 84 cm in diameter and filled with opaque water at 22°C. No visual cues were present within the pool. External cues were provided through the wall decoration of the room. Visuospatial learning was measured as latency to reach the hidden platform. Swim speed was recorded. One day after the fourth training session, mice performed a 90 s spatial memory trial in which no platform was present in the maze, which was scored as the percentage of time spent in the quadrant of the pool where the platform was positioned during training.
Water U-maze learning.
Cue-dependent learning, egocentric learning, and cognitive flexibility were measured using a U-maze water escape task (Darvas and Palmiter, 2009, 2011). The maze consisted of a stem that led to two backward bent arms (one white and one black) at the end of which an escape platform not visible from the end of stem could be placed. The right-left orientation of the white and black arm of the maze was alternated in a pseudorandom sequence every day so that either arm was equally located on both sides of the maze during daily sessions. Mice were always trained with 10 trials/d that were separated by 3–5 min of intertrial intervals. The percentage of correct trials and latencies to reach the platform were recorded. Mice remained in the maze until a correct turn was made to ensure an equal number of reinforced responses. For cue-dependent learning, one half of all mice had to learn that the escape platform was always located at the end of the black arm and the other half of the mice had to learn that it was always at the end of the white arm. Mice were trained on this procedure for 4 d. Another group of mice was trained under egocentric learning conditions, in which one half of all mice had to learn that the escape platform was always located in the right arm and the other half had to learn that it was always in the left arm of the maze. Mice were trained for 3 d. The same mice that learned the egocentric escape strategy were then used for the cognitive flexibility test. One day after the last reinforced egocentric water escape, the rules of the maze were now changed to cue-dependent conditions (as described above). Mice were trained to learn this strategy shift for 5 d.
Behavioral tests during amphetamine withdrawal
Wild-type mice were treated for 2 d with saline and then for 5 d with either saline or amphetamine (2 mg/kg) and locomotor ambulations were recorded. In amphetamine-treated mice, locomotor sensitization was tested by amphetamine challenge on experiment days 10 and 28. Behavioral experiments began on experiment day 20. Visuospatial function was assessed in half of the animals (8 sensitized and 7 nonsensitized) using the Morris water maze procedure. Six days after the Morris water maze test, all mice were injected with saline and motor coordination was tested 15 min later in a beam-walk test. One day later, the same mice were then injected with nicotine (0.25 mg/ kg) and subjected to the beam-walk test. The other half of the sensitized (n = 7) and nonsensitized (n = 8) mice were tested using the beam-walk test on experiment day 20. For these mice, the same procedure of saline treatment on the first day of motor testing and nicotine treatment on the second day of motor testing was used.
Histological analysis and acetylcholine measurement
The AAV1-Cre-GFP virus injection site was verified by detecting virus-mediated expression of GFP on 30 μm coronal brain sections from mice that were transcardially perfused with 4% paraformaldehyde. To measure acetylcholine, mice were injected with eserine (0.32 mg/kg i.p.; Sigma), their heads were removed 10 min later, microwaved at full power for 5 s, and then a punch containing the dorsal striatum was prepared and flash frozen. For some experiments, eserine was not used and microwave treatment occurred immediately upon decapitation. Acetylcholine content was determined by high-performance liquid chromatography by the Neurochemistry Core Laboratory at Vanderbilt University's Center for Molecular Neuroscience Research (Bamford et al., 2008).
Electrophysiology
Evoked EPSCs (eEPSCs) and miniature EPSCs (mEPSCs) were recorded in 371 MSNs and 85 TANs from 192 mice. Standard techniques were used to prepare 300 μm slices for electrophysiology (Bamford et al., 2004b). Experiments in the dorsal striatum were performed using coronal sections containing the motor cortex and dorsal striatum second to fourth frontal slice of caudate–putamen (bregma, +1.54 to +0.62 mm). Experiments in the nucleus accumbens shell used sagittal sections containing both the PFC and nucleus accumbens at an interaural distance range of ±0.72 mm to ±1.44 mm from midline (Paxinos and Franklin, 2005). Brains were dissected and submerged in ice-cold, carbogenated (95% O2, 5% CO2) cutting solution containing the following (in mm): 4 KCl, 26 NaHCO3, 1.25 NaH2PO4, 3.3 MgSO4, 1.7 MgCl2, 1 CaCl2, 236 sucrose, and 10 glucose, pH 7.2–7.4, 290–310 mOsm. Slices were prepared on a Vibratome and then transferred to an incubating chamber with carbogenated artificial CSF solution (aCSF) plus the following (in mm): 124 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, and 10 glucose, pH 7.2–7.4, 290–310 mOsm at room temperature. After 1 h, slices were placed on the stage of an upright Zeiss Axioskop FS or an Olympus BX51WI microscope and submerged in continuously flowing carbogenated aCSF (3 ml/min) warmed to 35°C.
Whole-cell patch-clamp recordings in voltage-clamp mode were obtained from MSNs visualized in slices with the aid of infrared videomicroscopy coupled with differential interference contrast optics. MSNs were identified by size (∼8–12 μm), membrane properties (Joshi et al., 2009), and fluorescence. Cell identification was confirmed by labeling with 1% biocytin according to published protocols (Joshi et al., 2009). The series resistance of each cell was <20 MΩ (14 ± 0.7 MΩ and 15 ± 0.5 MΩ for saline- and amphetamine-treated mice, respectively). Electrophysiological properties were monitored throughout the recording and cells were removed from further analysis if the series resistance changed by >20%. For voltage-clamp recordings, the patch pipette (4–6 MΩ) contained the following internal solution (in mm): 125 Cs-methanesulfonate, 3 KCl, 4 NaCl, 1 MgCl2, 5 MgATP, 5 EGTA, 8 HEPES, 1 Tris-GTP, 10 di-sodium phosphocreatine, 0.1 leupeptin, and 4 N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314), pH 7.2–7.3, 270–280 mOsm. In some experiments, the cAMP inhibitor Rp-cAMPS (400 μm) was added to the internal solution. EPSCs were isolated by blocking GABAA receptors with bicuculline (10 μm). Cells were held at −70 mV to further minimize the contribution of GABAA-mediated events and that of voltage-gated conductances. Amphetamine was used to elevate striatal dopamine concentrations to ∼3 μm after superfusion for 5–10 min (Bamford et al., 2004b) via reversal of the dopamine transporter (Sulzer, 2011).
Synaptic currents were evoked by electrical stimulation of the deep cortical layers at stimulation strengths adjusted to 1.5 × threshold. For recordings in coronal sections, a twisted tungsten bipolar electrode (Plastics One) was placed over the motor cortex. For recordings in sagittal sections, the electrode was placed over the dorsal PFC, which preferentially projects to the nucleus accumbens (Gorelova and Yang, 1997). Paired current pulses (200 μs) delivered at 20 Hz were presented every 30 s. Five paired pulses were delivered to establish a stable baseline. Each cell then received 10 paired pulses in background aCSF before receptor ligands were added to the bath. eEPSC amplitudes and paired-pulse ratios (PPR) were averaged and compared 5 min before and 5–7.5 min after the receptor ligand was applied. PPRs were determined by dividing the amplitude of the second pulse by that of the first pulse and then multiplying by 100. Cells demonstrating eEPSCs with variable latencies or prolonged durations suggesting polysynaptic responses were rejected from further analysis. Ligands were superfused or puff applied as indicated using a fast-step perfusion system (Warner Instruments).
To measure AMPAR and NMDAR response ratios, MSNs were voltage clamped at +40 mV and GABAA-receptor-mediated IPSCs were blocked with bicuculline. Layers V and VI of the motor cortex were stimulated with bipolar electrodes at 0.2 Hz, with the intensity adjusted to obtain stable eEPSC amplitudes between 150 and 250 pA. Mixed AMPAR/NMDAR responses were measured and averaged over 20 sweeps. The NMDAR antagonist DL-2-amino-5-phosphonopentoic acid (APV) was then bath applied and, 5 min later, AMPAR responses were measured and averaged over 20 sweeps. At the conclusion of each experiment, the AMPAR antagonist NBQX (2 μm) was applied to confirm that the remaining currents were AMPAR mediated. Membrane parameters were monitored throughout the recording and cells were removed from the analysis if the holding current changed by >20%. Currents were Bessel filtered at 2 kHz and digitized at 50 μs using an IBM computer equipped with Digidata 1440A data acquisition and pClamp10.2 software (Molecular Devices). Peak AMPAR-mediated currents were measured by averaging a 5 ms window around the eEPSC peak in the presence of APV; peak NMDAR-mediated currents were measured from a 5 ms window around the peak of the resulting trace created by subtracting the eEPSC in the presence of APV from the eEPSC without APV. Amplitudes were normalized by the peak NMDAR value for each respective pair of traces.
mEPSCs were recorded in gap-free mode both before and 10 min after beginning bath application of a receptor ligand (Wang et al., 2013). The Na+ channel antagonist TTX (1 μm) was added to block spontaneous cortically derived action potentials and to isolate presynaptic terminal activity. The membrane current was filtered at 1 kHz and digitized at 100 μs using Clampfit 10.2 (Molecular Devices). Spontaneous synaptic events were analyzed offline using Mini Analysis (Synaptosoft). The threshold amplitude for the detection of an event was adjusted to at least 2 times above root mean square noise level (∼2–3 pA at −70 mV). Synaptic events could be prevented by adding NBQX (10 μm) to the bath solution, indicating that they arose from activation of glutamatergic receptors.
For cell-attached recordings in TANs, cells were visualized in slices with the aid of infrared videomicroscopy coupled with differential interference contrast optics and were identified by size (∼18–25 μm) and repetitive firing in gap-free mode. Cell identification was confirmed after each experiment by measuring passive and active membrane properties in whole-cell configuration (with cells clamped at −70 mV) and by labeling with 1% biocytin. The internal solution contained the following (in mm): 119 KMeSO4, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 12 phosphocreatine, 2 Na2ATP, and 0.7 Na2GTP, pH 7.2, 280–300 mOsm/L (Bennett and Wilson, 1999; Maurice et al., 2004). Cells were allowed to stabilize for 5 min after achieving a seal resistance >1 GΩ, and their activity was then sampled for 5 min to establish a stable baseline before bath application of receptor ligands. Spontaneous interspike intervals were determined offline with Mini Analysis. Responses to ligands were determined using Clampfit subroutines by comparing average spiking frequencies during the 5 min baseline period with those obtained 5–7.5 min after application of the receptor ligand. Cells were removed from further analysis if the seal resistance or firing rate fell below 1 GΩ or 0.3 Hz, respectively (Bennett and Wilson, 1999).
FM1-43 loading and unloading
Optical recordings of cortical afferents in the dorsal motor striatum were performed on 151 slices from 31 mice, as described previously (Bamford et al., 2004a; Wang et al., 2012). Coronal sections were prepared as in the electrophysiology studies and were allowed to recover for 1 h in carbogenated aCSF containing the following (in mm): 109 NaCl, 5 KCl, 35 NaHCO3, 1.25 NaH2PO4, 20 HEPES, 1.2 MgCl2, 2 CaCl2, and 10 glucose, pH 7.3–7.4, 295–305 mOsm at room temperature. During the experiment, slices were held in a RC-27L incubation chamber (56 μl/mm; Warner Instruments) and perfused at 3 ml/min with aCSF warmed to 35°C. To ensure equilibrium, sections were exposed to pharmacological agents for 10 min before stimulation-mediated unloading. Bicuculline methiodide, APV, and NBQX were dissolved in dimethylsulfoxide and diluted to final concentrations in aCSF. Unless noted otherwise, drugs were obtained from either Sigma or Abcam.
The endocytic tracer N-[3-(triethylammonio)propyl]-4-(4-dibutylaminostyryl) pyridinium dibromide (FM1-43; Invitrogen) at 8 μm in aCSF was loaded into presynaptic terminals by stimulating cortical layers V-VI at 10 Hz for 10 min with 400 μA, 200 μs pulses. This loading method prevents provoked striatal dopamine release and ensures that changes in FM1-43 release are not due to inadequate FM1-43 loading of the recycling synaptic vesicle pool (Bamford et al., 2004b; Joshi et al., 2009). The stimulating electrode was of the same type and was placed in the same locations as that used for the electrophysiology studies. Similarly, the recording region was the same as that in the electrophysiological experiments. After terminal loading, slices were superfused in ADVESEP-7 at 1 mm in aCSF for 2 min to remove adventitious staining. For stimulation-dependent destaining, pulse trains were again delivered to the cortex. During unloading, aCSF was supplemented with ADVASEP-7 (100 μm) to prevent recurrent endocytosis of dye into synaptic terminals. Electrical stimulation was provided by a Grass stimulator through a stimulation isolator (AMPI) and monitored by a Tektronix TDS 3014B digital oscilloscope.
Optical imaging and data analysis
Fluorescent terminals in the dorsal striatum were visualized using an LSM 510 NLO multiphoton laser-scanning microscope equipped with a titanium-sapphire laser (excitation 810 nm/emission 625 nm) and a 40× inverted oil objective (both Zeiss). Images were captured in 8-bit, 123 × 123 μm regions of interest at 512 × 512 pixel resolution and acquired at 22.5 s intervals using Zeiss LSM 510 software. To compensate for z-axis shift, a z-series of 5 images separated by 1 μm in the z-axis, was obtained for each imaging period. The time series of images were analyzed for changes in presynaptic terminal fluorescence using ImageJ and custom software written in Interactive Data Language (IDL; Research Systems) (Zakharenko et al., 2001; Bamford et al., 2004b). Fluorescent puncta, 0.5–1.5 μm in diameter, were identified. The criteria for punctum inclusion were that they be spherical in shape, have fluorescence two SDs above the background, and exhibited stimulation-dependent destaining. The IDL software aligned and combined the five-image z-series for each time interval and the overall intensity of the FM1-43 fluorescence was measured over the course of the time series. ImageJ was used to subtract background fluorescence of the tissue from the fluorescence intensity of each individual punctum. The results were then normalized by the maximal puncta fluorescence just before application of destaining stimulation. The halftime of fluorescence intensity decay during destaining (t1/2) was determined using a software algorithm written in SigmaPlot software (SPSS).
Statistics
Values given in the text and in the figures are indicated as mean ± SE. Electrophysiological data were derived from 3–12 mice per experiment; optical data were derived from 3–8 mice per experiment. Differences in mean values were assessed with the nonparametric Mann–Whitney test, Student's t tests (two groups), or appropriate ANOVAs (multiple groups) followed by multiple comparisons using Bonferroni t tests. Changes in terminal subpopulations and testing for normally distributed data were determined graphically using normal probability plots. When individual terminal halftimes are presented in a normal probability plot, a straight line indicates a normally distributed population (Bamford et al., 2004b). Statistical analyses were performed with Statistica software (StatSoft) and differences were considered significant if p < 0.05.
Results
Exposure to repeated amphetamine causes a chronic presynaptic depression
Adaptations in glutamatergic signaling to MSNs in the dorsal striatum are required for cue-dependent behaviors (Darvas and Palmiter, 2011; Quintana et al., 2012) and locomotor sensitization (Bamford et al., 2008), but the mechanisms underlying this plasticity remain unclear. To determine whether repeated amphetamine causes plasticity at excitatory presynaptic corticostriatal terminals, mice were treated with saline or amphetamine (2 mg/kg/d; i.p.) for 5 consecutive days and electrophysiological experiments were performed in MSNs in withdrawal (Fig. 1A). Amphetamine produced locomotor sensitization; the locomotor response to the fifth amphetamine injection was approximately fivefold greater than the response to the first one (Fig. 1B). Coronal slices encompassing the motor cortex and striatum were prepared on withdrawal day (WD) 10 (Fig. 1C). Whole-cell voltage-clamp recordings in MSNs revealed typical passive membrane properties (Table 1), with no differences between saline-treated mice (n = 113 cells) and amphetamine-treated mice (n = 171 cells; membrane capacitance, 109 ± 5 and 111 ± 4 pF; membrane resistance, 139 ± 15 and 195 ± 35 MΩ; time constant 1.8 ± 0.1 and 1.9 ± 0.1 ms, respectively; p > 0.05, Student's t test).
Figure 1.

Repeated amphetamine causes CPD. A, Paradigm for testing synaptic plasticity after repeated amphetamine. Mice were treated with saline for 2 d, amphetamine for 5 d, and were killed for experiments on WD 10 or 21. B, Representative locomotor ambulations in mice treated with either saline or amphetamine (n = 4 each). C, Coronal corticostriatal slice, stained with FM1-43 and 3,3′-diaminobenzidine, shows the areas of stimulation and recording. D, Compared with saline-exposed mice, the stimulation current required to achieve the eEPSC threshold was greater in MSNs from amphetamine-treated mice on WD 10 and WD 21. **p < 0.01 compared with saline, Student's t test. E, Representative traces (top) and input-output curves show that cortical stimulation produced lower-amplitude eEPSCs in MSNs on WD 10 and WD 21. @p < 0.05, saline compared with either WD 10 or WD 21, 2-way ANOVA. F, Cortical stimulation intensity required to achieve 25%, 50%, and 75% of maximum eEPSC amplitude was greater in MSNs from amphetamine-treated mice on WD 10 and WD 21. *p < 0.05 compared with saline, Bonferroni t test. G, Example traces (top) and quantified AMPAR/NMDAR response ratios show equivalent isolated AMPAR (black) and NMDAR-mediated (gray) eEPSCs measured in MSNs from saline-treated mice (left) and amphetamine-treated mice (right) on WD 10. H, Representative traces (top) and graph show that the PPR is lower in MSNs from saline-exposed mice (left) compared with amphetamine-treated mice in withdrawal (right). **p < 0.01, Student's t test. I, Representative traces (top) show mEPSCs recorded in MSNs from saline- (left) and amphetamine-treated mice on WD 10 (right). Amphetamine in vitro did not change the mean mEPSC frequency (inset) or frequency distribution. J, The average amplitude (inset) and cumulative amplitude distribution of mEPSC from saline-exposed mice was similar to that of amphetamine-treated mice on WD 10. K, Representative traces (top) of mEPSCs obtained from MSNs in the nucleus accumbens shell 10 d after treatment with saline (left) or amphetamine (right). Exposure to repeated amphetamine reduced high-frequency, small-amplitude inward currents, but did not change the average mEPSC frequency (inset) on WD 10. **p < 0.01 compared with saline, paired t test. L, Exposure to amphetamine increased the amplitude of mEPSCs in the nucleus accumbens shell on WD 10. *p < 0.05, **p < 0.01 compared with saline, paired t test. Scale bars in C, 1 mm; E, H, 100 pA, 5 ms; G, 100 ms; I, K, 10 pA, 1 s. Curves were fit with a Hill equation.
Table 1.
Passive and active membrane properties of MSNs, TANs, and PLTS interneurons
| MSN | TAN | PLTS* | |
|---|---|---|---|
| Passive membrane properties | |||
| Ra, MΩ | 14.0 ± 0.7 (114) | 20.8 ± 0.7 (34) | 15.9 ± 1.4 (10) |
| Rm, MΩ | 139.3 ± 15.1 (114) | 337.2 ± 22.3 (50) | 693.6 ± 82.6 (10) |
| Cm, pF | 108.9 ± 4.6 (114) | 113.0 ± 5.1 (50) | 38.6 ± 4.5 (10) |
| RMP, mV | −73.2 ± 2.2 (14) | −57.8 ± 1.1 (50) | −58.6 ± 2.3 (15) |
| AP properties | |||
| #APs | 5.6 ± 0.8 (14) | 14.4 ± 1.1 (50) | 9.9 ± 1.2 (14) |
| Amplitude, mV | 77.7 ± 3.3 (14) | 59.6 ± 2.3 (50) | 41.5 ± 1.8 (14) |
| AP half-width, ms | 0.8 ± 0.1 (14) | 2.5 ± 0.1 (50) | 1.7 ± 0.1 (14) |
| AHP, mV | −11.1 ± 0.8 (14) | −8.2 ± 0.6 (50) | −7.1 ± 0.5 (14) |
| Input current at threshold, pA | 368 ± 29 (14) | 107 ± 18 (50) | 25 ± 4 (14) |
| Threshold, mV | −34.4 ± 0.7 (14) | −45.4 ± 1.6 (50) | −35.7 ± 1.4 (14) |
Properties of TANs are compared with MSNs and with persistent low-threshold spike (PLTS) striatal interneurons that can be spontaneously active in cell-attached configuration. Compared with PLTS cells, TANs have a lower membrane resistance (Rm), higher membrane capacitance (Cm), and longer action potential duration (AP) half-width. Values are means ± SE. Group n is shown in parentheses. Ra indicates access resistance; RMP, resting membrane potential; #APs, number of action potentials counted over 400 ms at the third trace exhibiting Aps; AHP, afterhyperpolarization potential.
*Data for PLTS cells were obtained from Wang et al., 2013.
Stimulation of the motor cortex produced eEPSCs in all cells examined, but the stimulation intensity required to reach threshold in MSNs from saline-exposed mice (0.28 ± 0.04 mA; n = 26 cells from 12 mice) was lower than in cells from amphetamine-treated mice (0.59 ± 0.12 mA; n = 29 cells from 10 mice; p = 0.01, compared with saline, Student's t test; Fig. 1D). This synaptic depression was long lasting, because similar responses were found on WD 21 (0.57 ± 0.11 mA; n = 28 cells from 6 mice; p = 0.01, compared with saline). Cortical stimulation intensities above threshold evoked larger eEPSC amplitudes in MSNs from saline-exposed mice (p = 0.04 compared with amphetamine-treated mice, 2-way ANOVA; Fig. 1E). In addition, the stimulation intensity required to achieve peak eEPSC amplitude was lower in MSNs from saline-exposed mice compared with amphetamine-treated mice on both WD 10 (F(10,300) = 2.13, p = 0.02, 2-way ANOVA) and WD 21 (F(10,360) = 2.42, p = 0.01, 2-way ANOVA; Fig. 1F).
To determine whether this reduction in synaptic strength after repeated amphetamine could occur through plasticity involving postsynaptic glutamatergic receptors, we measured AMPAR/NMDAR response ratios in MSNs from saline- and amphetamine-treated mice on WD 10. eEPSCs in MSNs voltage-clamped at +40 mV, measured both before and after bath application of the NMDAR antagonist APV (50 μm), showed no difference in the AMPAR/NMDAR ratio (0.68 ± 0.08 in amphetamine vs 0.64 ± 0.1 in aCSF; n = 8 cells from 3 mice each; p = 0.7, Student's t test; Fig. 1G).
As an index of presynaptic corticostriatal activity, the PPR (amplitude of the second eEPSC /amplitude of the first eEPSC) was measured in response to cortical stimulation with 50 ms paired pulses applied every 30 s. The observed synaptic depression was likely of presynaptic origin, because the PPR in MSNs from amphetamine-treated mice (1.35 ± 0.5; n = 58 cells from 21 mice) was higher than that in cells from saline-exposed mice (1.13 ± 0.07; n = 34 cells from 13 mice; p = 0.007, Student's t test; Fig. 1H) (Mennerick and Zorumski, 1995).
Presynaptic corticostriatal activity in the absence of cortical stimulation was measured in MSNs by recording mEPSCs in the presence of TTX (1 μm), in which alterations in mEPSC frequency and amplitude suggest changes in presynaptic activity and postsynaptic responsiveness, respectively (Van der Kloot, 1991). Interestingly, both the frequency (1.59 ± 0.21 for saline; n = 32 cells from 11 mice vs 1.78 ± 0.18 for withdrawal; n = 33 cells from 7 mice; p = 0.33; Fig. 1I) and the cumulative amplitude distribution (9.58 ± 0.31 for saline vs 9.17 ± 0.29 for withdrawal; p = 0.3, paired t test; Fig. 1J) of mEPSCs in MSNs from saline-exposed mice was similar to cells from amphetamine-treated mice in withdrawal, consistent with equivalent basal levels of glutamate measured in the dorsal striatum during cocaine withdrawal (Baker et al., 2003).
Repeated use of psychostimulants produces postsynaptic plasticity in the nucleus accumbens (Pascoli et al., 2012). The mEPSCs recorded from MSNs in the nucleus accumbens shell showed no overall change in frequency (3.2 ± 0.53 for saline vs 2.37 ± 0.6 for withdrawal; n = 6 cells from 4 mice each; p = 0.11; Fig. 1K), but higher mEPSC amplitudes after repeated amphetamine (9.7 ± 0.2 for saline vs 12.44 ± 1.43 for withdrawal; n = 6 each; p = 0.04, paired t test; Fig. 1L). Therefore, exposure to repeated amphetamine can cause anatomically dependent changes in psychostimulant-induced striatal plasticity characterized by stimulation-dependent chronic presynaptic depression (CPD) in the dorsal striatum.
Amphetamine challenge in withdrawal causes a paradoxical presynaptic potentiation
Plasticity in glutamatergic signaling promotes drug-seeking behaviors (Pierce et al., 1996; Bell et al., 2000; Baker et al., 2003; McFarland et al., 2003). Therefore, we tested the effect of an amphetamine challenge in vitro on corticostriatal activity by measuring eEPSCs in response to paired-pulse cortical stimulation on WD 10. In MSNs from saline-exposed mice, bath application of amphetamine (10 μm) decreased the amplitude of the first current of the pair by 12 ± 5% (−118 ± 23 pA in aCSF vs −106 ± 21 pA after amphetamine; n = 15 cells from 6 mice; p = 0.01) and the PPR increased by 17 ± 6% (1.05 ± 0.05 in aCSF vs 1.23 ± 0.09 in amphetamine; p = 0.01, paired t test; Fig. 2A), suggesting that amphetamine reduced glutamate release from corticostriatal terminals (Mennerick and Zorumski, 1995).
Figure 2.

Repeated amphetamine produces PPP. A, Representative traces (top) show the average responses of cortically evoked paired pulses in MSNs before (left) and 5 min after bath application of amphetamine (right). Graph shows the normalized amplitude of the first eEPSC (of the pair) and the normalized PPR. In saline-exposed mice, amphetamine in vitro decreased the amplitude of the first eEPSC and increased the PPR. Ten days (B) and 21 d (C) after repeated amphetamine in vivo, bath-applied amphetamine increased the eEPSC amplitude and decreased the PPR. D, Representative traces (top) show mEPSCs in MSNs from saline-exposed mice in aCSF (left) and 5 min after bath-applied amphetamine (right). Amphetamine in vitro decreased the frequency of mEPSCs (inset, left) primarily by inhibiting small amplitude (5–10 pA) spontaneous inward currents while having no effect on their cumulative amplitude distribution (inset, right). For D--F: *p < 0.05, **p < 0.01, paired t test. E, On WD 10 and F, WD 21 after repeated amphetamine, bath-applied amphetamine increased the frequency of mEPSCs (inset, left) by boosting the high-frequency, low-amplitude spontaneous inward currents while having no effect on their cumulative amplitude distribution (inset, right). Scale bars in A–C, 100 pA, 5 ms; D--F, 10 pA, 1 s.
In MSNs from amphetamine-treated mice, amphetamine in vitro paradoxically increased the amplitude of the first eEPSC by 46 ± 17% (−130 ± 23 pA in aCSF vs −165 ± 22 pA after amphetamine; n = 9 cells from 4 mice; p = 0.003) and the PPR decreased by 22 ± 3% (1.46 ± 0.1 in aCSF vs 1.12 ± 0.06 in amphetamine; p = 0.002, paired t test; Fig. 2B). Paradoxical presynaptic potentiation (PPP) was long-lasting, because bath-applied amphetamine on WD 21 also increased the amplitude of the first eEPSC by 41 ± 18% (−95 ± 17 pA in aCSF vs −133 ± 25 pA after amphetamine; n = 16 cells from 5 mice; p = 0.05) and reduced the PPR by 10 ± 7% (1.45 ± 0.11 in aCSF vs 1.25 ± 0.1 in amphetamine; p = 0.04, paired t test; Fig. 2C).
In the absence of cortical stimulation, amphetamine in vitro diminished the frequency of mEPSC in MSNs from saline-exposed mice by 17 ± 6% (1.32 ± 0.24 Hz in aCSF vs 1.04 ± 0.16 Hz in amphetamine; n = 12 cell from 5 mice; p = 0.05, paired t test) and selectively increased high-frequency, low-amplitude inward currents, but had no effect on their cumulative amplitude distribution (Fig. 2D). In cells from amphetamine-treated mice, however, bath-applied amphetamine increased high-frequency, low-amplitude mEPSCs on both WD 10 (107 ± 48%; 2.28 ± 0.51 Hz in aCSF vs 3.88 ± 0.95 Hz in amphetamine; n = 8 cells from 3 mice; p = 0.05; Fig. 2E) and WD 21 (28 ± 10%; 1.54 ± 0.24 Hz in aCSF vs 1.87 ± 0.26 Hz in amphetamine; n = 11 cells from 4 mice; p = 0.01, paired t test; Fig. 2F), but had no effect on their cumulative amplitude distribution.
PPP is independent of D2Rs
We also investigated whether D2Rs might participate in PPP because they modulate presynaptic corticostriatal activity of D2R-expressing MSNs in untreated mice (Yin and Lovinger, 2006; André et al., 2010; Wang et al., 2012). In MSNs from saline-exposed mice, the D2R agonist quinpirole (5 μm) decreased the amplitude of the first eEPSC by 20 ± 6% (−250 ± 47 pA in aCSF vs −203 ± 36 pA in quinpirole; n = 11 cells from 5 mice; p = 0.01) and increased the PPR by 12 ± 4% (0.94 ± 0.09 in aCSF vs 1.05 ± 0.08 in quinpirole; p = 0.02, paired t test; Fig. 3A). In cells from amphetamine-treated mice on WD 10, quinpirole also suppressed the amplitude of the first eEPSC (−13 ± 5%; −155 ± 41 pA in aCSF vs −139 ± 41 pA in quinpirole; n = 8 cells from 3 mice; p = 0.05) and the PPR increased (9 ± 4%; 1.31 ± 0.18 in aCSF to 1.42 ± 0.18 in quinpirole; p = 0.04, paired t test; Fig. 3B). Quinpirole decreased the frequency of mEPSCs in MSNs from both saline-treated mice (−15.5 ± 6%; 1.38 ± 0.22 Hz in aCSF vs 1.19 ± 0.23 Hz in quinpirole; n = 7 cells from 3 mice; p = 0.03) and amphetamine-treated mice (−11 ± 5%; 1.57 ± 0.16 Hz in aCSF vs 1.37 ± 0.11 Hz in quinpirole; n = 8 cells from 3 mice; p = 0.01, paired t test) by suppressing high-frequency, low-amplitude mEPSCs, whereas the cumulative amplitude distribution remained unchanged (Fig. 3C,D).
Figure 3.

D2Rs remain inhibitory after repeated amphetamine. A, Representative traces (top) show average responses to cortically evoked paired pulses in MSNs before (left) and 5 min after bath application of the D2R agonist quinpirole (right). In both saline-exposed (A) and amphetamine-exposed (B) mice, bath-applied quinpirole decreased the amplitude of the first eEPSC and increased the PPR. C, Representative traces (top) show mEPSCs in aCSF (left) and 5 min after bath-applied quinpirole (right). In both saline-exposed (C) and amphetamine-exposed (D) mice, quinpirole decreased the frequency of mEPSCs (inset, left) by inhibiting the number of high-frequency, low-amplitude spontaneous inward currents while having no effect on their cumulative amplitude distribution (inset, right). For C and D, *p < 0.05, **p < 0.01, paired t test. Scale bars in A, B, 100 pA, 5 ms; C, D, 10 pA, 1 s.
PPP is dependent on D1 receptors
Next, we explored whether amphetamine-induced PPP might occur through D1Rs. In MSNs from saline-exposed mice, the D1R agonist SKF81297 (1 μm) did not alter the eEPSC amplitude (4 ± 5%; −193 ± 29 pA in aCSF vs −198 ± 30 pA in SKF81297; n = 11 cells from 3 mice; p = 0.6) or the PPR (−3 ± 4%; 1.16 ± 0.09 in aCSF vs 1.1 ± 0.07 in SKF81297; p = 0.1, paired t test; Fig. 4A). Further, the D1R antagonist SCH23390 (10 μm) had no effect on the eEPSC amplitude (15 ± 10%; −129 ± 25 pA in aCSF vs −139 ± 18 pA in SCH23390, n = 7 cells from 3 mice; p = 0.4) or the PPR (−2 ± 6%; 1.32 ± 0.1 in aCSF vs 1.26 ± 0.08 in SCH23390; p = 0.6, paired t test; data not shown), consistent with the near absence of D1Rs (Dumartin et al., 2007) and their actions (Bamford et al., 2004a) on cortical afferents in the dorsal striatum.
Figure 4.
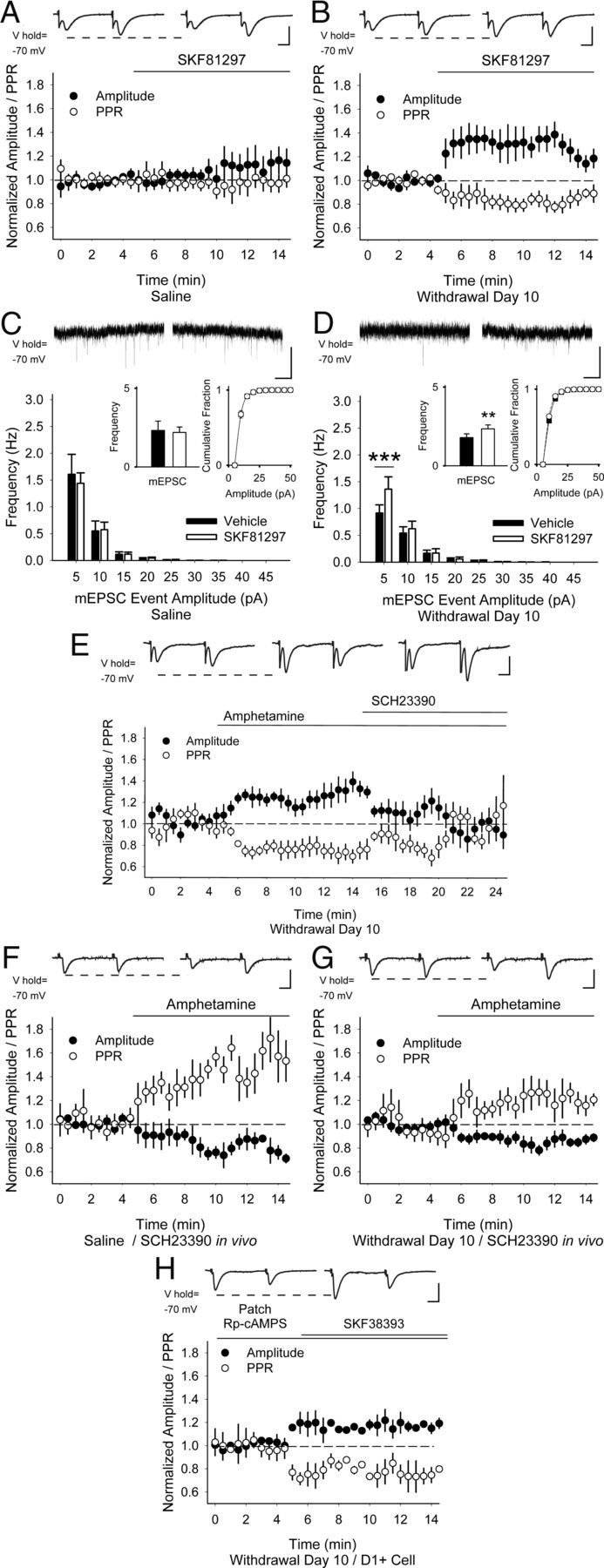
PPP is dependent on D1Rs. Representative traces (top) show the average responses to cortically evoked paired pulses in MSNs before (left) and 5 min after (right) bath application of the D1R agonist SKF81297. SKF81297 did not change the amplitude of the first eEPSC or the PPR in MSNs from saline-exposed mice (A), but increased the eEPSC amplitude and decreased the PPR 10 d after repeated amphetamine (B). C, Representative traces (top) show mEPSCs in aCSF (left) and 5 min after bath-applied SKF81297 (right). SKF81297 did not change the mEPSC frequency (inset, left) or amplitude (inset, right) in MSNs from saline-exposed mice. D, On WD 10, SKF81297 increased the frequency of mEPSCs (inset, left) by boosting the frequency of 5–10 pA inward currents while having no effect on their cumulative amplitude distribution (inset, right). **p < 0.01, ***p < 0.001, paired t test. E, Representative traces (top) show the average responses to paired pulses in MSNs on WD 10 in aCSF (left), 5 min after bath application of amphetamine (center), and 5 min after perfusion of both amphetamine and the D1R antagonist SCH23390 (right). The increase in eEPSC amplitude and the reduction in the PPR by amphetamine in vitro was blocked by SCH23390. F, Representative traces (top) show the average responses to paired pulses in MSNs before (left) and 5 min after bath application of amphetamine (right). In MSNs from both saline-treated (F) and amphetamine-treated (G) mice, SCH23390 in vivo followed by amphetamine in vitro decreased the eEPSC amplitude and increased the PPR. H, When Rp-cAMPS was applied via the patch electrode in D1+ MSNs, SKF81297 remained excitatory, because the eEPSC amplitude increased and the PPR decreased. Scale bars in A, B, and E–H, 100 pA, 5 ms; C, D, 10 pA, 1 s.
In MSNs from amphetamine-treated mice on WD 10, bath-applied SKF81297 increased the eEPSC amplitude by 33 ± 10% (−144 ± 36 pA in aCSF vs −184 ± 47 pA; n = 8 cells from 3 mice; p = 0.04) and reduced the PPR by −19 ± 5% (1.49 ± 0.16 in aCSF vs 1.18 ± 0.08 in amphetamine; p = 0.02, paired t test; Fig. 4B). Similarly, although SKF81297 had no effect on mEPSC frequency (22 ± 22%; 2.32 ± 0.59 Hz in aCSF vs 2.19 ± 0.34 Hz in SKF81297; n = 9 cells from 3 mice; p = 0.8) or their cumulative amplitude distribution in MSNs from saline-exposed mice (Fig. 4C), SKF81297 increased high-frequency, low-amplitude inward currents by 44 ± 16% (1.79 ± 0.34 Hz in aCSF vs 2.25 ± 0.35 Hz in SKF81297; n = 8 from 3 mice; p = 0.001, paired t test) 10 d after repeated amphetamine and had no effect on their cumulative amplitude distribution (Fig. 4D). PPP was dependent on D1Rs, because the increase in eEPSC amplitude by amphetamine (18 ± 4%; −90 ± 14 pA in aCSF vs −106 ± 16 pA in amphetamine; n = 7 cells from 3 mice; p = 0.02) was blocked by the D1R antagonist SCH23390 (−2 ± 12%; −93 ± 21 pA in amphetamine with SCH23390; p = 0.8, compared with aCSF; Fig. 4E). Likewise, the reduction in the PPR after amphetamine (−15 ± 6%; 1.46 ± 0.13 in aCSF vs 1.23 ± 0.11 in amphetamine; p = 0.04) was blocked by SCH23390 (−3 ± 8%; 1.4 ± 0.17 in amphetamine with SCH23390; p = 0.7, compared with aCSF, paired t test).
To further test the dependence of D1Rs on PPP, mice were treated with the D1R antagonist SCH23390 in vivo (20 μg/kg i.p) 30 min before killing. SCH23390 in vivo had little effect in saline-exposed mice, because bath-applied amphetamine reduced the eEPSC amplitude (−18 ± 9%; −45 ± 4 pA in aCSF vs −37 ± 9 pA in amphetamine; n = 7 cells from 3 mice; p = 0.04) while increasing the PPR (34 ± 9%; 1.18 ± 0.6 in aCSF vs 1.49 ± 0.6 in amphetamine; p = 0.04; Fig. 4F). However, SCH23390 in vivo blocked PPP in amphetamine-treated mice, because an amphetamine challenge also reduced the eEPSC amplitude (−15 ± 2%; −59 ± 7 pA in aCSF vs −40 ± 7 pA in amphetamine; n = 7 cells from 3 mice; p < 0.001) while increasing the PPR (17 ± 5%; 0.89 ± 0.07 in aCSF vs 1.1 ± 0.09 in amphetamine; p = 0.02; Fig. 4G).
Intracellular signaling by G-protein-coupled receptors requires activation of adenylyl cyclase, cAMP, and protein kinase A (Memo et al., 1986). To confirm that presynaptic mechanisms were involved in PPP, we made recordings from D1R-expressing MSNs with internal solutions containing the protein kinase A inhibitor Rp-cAMPS (400 μm). When Rp-cAMPS was applied via the patch electrode, SKF81297 remained excitatory, the eEPSC amplitude increased by 18 ± 4% (−139 ± 6 pA in aCSF vs −165 ± 10 pA after SKF81297; n = 6 cells from 3 mice; p = 0.03, paired t test), and the PPR decreased (−23 ± 7%; 0.85 ± 0.09 in aCSF to 0.64 ± 0.04 in SKF81297; p = 0.04, paired t test; Fig. 4H), suggesting that PPP caused by SKF81297 did not arise through mechanisms intrinsic to the neuron under evaluation.
PPP boosts corticostriatal activity predominantly in striatonigral pathway neurons
The striatum contains D1R-expressing MSNs that initiate movements and behaviors via the “direct” striatonigral pathway and D2R-expressing MSNs that suppress competing actions via the “indirect” striatopallidal pathway (Pennartz et al., 1994; Nicola et al., 2000). To determine whether PPP excites D1R-expressing (D1+) and/or D2R-expressing (D2+) MSNs, hemizygotic BAC transgenic mice expressing the reporter EGFP under the control of the D1R and D2R promoters (Drd1-EGFP and Drd2-EGFP, respectively) were treated with saline or amphetamine for 5 d and the effect of an amphetamine challenge in vitro was determined on WD 10 (Fig. 1A). As reported (Nelson et al., 2012), there were no significant differences in locomotor responses of Drd1-EGFP and Drd2-EGFP mice (n = 4 each) to repeated amphetamine (Fig. 5A). However, due to overexpression of D2R mRNA in Drd2-EGFP mice (Kramer et al., 2011) and behavioral deficits in both Drd1-EGFP and Drd2-EGFP mice (Bagetta et al., 2011; Kramer et al., 2011), recordings were made in both fluorescent and nonfluorescent cells.
Figure 5.

Amphetamine challenge in withdrawal causes PPP in D1+ MSNs. A, Locomotor ambulations in response to repeated amphetamine in Drd1- and Drd2-EGFP mice was not significant (n.s.), p = 0.09, Student's t test. B, Representative traces (top) show the average responses to cortically evoked paired pulses in MSNs before (left) and 5 min after bath application of amphetamine (right). In D1+ MSNs from saline-exposed mice, amphetamine in vitro had no effect on the amplitude or PPR. C, In D1+ MSNs from amphetamine-treated mice on WD 10, amphetamine increased the eEPSC amplitude and reduced the PPR. D, In D2+ MSNs from saline-exposed mice, amphetamine decreased the amplitude and increased PPR. E, In D2+ MSNs from amphetamine-treated mice, amphetamine did not change the amplitude or the PPR. Amphetamine increased the eEPSC amplitude and reduced the PPR in a subset of D2+ MSNs from amphetamine-treated mice (F), but reduced the amplitude and increased the PPR in the remainder (G). H, Representative traces (top) show mEPSCs in aCSF (left) and 5 min after bath-applied amphetamine (right). In D1+ MSNs from saline-exposed mice, amphetamine in vitro did not change mEPSC frequency (inset, left) or the cumulative amplitude distribution (inset, right). I, In D1+ MSNs on WD 10, amphetamine in vitro increased the frequency of mEPSCs by augmenting 5–10 pA inward currents, but had no effect on their cumulative amplitude distribution. For I–L, *p < 0.05, **p < 0.01, ***p < 0.001, paired t test. J, In D2+ MSNs from saline-exposed mice, amphetamine in vitro decreased the frequency of mEPSCs, but had no effect on the cumulative amplitude distribution. In a subset of D2+ MSNs examined on WD 10, bath-applied amphetamine increased the frequency of mEPSCs (K), but decreased the frequency of mEPSCs in the remaining cells while having no effect on their cumulative amplitude distributions (L). Scale bars in B–D, F, G, 100 pA, 5 ms; H–L, 10 pA, 1 s.
In D1+ MSNs from saline-exposed mice, amphetamine in vitro did not change the amplitude for the first eEPSC of the pair (−12 ± 9%; −97 ± 31 pA in aCSF vs −84 ± 27 pA in amphetamine; n = 6 cells from 3 mice; p = 0.1) or the PPR (8 ± 7%; 1.36 ± 0.23 in aCSF vs 1.5 ± 0.32 in amphetamine; p = 0.3, paired t test; Fig. 5B). In amphetamine-treated mice on WD 10, amphetamine in vitro potentiated corticostriatal activity in D1+ MSNs, because the eEPSC amplitude increased (21 ± 5%; −146 ± 30 pA in aCSF vs −171 ± 31 pA in amphetamine; n = 7 cells from 5 mice; p = 0.006) but the PPR decreased (−17 ± 4%; 1.12 ± 0.1 in aCSF vs 0.92 ± 0.07 in amphetamine; p = 0.02, paired t test; Fig. 5C).
In D2+ MSNs from saline-exposed mice, amphetamine in vitro decreased the eEPSC amplitude (−27 ± 9%; −120 ± 37 pA in aCSF vs −92 ± 26 pA in amphetamine; n = 8 cells from 3 mice; p = 0.02) and the PPR increased (26 ± 8%; 1.1 ± 0.1 in aCSF vs 1.3 ± 0.2 in amphetamine; p = 0.01, paired t test; Fig. 5D). Surprisingly, recordings in D2R+ MSNs 10 d after repeated amphetamine showed no significant change in either the amplitude (−7 ± 9%; −83 ± 11 pA in aCSF vs −71 ± 10 pA in amphetamine; n = 14 from 9 mice; p = 0.2) or the PPR (6 ± 8%; 1.33 ± 0.2 in aCSF vs 1.3 ± 0.12 in amphetamine; p = 0.8, paired t test; Fig. 5E) after amphetamine in vitro. In approximately half the D2+ MSNs examined (n = 6 from 3 mice), amphetamine in vitro increased the eEPSC amplitude (25 ± 7%; −72 ± 17 pA in aCSF vs −87 ± 18 pA in amphetamine; p = 0.002) and reduced the PPR (−24 ± 5%; 1.66 ± 0.29 in aCSF vs 1.23 ± 0.17 in amphetamine; p = 0.03, paired t test; Fig. 5F). In the remaining D2+ MSNs (n = 8 from 6 mice), amphetamine decreased the amplitude (−31 ± 6%; −90 ± 16 pA in aCSF vs −60 ± 11 pA in amphetamine; p = 0.006) and the PPR increased (28 ± 7%; 1.08 ± 0.17 in aCSF vs 1.36 ± 0.18 in amphetamine; p = 0.002, paired t test; Fig. 5G). Therefore, a drug challenge potentiated corticostriatal activity onto D1+ MSNs and in a subpopulation of D2+ MSNs. Similar mercurial responses were found in both nonfluorescent cells from Drd1-EGFP mice (n = 9) and in fluorescent cells from Drd2-EGFP mice (n = 5; data not shown), so the results were pooled.
Under nonstimulated conditions, amphetamine in vitro did not change the frequency of mEPSC in D1+ MSNs from saline-exposed mice (4 ± 6%; 4.48 ± 0.65 Hz in aCSF vs 4.66 ± 0.72 Hz in amphetamine; n = 6 cells from 3 mice; p = 0.5; Fig. 5H), but increased the frequency of mEPSC in amphetamine-treated mice (20 ± 5%; 5.89 ± 0.4 Hz in aCSF vs 7.09 ± 0.71 Hz in amphetamine; n = 5; p = 0.03; paired t test; Fig. 5I) by selectively increasing high-frequency, low-amplitude inward currents while having no effect on their cumulative amplitude distribution.
In D2+ MSNs from saline-exposed mice, amphetamine in vitro reduced the frequency of mEPSC (−23 ± 5%; 5.02 ± 0.45 Hz in aCSF vs 4.01 ± 0.25 Hz in amphetamine; n = 6 cells from 4 mice; p = 0.003; paired t test; Fig. 5J), but not their cumulative amplitude distribution. In D2+ MSNs from amphetamine-treated mice, bath-applied amphetamine had no effect on mEPSC frequency (n = 10 cells from 4 mice; 22 ± 16%; 4.09 ± 0.5 Hz in aCSF vs 4.7 ± 0.55 Hz in amphetamine; p = 0.2; paired t test) or amplitude (data not shown). However, similar to eEPSCs in these cells, results were dichotomous, because amphetamine increased high-frequency, low-amplitude mEPSCs in half the cells examined (n = 5 cells from 3 mice; 59 ± 15%; 3.41 ± 0.3 Hz in aCSF vs 5.36 ± 0.75 Hz in amphetamine; p = 0.02; Fig. 5K), but reduced the frequency of mEPSCs in the remainder (n = 5 cells from 4 mice; −15 ± 2%; 4.76 ± 0.6 Hz in aCSF vs 4.06 ± 0.4 Hz in amphetamine; p = 0.01, paired t test; Fig. 5L) while having no effect on their amplitude distribution. Divergent responses were found in both nonfluorescent cells from Drd1-EGFP mice (n = 5) and in fluorescent cells from Drd2-EGFP mice (n = 5; data not shown), so the results were pooled.
CPD is frequency dependent and PPP excites subsets of corticostriatal terminals
To explore whether repeated amphetamine can promote presynaptic corticostriatal plasticity at physiologically relative frequencies (Cowan and Wilson, 1994; Kasper et al., 1994; Stern et al., 1997; Charpier et al., 1999; Fellous et al., 2003), we measured activity at corticostriatal terminals directly using the endocytic tracer FM1-43 combined with multiphoton confocal microscopy (Bamford et al., 2004b). In these experiments, mice received saline or amphetamine (2 mg/kg/d; i.p.) for 5 consecutive days and were challenged with amphetamine on experiment days 10 and 28. To confirm that both CPD and PPP were long lasting, some mice were killed and brain slices were prepared on experiment day 57 (WD 50; Fig. 6A). Stimulation of the motor cortex resulted in endocytosis of FM1-43 dye into recycling synaptic vesicles in the dorsal striatum, revealing fluorescent puncta distinctive of corticostriatal afferents (Bamford et al., 2004b). After dye loading, cortical restimulation produced exocytosis of FM1-43 dye from terminals, characteristic of synaptic vesicle fusion (Fig. 6B; Bamford et al., 2004b). Because FM1-43 destaining follows first-order kinetics (Joshi et al., 2009), corticostriatal release was characterized by the halftime of destaining (t1/2), defined as the time required for terminal fluorescence to decay to half of its initial value.
Figure 6.
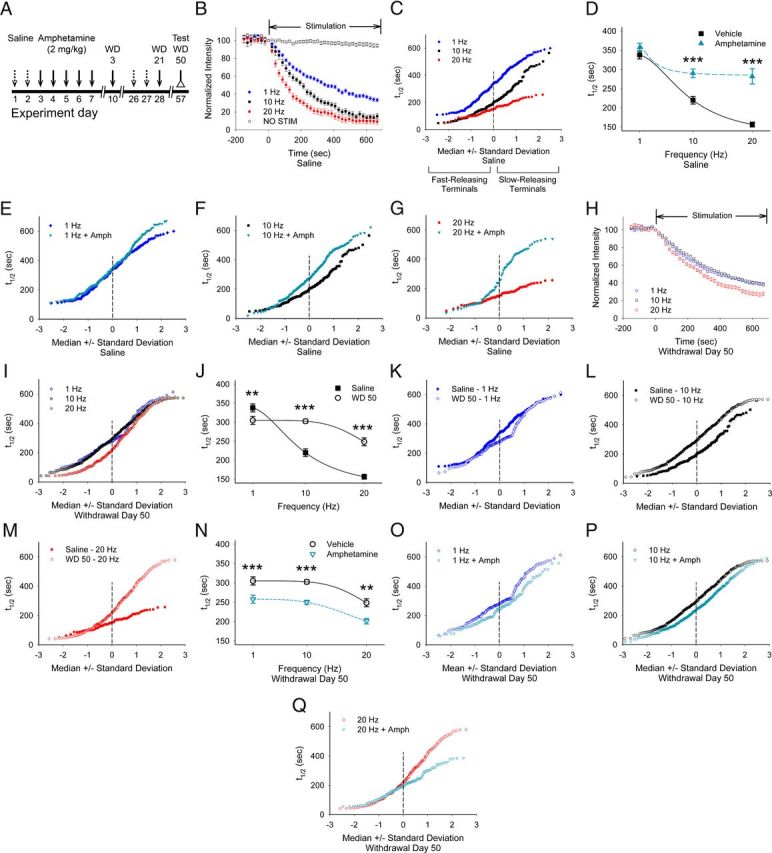
Repeated amphetamine produces frequency-dependent CPD and frequency-independent PPP. A, Paradigm for measuring the long-term effects of repeated amphetamine. Amphetamine-treated mice received saline injections on days 1, 2, 26, and 27 and amphetamine on days 3–7, 10, and 28, whereas saline-exposed mice received saline on those days. B, In saline-exposed mice, stimulation of the motor cortex at 1, 10, and 20 Hz produced a frequency-dependent increase in FM1-43 release, as reflected by a decrease in the destaining halftime (t½). Little FM1-43 destaining occurred when no stimulation was applied (NO STIM). C, Normal probability plot in which a straight line indicates a normally distributed population (Bamford et al., 2004b) shows the release kinetics of individual cortical terminals in slices from saline-exposed mice after cortical stimulation at 1, 10, and 20 Hz. D, Comparisons of the average halftimes of release in slices from saline-exposed mice stimulated at 1, 10, and 20 Hz with and without amphetamine in vitro. Amphetamine inhibited release at 10 and 20 Hz. For D, J, N, **p < 0.01, ***p < 0.001, Mann–Whitney. E–G, Normal probability plots of destaining halftimes of individual cortical terminals in slices from saline-exposed mice show that bath-applied amphetamine reduces exocytosis from terminals with a low probability of release at 1 Hz (E), 10 Hz (F), and 20 Hz (G). H, FM1-43 destaining curves for stimulation frequencies of 1–20 Hz from amphetamine-treated mice on WD 50. I, FM1-43 release kinetics from individual terminals in slices from amphetamine-treated mice on WD 50 after cortical stimulation at 1, 10, and 20 Hz. J, Comparison of release halftimes after cortical stimulation at 1, 10, and 20 Hz in slices from saline and amphetamine-treated mice on WD 50. K–M, Comparison of release halftimes in slices from saline- and amphetamine-treated mice at 1 Hz (K), 10 Hz (L), and 20 Hz (M). N, Average halftimes of release in slices from amphetamine-treated mice on WD 50 after stimulation at 1, 10, and 20 Hz. Bath-applied amphetamine boosted FM1-43 release at all cortical stimulation frequencies. O–Q, Release kinetics from individual terminals in slices from amphetamine-treated mice on WD 50 with and without bath-applied amphetamine with stimulation frequencies of 1 Hz (O), 10 Hz (P), and 20 Hz (Q).
In slices from saline-exposed mice, an increase in cortical stimulation frequency from 1 Hz to 20 Hz produced a corresponding increase in glutamate release from most cortical terminals (Fig. 6C), which was reflected by a decrease in the average t1/2 of FM1-43 destaining (t1/2 = 337 ± 10 s at 1 Hz, t1/2 = 220 ± 10 s at 10 Hz, and t1/2 = 157 ± 6 s at 20 Hz; n = 181, 139, and 66 puncta, respectively; F(2,400) = 77, p = 0.001, repeated-measures (rm)-ANOVA; Figure 6D). In the presence of bath-applied amphetamine, FM1-43 release also increased with the frequency of cortical stimulation (t1/2 = 357 ± 11 s at 1 Hz, t1/2 = 289 ± 11 s at 10 Hz, and t1/2 = 282 ± 20 s at 20 Hz; n = 182, 167, and 67 puncta, respectively; F(2,413) = 11, p = 0.001, rm-ANOVA; Fig. 6D). Compared with aCSF, amphetamine in vitro had little effect on terminal release at 1 Hz, but decreased exocytosis at 10 and 20 Hz (F(2,776) = 8, p = 0.001, 2-way ANOVA for interaction between frequency and amphetamine; Fig. 6D) and produced a low-pass-frequency filter with filtering applied specifically to a subset of terminals with a low probability of release (e.g., those with the highest t1/2; Fig. 6E–G).
In slices from amphetamine-treated mice, an increase in cortical stimulation frequency increased corticostriatal release (t1/2 = 305 ± 10 s at 1 Hz, t1/2 = 302 ± 5 s at 10 Hz, and t1/2 = 248 ± 10 s at 20 Hz; n = 158, 554, and 199 puncta, respectively; F(2,908) = 13, p = 0.001, rm-ANOVA; Fig. 6H), but did so by boosting exocytosis from terminals with a high probability of release (e.g., those with the lowest t1/2; Fig. 6I). Compared with slices from saline-exposed mice, corticostriatal release in withdrawal was increased at 1 Hz (p < 0.01), but was reduced at 10 and 20 Hz (p < 0.001; Fig. 6J). Withdrawal from repeated amphetamine increased release from terminals with intermediate kinetics at 1 Hz, reduced exocytosis from most terminals at 10 Hz, and diminished destaining of terminals with the lowest probability of release at 20 Hz (Fig. 6K–M), suggesting that CPD was dependent on stimulation frequency. PPP, however, was independent of cortical stimulation frequencies, because amphetamine in vitro during withdrawal accelerated exocytosis at all stimulation frequencies (t1/2 = 258 ± 10 s at 1 Hz, t1/2 = 250 ± 5 s at 10 Hz, and t1/2 = 201 ± 7 s at 20 Hz; n = 128, 530, and 144 puncta, respectively; F(2, 1707) = 4, p = 0.01, 2-way ANOVA for interaction between frequency and amphetamine; Fig. 6N) and boosted release from the majority of terminals at 1 and 10 Hz, but only from the slower-releasing terminals at 20 Hz (Fig. 6O–Q).
Locomotor sensitization is encoded by PPP
Because amphetamine-induced adaptations in glutamatergic signaling may underlie locomotor sensitization (Cornish et al., 1999; Li et al., 1999; Ghasemzadeh et al., 2003; Knackstedt and Kalivas, 2009), we compared optical measurements of corticostriatal release with locomotor behavior in individual mice to determine whether striatal excitation via PPP might parallel the amphetamine-dependent increase in locomotor activity. For these experiments, mice (n = 7) received amphetamine (2 mg/kg/d; i.p.) for 5 consecutive days, were challenged with amphetamine on experiment days 10 and 28, and were killed on experiment day 57 (WD 50; Fig. 6A). Amphetamine produced locomotor sensitization (Fig. 7A), with a variable increase in ambulations after each amphetamine injection (Fig. 7B). The percent increase in ambulations for each mouse, determined by comparing locomotor ambulations with those obtained after the first amphetamine injection (Pierce et al., 1996), also varied and generally increased after each injection of amphetamine (297 ± 34%; range, 45–657%; Fig. 7C).
Figure 7.

PPP is proportional to the magnitude to locomotor sensitization. A, Locomotor ambulations in mice treated with either saline or amphetamine. B, Locomotor ambulations of individual mice after each amphetamine challenge from experiments shown in A. C, Percent increase in locomotor ambulations for individual mice shown in B, when normalized to ambulations after their first exposure to amphetamine (experiment day 3). D, Percent increase in corticostriatal release (PPP) as measured in slices from mice in A after killing on WD 50. E, Percent increase in ambulations of individual mice after amphetamine in vivo on experiment days 4–7 compared with PPP. F–I, PPP compared with the percent increase in ambulations on experiment days 4 (F), 5 (G), 6 (H), and 7 (I). J, Percent increase in ambulations after an amphetamine challenge in vivo on experiment days 10 and 28 compared with PPP. K, L, PPP compared with the percent increase in ambulations on experiment days 10 (K) and 28 (L).
Upon killing, presynaptic corticostriatal release was determined optically. The increase in corticostriatal release after an amphetamine challenge in vitro varied widely (22 ± 5.1%; range, 7.6%–44.7%; Fig. 7D). Linear regression comparisons between the change in corticostriatal release (i.e., PPP) and locomotor ambulations for each mouse revealed a significant correlation during each of the initial 5 d of treatment (R2 = 0.82, F(1,26) = 119, p = 0.001; Fig. 7E–I) and when challenged with amphetamine in withdrawal (R2 = 0.77, F(1,12) = 40, p = 0.001; Fig. 7J–L).
Locomotor sensitization is promoted by D1Rs
To confirm that D1Rs promote behavioral sensitization, we measured locomotor responses in amphetamine-sensitized mice after treatment with the D1R antagonist SCH23390 in vivo. Mice were treated with saline or amphetamine (2 mg/kg/d i.p.) for 5 consecutive days and again on experiment days 10 and 28 (Fig. 6A). On experiment day 57 (WD 50), SCH23390 (20 μg/kg i.p.) had no effect on the open-field ambulations in saline-exposed mice (n = 5; F(17,153) = 1.1, p = 0.38) compared with saline-exposed mice without SCH23390 (n = 8, rm-ANOVA; Figure 8A), but suppressed ambulations in amphetamine-treated mice (n = 8; F(17,221) = 4.6, p < 0.001) compared with amphetamine-treated mice without SCH23390 (n = 7, rm-ANOVA; Figure 8B), suggesting that repeated amphetamine enhanced D1R-medicated locomotor responses in withdrawal.
Figure 8.

Locomotor sensitization is dependent on D1Rs. A, B, Interval and mean ± SE (inset) locomotor ambulations over 90 min of saline-treated (A) and amphetamine-treated (B) mice on experiment day 57 (WD 50) tested with either SCH23390 or saline. For all panels, *p < 0.05, Student's t test; ##p < 0.01, ###p < 0.001, ANOVA. C, Interval ambulations over 90 min of amphetamine-treated mice on experiment day 57 tested with amphetamine (Amph) or with amphetamine and SCH23390 (Amph+SCH23390) compared with ambulations after their first dose of amphetamine (experiment day 3) and with ambulations of saline-exposed mice treated with saline. Inset: Mean ± SE ambulations of amphetamine-treated mice after a test dose of amphetamine (left) or after amphetamine and SCH23390 (right). D, E, Interval and mean ± SE (inset) ambulations over 90 min of saline-treated (D) and amphetamine-treated (E) mice on experiment day 57 tested with a higher dose of SCH23390 or saline. F, Interval ambulations of amphetamine-treated mice on experiment day 57 tested with amphetamine or with amphetamine and SCH23390 compared with locomotor responses after their first dose of amphetamine and to saline-exposed mice tested with saline. Inset: Mean ± SE ambulations for amphetamine-treated mice after a test dose of amphetamine (left) or after both amphetamine and SCH23390 (right).
We tested the dependence of D1Rs on ambulations after a drug challenge. In amphetamine-sensitized mice (n = 15), an amphetamine challenge (2 mg/kg/d i.p.) on experiment day 57 caused a 104 ± 37% increase in ambulations compared with those obtained after their first dose of amphetamine (experiment day 3; F(17,442) = 10.8, p < 0.001, rm- ANOVA) and a 1160 ± 155% increase in locomotor ambulations compared with saline-exposed mice (n = 15; F(17,459) = 81, p < 0.001, rm-ANOVA; Fig. 8C). Locomotor sensitization was blocked by the D1R antagonist, because SCH23390 (20 μg/kg i.p.) reduced ambulations by 60 ± 8% after an amphetamine challenge on experiment day 57 (n = 8; F(17,459) = 9.8, p < 0.001, rm-ANOVA), with locomotor responses similar to those recorded after the first day of amphetamine treatment (experiment day 3; −1 ± 28%; F(17,459) = 2.7, p = 0.07, rm-ANOVA).
A higher dose of SCH23390 (40 μg/kg i.p.) also had no effect on locomotor activity in saline-exposed mice (n = 7; F(17,187) = 1, p = 0.46) compared with saline-exposed mice without SCH23390, n = 8; Figure 8D), but depressed locomotor ambulations in withdrawal (n = 7; F(17,221) = 2, p = 0.01) compared with amphetamine-treated mice without SCH23390 (n = 8, rm-ANOVA; Figure 8E) and further reduced ambulations after an amphetamine challenge (n = 8; 70 ± 6%; Fig. 8F).
Repeated amphetamine promotes PPP in D1R-expressing MSNs through D1R-dependent plasticity in striatal acetylcholine-releasing interneurons
Psychostimulant exposure is known to cause long-lasting plasticity in striatal TANs, which promote behavioral sensitization (Bickerdike and Abercrombie, 1997), cocaine conditioning (Witten et al., 2010), and downstream changes in corticostriatal activity (Bamford et al., 2008; Witten et al., 2010). To determine whether TAN activity was modified during amphetamine withdrawal, mice were treated with repeated amphetamine or saline for 5 d and TAN firing frequencies were measured using cell-attached recordings in acute striatal slices (Fig. 1A, Fig. 9A). Whole-cell recordings after each experiment revealed passive and active membrane responses (Fig. 9B,C, Table 1), which are typical of TANs (Kawaguchi, 1993; Lee et al., 1998).
Figure 9.

TAN firing is reduced in withdrawal and increases via D1Rs after an amphetamine challenge in withdrawal. A, Differential interference contrast image showing a cell-attached TAN adjacent to a smaller MSN. B, Typical response of TANs to depolarizing and hyperpolarizing current injections. Arrows show characteristic “sag” (left) and after-hyperpolarization (right). C, Current–voltage graph comparing nonrectifying responses in TANs with quiescent, rectifying MSNs and with sometimes spontaneously active persistent low-threshold spike (PLTS) GABA interneurons (note that data for PLTS cells was obtained from Wang et al., 2013). D, Representative traces from experimental results in E showing cell-attached recordings in saline- and amphetamine-treated TANs. Baseline firing frequencies were reduced in withdrawal, but increased after an amphetamine challenge. E, Baseline firing frequencies in saline- and amphetamine-treated TANs. Amphetamine had little effect on saline-exposed mice, but increased firing frequencies in TANs from amphetamine-treated mice. $$p < 0.01, Student's t test and ***p < 0.001, paired t test. F, Normalized TAN firing rates in saline- and amphetamine-treated mice on WD 10 showing response to amphetamine in vitro and after treatment with reserpine. G, Normalized firing frequencies of TANs from saline- and amphetamine-treated mice on WD 10 in response to the D2R agonist quinpirole. Both the depression and rebound increase in cell firing was blocked by the D2R antagonist sulpiride. H, Normalized firing frequencies of TANs from saline- and amphetamine-treated mice on WD 10 in response to the D1R agonist SKF81297. I, Acetylcholine content was reduced in striatal tissue from amphetamine-sensitized mice on WD 10. Amphetamine in vivo on WD 10 reduced acetylcholine content in saline-exposed mice, but increased acetylcholine content in amphetamine-treated mice. $p < 0.05, $$$p < 0.001, Student's t test. J, Normalized amplitude and PPR in response to cortically evoked paired pulses. In MSNs from saline-exposed mice, nicotine (5 nm, n = 6, top; 10 nm, n = 4, bottom) applied by puff pipette reduced the amplitude (of the first pulse) and increased the PPR. K, In MSNs from amphetamine-treated mice, nicotine (5 nm, n = 6, top; 10 nm, n = 9, bottom) increased the eEPSC amplitude and reduced the PPR. L, Representative traces (top) in D1+ and M, D2+ MSNs on WD 10 show the average responses of paired pulses before (left) and 5 min after puff application of nicotine (5–10 nm; right). Nicotine increased the amplitude of the first eEPSC and reduced the PPR in D1+ MSNs, but had no effect on the eEPSC amplitude or the PPR in D2+ MSNs. Scale bars in A, 10 μm; B, 50 mV, 100 ms; D, 100 pA, 5 s; L, M, 100 pA, 5 ms.
Ten days after repeated amphetamine, the baseline firing frequency of amphetamine-exposed TANs (1.5 ± 0.1 Hz; n = 47 cells from 28 mice) was lower than TANs from saline-exposed mice (2 ± 0.2 Hz; n = 38 from 25 mice; p = 0.03, Student's t test). An amphetamine challenge in vitro did not significantly change the firing frequency of TANs from saline-exposed mice (−4 ± 23%; 3.13 ± 0.53 Hz in aCSF vs 2.56 ± 0.53 Hz with amphetamine; n = 7 cells from 5 mice), but increased the firing frequency of TANs from amphetamine-sensitized mice (88 ± 28%; 1.63 ± 0.25 Hz in aCSF vs 2.45 ± 0.24; n = 13 cells from 9 mice; p < 0.001, Student's t test; Fig. 9D–F). The increase in firing frequency during withdrawal was dependent on dopamine release, because it was blocked (17 ± 10%; 2.06 ± 0.21 Hz in aCSF vs 2.36 ± 0.26 Hz with amphetamine; n = 6 cells from 3 mice) by the dopamine depleter reserpine (5 mg/kg i.p. 12 h before killing; Fig. 9F).
The D2R agonist quinpirole caused a reduction in the firing frequency of most TANs (17/23 cells) from both saline-treated mice (n = 10 from 6 mice) and amphetamine-treated mice (n = 13 from 8 mice; Fig. 9G), consistent with prior studies (Yan and Surmeier, 1996, 1997). This reduction in firing frequency varied widely between cells and was generally followed by a rebound increase in firing (71 ± 43% for saline and 68 ± 35% for withdrawal). The depression and rebound in firing after quinpirole were similar in TANs from saline- and amphetamine-treated mice and both were blocked by the D2R antagonist sulpiride (8 ± 8%; n = 4 cells).
The D1R agonist SKF81297 had no effect on the firing frequencies of TANs from saline-exposed mice (25 ± 18%; 2.11 ± 0.46 Hz in aCSF vs 2.76 ± 0.62 Hz with amphetamine; n = 8 from 5 mice), but increased firing rates in cells from amphetamine-treated mice on WD 10 (51 ± 14%; 1.83 ± 0.26 Hz in aCSF vs 2.57 ± 0.3; n = 14 from 7 mice; p = 0.001, Student's t test; Fig. 9H). SKF81297 was sufficient to increase TAN activity in withdrawal, because the D1R antagonist SCH23390 had no effect on firing frequencies in saline-exposed TANs (15 ± 12%; 1.72 ± 0.49 Hz in aCSF vs 1.98 ± 0.58 with SCH23390; n = 4 from 3 mice), but reduced firing frequencies when combined with amphetamine on WD 10 (−38 ± 10%; 1.4 ± 0.31 Hz in SCH23390 vs 0.94 ± 0.29 with amphetamine and SCH23390; n = 6 from 4 mice; data not shown). Therefore, TAN activity was depressed after repeated amphetamine but increased through a D1R-dependent mechanism after an amphetamine challenge in withdrawal.
TAN activity is proportional to acetylcholine efflux (Witten et al., 2010) and we investigated whether these changes in TAN activity were reflected in striatal acetylcholine content as measured by HPLC. In mice treated with repeated amphetamine for 5 d (Fig. 1A), acetylcholine content in the dorsal striatum was reduced on WD 10 (0.156 ± 0.027 nmol/mg protein vs 0.339 ± 0.019 nmol/mg protein for saline-exposed mice; n = 8 each; p < 0.001, t test; Fig. 9I). Acute amphetamine (2 mg/kg i.p 30 min before killing) reduced striatal acetylcholine content in saline-exposed mice (0.249 ± 0.029 nmol/mg protein; p = 0.02, t test), but increased acetylcholine content in withdrawal (0.247 ± 0.023 nmol/mg protein; n = 8 each; p = 0.02, t test), with changes parallel to firing rates in TANs (compare with Fig. 9E).
We also determined whether these changes in acetylcholine availability might modulate corticostriatal activity. Activation of excitatory synapses (McGehee et al., 1995), including corticostriatal projections (Bamford et al., 2008), by acetylcholine is dependent on presynaptic nicotinic receptors that contain both α7* and β2* subunits (Pakkanen et al., 2005). In saline-exposed mice, puff-applied nicotine (5–10 nm) reduced the eEPSC amplitude (−12 ± 3%; −77 ± 10 pA in aCSF vs −68 ± 11 pA after nicotine; n = 10 cells from 6 mice; p = 0.003, paired t test) and increased the PPR (12 ± 2%; 1.15 ± 0.08 in aCSF to 1.29 ± 0.1 in nicotine; p = 0.002, paired t test; Fig. 9J), suggesting β2* nicotinic receptor desensitization (Bamford et al., 2008). In amphetamine-treated mice, nicotine (5–10 nm) boosted the eEPSC amplitude (26 ± 4%; −73 ± 9 pA in aCSF vs −91 ± 11 pA after nicotine; n = 15 cells from 10 mice; p = 0.001, paired t test) and reduced the PPR (−25 ± 2%; 1.28 ± 0.1 in aCSF to 0.9 ± 0.07 in nicotine; p < 0.001, paired t test; Fig. 9K).
PPP was generated in D1R+ MSNs, because nicotine (5–10 nm) increased the eEPSC amplitude (30 ± 9%; −69 ± 14 pA in aCSF vs −87 ± 15 pA after nicotine; n = 7 from 4 mice; p = 0.02, paired t test) and reduced the PPR (−26 ± 4%; 1.29 ± 0.14 in aCSF to 0.94 ± 0.1 in nicotine; p = 0.003, paired t test) in fluorescent MSNs from amphetamine-treated Drd1-EGFP mice on WD 10 (Fig. 9L). Although nicotine (5–10 nm) excited a subset of D2R-expressing MSNs (5/9 cells from 3 mice sampled from both fluorescent MSNs from amphetamine-treated Drd2-EGFP mice and nonfluorescent MSNs from amphetamine-treated Drd1-EGFP mice; data not shown), the ligand had no overall effect on indirect pathway neurons, because the amplitude (−5 ± 10%; −82 ± 12 pA in aCSF vs −82 ± 11 pA after nicotine; n = 9) and the PPR (3 ± 7%; 1.38 ± 0.09 in aCSF to 1.4 ± 0.26 in nicotine) remained unchanged (Fig. 9M).
Locomotor sensitization is dependent on striatal acetylcholine
To assess whether acetylcholine can modify behavioral sensitization through corticostriatal synapses in vivo, we reduced acetylcholine synthesis selectively in conditional choline acetyltransferase (Chatlox/lox) mice (vChAT-KO) through bilateral injection of AAV1-Cre-GFP into the dorsal striatum. Results were compared with sham-KO mice, wild-type littermates that were similarly injected with AAV1-Cre-GFP. Expression analysis of virus-mediated GFP revealed the placement of our injection site within the dorsal striatum (Fig. 10A). Quantitative analysis of acetylcholine by HPLC-electrochemical detection showed that acetylcholine in the dorsal striatum of vChAT-KO mice (0.368 ± 0.056 nmol/mg protein; n = 6 mice) was significantly reduced to ∼51% of sham-KO levels (0.179 ± 0.032 nmol/mg protein; n = 10 mice; p < 0.05, Student's t test).
Figure 10.

Effects of partial acetylcholine depletion in the dorsal striatum. A, Coronal corticostriatal slice showing the areas of stimulation and recording with histology and the typical location of AAV1-Cre-GFP. Inset: Tissue acetylcholine content in the dorsal striatum of sham-KO and vChAT-KO injected with AAV1-Cre-GFP. *p < 0.05, Student's t test. B, Locomotor ambulations in sham-KO and vChAT-KO mice after bilateral injections with AAV1-Cre-GFP. *p < 0.05, Student's t test; @p < 0.05, 2-way ANOVA. C, Locomotor ambulations in sham-KO and vChAT-KO mice after unilateral injections with AAV1-Cre-GFP. D, Representative traces (top) show the average responses of cortically evoked paired pulses before (left) and 5 min after bath application of amphetamine (right). Amphetamine reduced the eEPSC amplitude and increased the PPR in MSNs from saline-exposed, bilaterally injected sham-KO mice. E, F, In saline-exposed, bilaterally injected vChAT-KO mice, amphetamine reduced the eEPSC amplitude and increased the PPR in both GFP+ MSNs (E) and in nonfluorescent MSNs (F). G, Amphetamine increased the eEPSC amplitude and decreased the PPR in MSNs from amphetamine-treated, bilaterally injected sham-KO mice. H, Amphetamine reduced the eEPSC amplitude and increased the PPR in GFP+ MSNs from amphetamine-treated, bilaterally injected vChAT-KO mice. I, Amphetamine increased the eEPSC amplitude and reduced the PPR in nonfluorescent MSNs from amphetamine-treated, bilaterally injected vChAT-KO mice. Scale bars in A, 1 mm; D–I, 100 pA, 5 ms.
Analysis of locomotor activity of vChAT-KO mice (n = 8) and sham-KO mice (n = 13) during our sensitization protocol (Fig. 1A) showed a significant main effect of time (F(5,70) = 16.96, p < 0.01, 2-way ANOVA), indicating increasing locomotor activity after repeated amphetamine stimulation. We found a significant main effect of genotype (F(1,70) = 5.58, p < 0.05, 2-way ANOVA) and a significant time × genotype interaction effect (F(5,70) = 2.97, p = 0.02, 2-way ANOVA), indicating that partial acetylcholine depletion in the dorsal striatum impacted both the stimulatory effect of amphetamine and locomotor sensitization (Fig. 10B).
We generated unilateral vChAT-KO mice (n = 8) by injecting AAV1-Cre-GFP into the dorsal striatum only in one hemisphere. Again, sham-KO animals (n = 8) were generated by viral injections into wild-type littermates of conditional KO mice. Analysis of locomotor activity during our sensitization protocol showed a significant main effect of time (F(5,70) = 26.78, p < 0.001, 2-way ANOVA), indicating increasing locomotor activity after repeated amphetamine stimulation. We found no significant main effects of genotype (F(1,14) = 2.04, p = 0.2, 2-way ANOVA) or time × genotype interaction (F(5,70) = 0.30, p = 0.9, 2-way ANOVA), suggesting that partial acetylcholine depletion in the dorsal striatum of one hemisphere had no effect on amphetamine-induced locomotion or sensitization (Fig. 10C).
Acetylcholine depletion prevents PPP
We tested the effect of acetylcholine depletion on corticostriatal activity in sham-KO and vChAT-KO mice after unilateral injection of AAV-Cre-GFP. Mice were treated with saline or amphetamine for 5 d and eEPSCs in response to paired-pulse cortical stimulation on WD 10 were measured in GFP-expressing MSNs lying adjacent to GFP-expressing cholinergic interneurons. In MSNs from saline-exposed sham-KO mice, amphetamine in vitro decreased the eEPSC amplitude by 26 ± 4% (−120 ± 24 pA in aCSF vs −85 ± 13 pA after amphetamine; n = 6 cells from 4 mice; p = 0.04) and the PPR increased by 26 ± 11% (1.03 ± 0.09 in aCSF vs 1.44 ± 0.14 in amphetamine; Fig. 10D; p = 0.01, paired t test). In these mice, amphetamine reduced corticostriatal activity in both GFP+ (n = 4) and nonfluorescent MSNs (n = 2), so results were pooled.
In GFP+ MSNs from saline-exposed vChAT-KO mice, amphetamine reduced the eEPSC amplitude (−31 ± 8%; −104 ± 13 pA in aCSF vs −71 ± 12 pA after amphetamine; n = 6 from 3 mice; p = 0.02) and increased the PPR (36 ± 14%; 1.18 ± 0.05 in aCSF vs 1.56 ± 0.2 in amphetamine; Fig. 10E; p = 0.02, paired t test). Amphetamine also suppressed corticostriatal activity in both non-GPF-expressing MSNs in saline-exposed vChAT-KO mice and in MSNs from the opposite, noninjected hemisphere, because the eEPSC amplitude decreased by 37 ± 9% (−127 ± 29 pA in aCSF vs −81 ± 24 pA after amphetamine; n = 8 from 4 mice; p = 0.02) and the PPR increased by 58 ± 11% (1.01 ± 0.07 in aCSF vs 1.67 ± 0.21 in amphetamine; p = 0.01, paired t test; Fig. 10F).
In MSNs from amphetamine-treated sham-KO mice, amphetamine in vitro increased the eEPSC amplitude by 18 ± 3% (−87 ± 19 pA in aCSF vs −102 ± 23 pA after amphetamine; n = 10 from 6 mice; p = 0.02) and the PPR decreased by 16 ± 6% (1.16 ± 0.12 in aCSF vs 0.88 ± 0.1 in amphetamine; p = 0.03, paired t test; Fig. 10G), consistent with PPP. Amphetamine potentiated corticostriatal activity in both GFP+ and nonfluorescent MSNs (n = 6) from amphetamine-treated sham-KO mice and also after bihemispheric injection (n = 4), so the results were pooled.
The increase in corticostriatal activity after an amphetamine challenge was prevented by acetylcholine depletion. In GFP+ MSNs from amphetamine-treated vChAT-KO mice, amphetamine decreased the eEPSC amplitude (−27 ± 5%; −137 ± 44 pA in aCSF vs −100 ± 32 pA after amphetamine; n = 7 from 6 mice; p = 0.02) and the PPR increased (36 ± 5%; 1.3 ± 0.12 in aCSF vs 1.78 ± 0.19 in amphetamine; p < 0.001, paired t test; Fig. 10H). Amphetamine caused a similar reduction in corticostriatal activity in MSNs from vChAT-KO mice after bihemispheric injection (n = 3), so results were pooled.
As anticipated, amphetamine in vitro augmented corticostriatal activity in nonfluorescent MSNs from amphetamine-exposed vChAT-KO mice and also in nonfluorescent MSNs from the opposite, noninjected hemisphere, because the eEPSC amplitude increased by 20 ± 6% (−96 ± 16 pA in aCSF vs −121 ± 26 pA after amphetamine; n = 8 from 4 mice; p = 0.04) and the PPR decreased by 16 ± 4% (1.16 ± 0.12 in aCSF vs 0.97 ± 0.12 in amphetamine; p = 0.01, paired t test; Fig. 10I). Amphetamine also potentiated corticostriatal activity in nonfluorescent MSNs from vChAT-KO mice after bihemispheric injection (n = 2), so the results were pooled. Overall, the PPR in MSNs from saline-exposed sham-KO mice (1.01 ± 0.05; n = 14) was less than that in MSNs from saline-exposed vChAT-KO mice (1.18 ± 0.05; n = 6; p = 0.04, Student's t test). Therefore, acetylcholine depletion produced CPD in MSNs adjacent to cholinergic-depleted TANs from vChAT-KO mice while preventing PPP in these MSNs.
Acetylcholine promotes locomotion and coordination
We assessed the impact of reduced acetylcholine content in the dorsal striatum on motor behaviors. Novelty-induced locomotion was measured in bilateral sham-KO mice (n = 7) and vChAT-KO mice (n = 15; Fig. 11A). Analysis of ambulations in the novel environment showed a significant main effect of time (F(11,220) = 21.42, p < 0.01, 2-way ANOVA), indicating decreasing locomotor activity after animals habituate to the novel environment. Although we found no significant main effect of genotype (F(1,220) = 0.30, p > 0.05, 2-way ANOVA), there was a significant time × genotype interaction effect (F(11,220) = 1.88, p < 0.05, 2-way ANOVA).
Figure 11.

Effects of partial acetylcholine depletion in the dorsal striatum on locomotion, coordination, cue-dependent learning, egocentric learning, and cognitive flexibility. A, Novelty-induced locomotor responses by bilaterally injected sham-KO and vChAT-KO mice. B, Number of slips during the balance beam test for motor coordination by bilaterally injected sham-KO and vChAT-KO mice (left) and in saline and amphetamine-treated mice on WD 10. *p < 0.05, **p < 0.01, Student's t test. C, Latency to fall from an accelerating rotarod by bilaterally injected sham-KO and vChAT-KO mice. D, Escape latency in a Morris water maze by bilaterally injected sham-KO and vChAT-KO mice. **p < 0.01, Student's t test; @p < 0.05, 2-way ANOVA. E, Time spent by sham-KO mice (left) and vChAT-KO mice (right) searching in the quadrants of the Morris water maze during a 90 s spatial memory trial 24 h after the last training session. **p < 0.01, post hoc t test against time spent in nontarget quadrants. F, Escape latencies (left) and correct choices (right) of bilaterally injected sham-KO and vChAT-KO mice during a cue-dependent water escape task. G, Escape latencies (left) and correct choices (right) of bilaterally injected sham-KO and vChAT-KO mice during an egocentric water escape task. H, Escape latencies (left) and correct choices (right) of bilaterally injected sham-KO and vChAT-KO mice during a strategy-shift water escape task (mice had previously learned egocentric responses and were then required to adapt to cue-dependent responses). **p < 0.01, Bonferroni's multiple-comparisons post hoc t test and @p < 0.05, 2-way ANOVA. I, Locomotor ambulations in saline- and amphetamine-treated mice used for behavioral experiments. J, Escape latency in a Morris water maze by saline- and amphetamine-treated mice on WD 10. @p < 0.05, 2-way ANOVA. K, Time spent by saline-treated mice (left) and amphetamine-treated mice (right) on WD 10 searching in the quadrants of the Morris water maze during a 90 s spatial memory trial 24 h after the last training session. **p < 0.01, post hoc t test against time spent in nontarget quadrants.
Motor coordination on the balance beam was assessed in sham-KO mice (n = 8) and vChAT-KO mice (n = 10; Fig. 11B, left). Analysis of the number of slips made while traversing the beam revealed a significant difference between sham and acetylcholine-depleted mice (p < 0.05, Student's t test), suggesting a deficit in learning-independent motor coordination.
Motor skill learning on a rotarod was recorded in sham-KO mice (n = 8) and vChAT-KO mice (n = 10; Fig. 11C). The latency to fall off the rod increased with time (F(11,176) = 16.22, p < 0.01, 2-way ANOVA), indicating increasing motor skill during training, but there was no significant main effect of genotype (F(1,176) = 0.55, p > 0.05, 2-way ANOVA) or time × genotype interaction effect (F(11,176) = 1.51, p > 0.05, 2-way ANOVA).
Acetylcholine promotes visuospatial function and spatial reference memory
Visuospatial performance was analyzed in sham-KO mice (n = 9) and vChAT-KO mice (n = 10) using the Morris water maze. Analysis of the latencies to escape from the water to a submerged platform showed a significant main effect of time (F(3,51) = 17.66, p < 0.01, 2-way ANOVA, Fig. 11D), indicating platform position learning after repeated training. We found significant main effects of genotype (F(1,51) = 5.69, p < 0.05, 2-way ANOVA), but not of the time × genotype interaction (F(3,51) = 1.05, p > 0.05, 2-way ANOVA), suggesting that partial acetylcholine depletion produced impaired visuospatial learning. Although vChAT-KO mice had reduced swim speeds (p < 0.05, Student's t test), estimates relating swim speeds with escape latencies showed that the reduction in swim speed in vChAT-KO mice might account for abnormal visuospatial performance on the first 2 d of training, but not on days 3 and 4.
Preference for the pool quadrant where the hidden platform was located during training was ascertained within each group by one-way ANOVA of the time spent in the four pool quadrants during the 90 s spatial memory trial on the last day of training (Fig. 11E). For sham-KO animals, we found a significant effect of quadrant location on the time spent in each quadrant (F(3,32) = 10.60, p < 0.01, ANOVA) and Bonferroni's multiple-comparisons post hoc test confirmed that sham animals spent significantly more time in quadrant 4, where the platform was located during training, than in any other quadrant (p < 0.01, Student's t test). For acetylcholine-depleted animals, however, the analysis could not confirm a significant effect of quadrant location on the time spent in each quadrant (F(3,39) = 1.87, p > 0.05, ANOVA). Depleted animals did not show a quadrant preference, suggesting a deficit of spatial reference memory.
Acetylcholine enables cognitive flexibility
Cue-dependent learning and cognitive flexibility are dependent on dopamine availability in the dorsal striatum (Darvas and Palmiter, 2011) and become impaired after psychostimulant exposure (Stalnaker et al., 2009). To determine whether partial acetylcholine depletion alters these behaviors, cue-dependent learning was measured in sham-KO mice (n = 6) and vChAT-KO mice (n = 6; Fig. 11F). Analysis of escape latencies showed a significant main effect of time (F(3,30) = 6.22, p < 0.01, 2-way ANOVA), but no significant main effects of genotype (F(1,30) = 0.07, p > 0.05) or of the time × genotype interaction (F(3,30) = 0.29, p > 0.05). Similarly, an analysis of correct choices showed a significant main effect of time (F(3,30) = 68.55, p < 0.01, 2-way ANOVA), indicating learning of the cue-based water escape strategy after repeated training. However, we found no significant main effects of genotype (F(1,30) = 0.32, p > 0.05, 2-way ANOVA) or of the time × genotype interaction (F(3,30) = 0.58, p > 0.05, 2-way ANOVA), suggesting that partial acetylcholine depletion had no impact on cue-dependent learning.
Egocentric (turn-based) learning was measured in sham-KO mice (n = 11) and vChAT-KO mice (n = 11; Fig. 11G). Analysis of escape latencies showed a significant main effect of time (F(2,40) = 12.15, p < 0.01, 2-way ANOVA). We found no significant main effects of genotype (F(1,40) = 1.18, p > 0.05, 2-way ANOVA) or of the time × genotype interaction (F(2,40) = 1.25, p > 0.05, 2-way ANOVA). Similarly, analysis of correct choices showed a significant main effect of time (F(2,40) = 36.07, p < 0.01, 2-way ANOVA), indicating learning of the egocentric escape strategy after repeated training. However, we found no significant main effects of genotype (F(1,40) = 0.05, p > 0.05, 2-way ANOVA) or of the time × genotype interaction (F(2,40) = 0.32, p > 0.05, 2-way ANOVA), suggesting that partial acetylcholine depletion had no impact on egocentric learning.
Cognitive flexibility was measured in sham-KO mice (n = 11) and vChAT-KO mice (n = 11) that had previously learned the egocentric escape strategy and were then forced to use a cue-based strategy (Fig. 11H). Analysis of escape latencies showed a significant main effect of time (F(4,80) = 7.51, p < 0.01, 2-way ANOVA), but we found no significant main effects of genotype (F(1,80) = 0.15, p > 0.05, 2-way ANOVA) or of the time × genotype interaction (F(4,80) = 0.25, p > 0.05, 2-way ANOVA). In contrast, analysis of correct choices showed significant main effects of time (F(4,80) = 56.81, p < 0.01, 2-way ANOVA), genotype (F(1,80) = 4.51, p < 0.05, 2-way ANOVA), and the time × genotype interaction (F(4,80) = 3.22, p < 0.05, 2-way ANOVA), suggesting that partial acetylcholine depletion impaired performance of the strategy-shifting task. Post hoc pairwise comparisons of correct choices confirmed a significant difference between sham and acetylcholine-depleted animals on the third day of cognitive flexibility testing (Bonferroni, p < 0.01).
Amphetamine withdrawal modifies visuospatial function, spatial reference memory and motor coordination
To determine whether behaviors that were affected by acetylcholine depletion in the dorsal striatum were also sensitive to changes occurring during amphetamine withdrawal, we sensitized a group of wild-type animals and tested their visuospatial function, spatial reference memory, and motor coordination during withdrawal. Results were compared with nonsensitized, saline-treated animals, wild-type mice that underwent the same series of locomotor recordings but always received saline injections instead of amphetamine. In addition, motor coordination was tested in the presence of either saline or the acetylcholine receptor agonist nicotine at 0.25 mg/kg, a dose previously found to promote behavioral change in mice (Darvas et al., 2009).
Analysis of locomotor activity (Fig. 11I; n = 15 each) during the sensitization procedure (Fig. 6A) revealed significant effects of time (F(5,140) = 12.53, p < 0.01, ANOVA), treatment (F(1,140) = 76.54, p < 0.01), and the time × treatment interaction (F(5,140) = 10.32, p < 0.01, ANOVA), confirming the stimulatory effects of amphetamine versus saline treatment and increasing locomotor activity after repeated amphetamine administration.
Visuospatial performance was analyzed in a subset of saline-treated mice (n = 7) and amphetamine-treated mice (n = 8) on WD 10 (experiment day 20) using the Morris water maze. Analysis of escape latencies to the submerged platform (Fig. 11J) showed a significant effect of time (F(3,39) = 18.26, p < 0.01), but not of treatment (F(1,39) = 0.98, p > 0.05, ANOVA). However, there was a significant effect of the time × treatment interaction (F(3,39) = 2.90, p < 0.05, ANOVA), suggesting an impact of amphetamine sensitization on visuospatial learning. Analysis of the memory probe trial (Fig. 11K) revealed a significant main effect of quadrant location for saline-exposed mice (F(3,27) = 13.76, p < 0.01, ANOVA) and Bonferroni's multiple-comparisons post hoc test confirmed that nonsensitized animals spent more time in the pool quadrant where the hidden platform was located during training (Student's t test, p < 0.01). For sensitized animals, analysis of the memory probe trial revealed no significant main effect of quadrant location (F(3,31) = 2.82, p > 0.05, ANOVA), suggesting impaired memory retention in withdrawal.
We also tested the effects of nicotine (0.25 mg/kg) on motor coordination in sensitized (n = 15) and nonsensitized (n = 15) mice. Although analysis of number of slips made while crossing the balance beam (Fig. 11B, right) revealed no significant effect of group (nonsensitized vs sensitized) factor (F(1,56) = 0.36, p > 0.05, two-way ANOVA), we found a significant effect of drug treatment (nicotine vs saline) during the balance beam procedure (F(1,56) = 11.09, p < 0.01, two-way ANOVA), but not of the group × treatment interaction (F(1,56) = 2.08, p > 0.05, two-way ANOVA). Bonferroni's multiple-comparisons post hoc test confirmed that only performance by sensitized animals was significantly altered by nicotine administration (Student's t test, p < 0.01), suggesting changes in sensitivity to cholinergic signaling during withdrawal.
Discussion
Motor movements and behaviors are putatively driven by complex interactions at cortical synapses on MSNs (Bamford and Cepeda, 2009; Cepeda et al., 2010), where short-term changes in dopamine availability provoke persistent functional and structural changes in striatal circuitry (Robinson and Berridge, 2003; Kalivas, 2009; Sulzer, 2011). Our data show that repeated exposure to amphetamine, when delivered in a paradigm that releases dopamine (Bamford et al., 2004b) while allowing simultaneous measurements of behavior (Beutler et al., 2011), promotes long-lasting, yet reversible changes in glutamate release from corticostriatal afferents in the dorsal striatum (Fig. 12). Repeated amphetamine caused a frequency-dependent CPD in corticostriatal activity during withdrawal, whereas an amphetamine challenge caused a D1R-dependent and frequency-independent PPP that reversed CPD. Although the development of CPD was dependent on repeated dopamine release, its long-term maintenance was supported by a reduction in acetylcholine availability from TANs. Acetylcholine appears central to this form of plasticity, because a rebound in TAN firing with an accompanying increase in acetylcholine availability after drug readministration promoted PPP. PPP caused synaptic normalization of specific subtypes of cortical terminals, promoted an imbalance in striatal output networks by potentiating corticostriatal activity through D1R-expressing MSNs, and was proportional to the expression of locomotor sensitization in individual animals. In addition to CPD and PPP, TANs also supported locomotion, motor coordination, and cognitive flexibility, suggesting that the dorsal striatum contributes to normal motor and cognitive function and participates in addictive behaviors (Robinson and Berridge, 2003) by promoting allostasis and sensitized responses (Ahmed and Koob, 2005).
Figure 12.

Proposed mechanism of presynaptic corticostriatal plasticity in the dorsal striatum. A, Simplified microcircuit showing that glutamatergic afferents from the motor cortex excite GABAergic D1+ and D2+ MSNs through AMPA and NMDA receptors. Glutamate release is modulated by dopamine from nigrostriatal afferents and by acetylcholine (ACh) released by TANs, which express both D1R and D2Rs. B1, B2, In drug-naive mice, ACh provides tonic excitation at corticostriatal terminals through α7* and β2* nicotinic receptors (B1) and dopamine (DA) released by amphetamine reduces excitation of D2+ MSNs through D2 and cannabinoid CB1 receptors (B2). C1, Changes in corticostriatal activity during withdrawal from repeated amphetamine. Presynaptic excitation of D1+ and D2+ MSNs is suppressed by a chronic reduction in TAN firing and acetylcholine efflux. C2, Amphetamine challenge in withdrawal preferentially increases presynaptic activity at D1+ MSNs by boosting TAN firing and acetylcholine availability at corticostriatal terminals.
Consistent with prior results (Bamford et al., 2004a), recordings from saline-exposed mice revealed that dopamine provides low-pass-frequency filtering of cortical information, with filtering specific to those terminals with a low probability of release. Dopamine filtering of corticostriatal activity was represented in postsynaptic MSNs by a reduction in high-frequency, low-amplitude inward currents. Repeated amphetamine treatment caused a CPD during withdrawal lasting over 50 d and redefined presynaptic filtering by inhibiting cortical terminals with a low probability of release. Although D2Rs encouraged further presynaptic inhibition of those synapses in withdrawal, a drug challenge provoked a D1R-dependent PPP with a corresponding increase in glutamate release. Therefore, the boost in glutamate release after an amphetamine challenge normalized subpopulations of individual synapses and at least transiently returned corticostriatal activity to a more stable, normalized state.
Neurotransmitter release from subpopulations of presynaptic corticostriatal terminals is contingent on neuronal firing frequencies (Bamford et al., 2004b; Wang et al., 2012) and this dependence is altered in diseases that affect corticostriatal function (Bamford et al., 2004a; Joshi et al., 2009). The detection of CPD at corticostriatal synapses was dependent on the frequency of cortical stimulation, because cortical stimulation at 1 Hz enhanced presynaptic release, whereas higher frequencies produced CPD. However, PPP was frequency independent and the boost in presynaptic activity from a depressed state to a potentiated state may be one mechanism involved in the initiation and maintenance of behavioral sensitization. PPP was not dependent on dopamine release kinetics (Bamford et al., 2008) and could not be dependent on changes in dopamine neuronal firing because it was measured in the striatal slice from which dopamine cell bodies were absent. Rather, PPP was dependent on the excitatory actions of D1Rs and occurred only in those mice with a history of drug exposure. Although PPP potentiated corticostriatal activity in some D2R-expressing MSNs, its main effect was seen in D1R-expressing MSNs, suggesting that repeated dopamine promotes locomotor sensitization by redistributing corticostriatal signals through the direct basal ganglia pathway. Unlike corticostriatal plasticity in the nucleus accumbens shell (Pascoli et al., 2012), we detected no differences in postsynaptic mEPSC amplitudes in the dorsal striatum and the ratio of AMPA and NMDA receptors was unchanged during withdrawal, suggesting that any postsynaptic metaplasticity in response to CPD (Moussawi et al., 2009) modifies the expression of these receptors in similar directions.
These data are consistent with and extend observations of cocaine-induced glutamatergic neuroadaptations in the nucleus accumbens, where drug withdrawal is accompanied by a marked reduction in glutamate availability both in the extracellular space via the cystine-glutamate exchange (Kalivas, 2004) and from synaptic terminals (Kozell and Meshul, 2003). Such changes in the gradient between synaptic and extrasynaptic glutamate would likely alter the induction of long-term potentiation and depression (Kalivas, 2009). Compared with measures of decreased extrasynaptic glutamate in the nucleus accumbens (McFarland et al., 2003), we found that synaptic glutamate during drug withdrawal in the dorsal striatum depends on cortical activity, with higher cortical frequencies favoring a comparatively larger reduction in glutamate release. The frequency dependence of CPD may be mediated secondarily through glutamate modulation of TANs (Aosaki et al., 2010), through endocannabinoid release (Wang et al., 2012), or by other means, including activity-dependent presynaptic group II metabotropic receptors that are potent inhibitors of glutamate release (Carlton et al., 2011; Wang et al., 2012) and play an important role in glutamate homeostasis during drug withdrawal (Kalivas, 2009).
In the nucleus accumbens, cocaine reinstatement or cortical activation during drug-seeking behavior (Moussawi et al., 2009) significantly increases glutamate availability (Pierce et al., 1996) from glutamatergic afferents (McFarland et al., 2003). Similarly, we found that acute amphetamine caused presynaptic inhibition of D2R-expressing MSNs, likely due to stimulation of presynaptic D2 and endocannabinoid CB1 receptors (Yin and Lovinger, 2006; André et al., 2010; Wang et al., 2012), whereas an amphetamine challenge caused PPP. The paradoxical boost of glutamate from cortical terminals on drug readministration likely participates in locomotor sensitization, because PPP was proportional to locomotor responses after a drug challenge and both PPP and locomotor sensitization could be blocked by D1R antagonists, as well as by acetylcholine depletion in choline acetyltransferase KO mice. Therefore, a net shift from D2R-generated depression to D1R-mediated excitation after an amphetamine challenge in withdrawal might increase locomotor activity by enhancing corticostriatal glutamate release, thereby activating MSNs that have a diminished capacity to respond (Gass and Olive, 2008) and explain why high doses of amphetamine reduce spontaneous nocturnal motor activity in withdrawal and enhance locomotor responses after drug reinstatement (Robinson and Camp, 1987). Sensitization models of incentive salience (Robinson and Camp, 1987; Robinson and Berridge, 2003) suggest that supraphysiological glutamatergic drive promotes compulsive drug seeking in addicts by decreasing the value of natural rewards (Kalivas and Volkow, 2005). Therefore, although CPD and PPP may play key roles in sensitization, further experiments are needed to determine if acetylcholine-dependent alterations in corticostriatal activity overlap with the plasticity and learning associated with self-administration and reinstatement.
Presynaptic plasticity is dependent on tonic excitation and inhibition by acetylcholine at both α7* and β2* nicotinic receptor subunits (Pakkanen et al., 2005; Bamford et al., 2008), indicating that dopamine modulation of acetylcholine release (Bickerdike and Abercrombie, 1997) may provide a set point for glutamate availability (Ahmed and Koob, 1998) that extends to escalation of drug intake (Hansen and Mark, 2007). CPD was dependent on D2Rs that depress TAN firing and acetylcholine efflux, whereas PPP was contingent on a D1R-mediated increase in TAN firing and acetylcholine efflux. Interestingly, D2Rs remained inhibitory in withdrawal, suggesting that although the set point of dynamic TAN firing was reduced, their lower limit had not been reached. Consistent with these findings, D2Rs (and CPD) may establish the degree of sensitization and D1Rs (and PPP) may promote its acquisition (Kuribara, 1995; Kelly et al., 2008). It is possible that therapeutic approaches aimed at elevating synaptic glutamate in withdrawal would counteract CPD and facilitate extinction of drug-seeking behavior, whereas those drugs that counteract drug-induced PPP would act to disrupt the reinforcing effects of drugs in addicts (Gass and Olive, 2008; Reissner and Kalivas, 2010). Because dopamine provokes state changes in synaptic activity, our findings may explain how salient experiences encode behaviors and automatic movements, extending to other neuropsychological disorders, including Parkinson's disease, where dopamine depletion disrupts striatal activity by modifying subpopulations of corticostriatal terminals (Bamford and Cepeda, 2009; Cepeda et al., 2010).
Because PPP may play a key role in learning and dependence, we investigated learning in mice with reduced acetylcholine and PPP. Partial depletion of striatal acetylcholine from TANs depressed corticostriatal activity in adjacent MSNs from saline-exposed mice, prevented PPP after amphetamine challenge in withdrawal, and partially reduced the magnitude of locomotor sensitization. Mice with acetylcholine depletion showed a modest deficit, consistent with impaired cognitive flexibility. Limited cognitive flexibility occurs in a number of neuropsychiatric disorders, including attention deficit disorder, obsessive-compulsive disorder, and substance dependence, where shifts in attention, inhibition of unwanted actions, and responses to psychostimulants become restricted (Etchepareborda and Mulas, 2004; Chamberlain et al., 2005; Stalnaker et al., 2009). Mice with partial acetylcholine depletion and control mice undergoing withdrawal from repeated amphetamine also had deficits in motor coordination, visuospatial learning, and exploratory behaviors. Because the motor performance of sensitized animals was significantly improved by enhancement of cholinergic signaling with nicotine, these results encourage the possibility of cognitive restoration and the reduction of drug seeking and craving by nicotine or cholinergic ligands in amphetamine-withdrawn animals and humans.
Footnotes
This work was supported by the National Institute on Drug Abuse–National Institutes of Health (Grant #5T32DA007278 to G.P.S), Pacific Northwest Udall Center (Grant #P50NS062684 to M.D. and R.D.P.), University of Washington Alcohol and Drug Abuse Institute (N.S.B.), National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grant #K02NS052536 and Grant #R01NS060803 to N.S.B.), and National Institute of Child Health and Human Development–National Institutes of Health (Grant #P30HD002274 to N.S.B.), University of Washington Vision Research Center and Seattle Children's Hospital (Seattle, WA). We thank Michael S. Levine and Carlos Cepeda at the University of California–Los Angeles for providing breeding pairs of Drd1 and Drd2 EGFP BAC transgenic mice and David Sulzer for careful reading and pertinent comments on the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interests.
References
- Abercrombie ED, DeBoer P. Substantia nigra D1 receptors and stimulation of striatal cholinergic interneurons by dopamine: a proposed circuit mechanism. J Neurosci. 1997;17:8498–8505. doi: 10.1523/JNEUROSCI.17-21-08498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Koob GF. Transition to drug addiction: a negative reinforcement model based on an allostatic decrease in reward function. Psychopharmacology (Berl) 2005;180:473–490. doi: 10.1007/s00213-005-2180-z. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- André VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010;31:14–28. doi: 10.1111/j.1460-9568.2009.07047.x. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int. 2010;10:S148–S157. doi: 10.1111/j.1447-0594.2010.00588.x. [DOI] [PubMed] [Google Scholar]
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampà C, Calabresi P. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson's disease. J Neurosci. 2011;31:12513–12522. doi: 10.1523/JNEUROSCI.2236-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Cepeda C. The corticostriatal pathway in Parkinson's disease. In: Tseng KY, editor. Cortico-subcortical dynamics in Parkinson's disease. New York: Humana; 2009. pp. 87–104. [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004a;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004b;42:653–663. doi: 10.1016/S0896-6273(04)00265-X. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Joyce JA, Scarlis CA, Hanan W, Wu NP, André VM, Cohen R, Cepeda C, Levine MS, Harleton E, Sulzer D. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008;58:89–103. doi: 10.1016/j.neuron.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell K, Duffy P, Kalivas PW. Context-specific enhancement of glutamate transmission by cocaine. Neuropsychopharmacology. 2000;23:335–344. doi: 10.1016/S0893-133X(00)00100-7. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler LR, Wanat MJ, Quintana A, Sanz E, Bamford NS, Zweifel LS, Palmiter RD. Balanced NMDA receptor activity in dopamine D1 receptor (D1R)- and D2R-expressing medium spiny neurons is required for amphetamine sensitization. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1101424108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickerdike MJ, Abercrombie ED. Striatal acetylcholine release correlates with behavioral sensitization in rats withdrawn from chronic amphetamine. J Pharmacol Exp Ther. 1997;282:818–826. [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Govea R, Du J. Group II/III metabotropic glutamate receptors exert endogenous activity-dependent modulation of TRPV1 receptors on peripheral nociceptors. J Neurosci. 2011;31:12727–12737. doi: 10.1523/JNEUROSCI.6558-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS. Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur J Neurosci. 2008;27:671–682. doi: 10.1111/j.1460-9568.2008.06038.x. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Bamford NS, André VM, Levine MS. Alterations in corticostriatal synaptic function in Huntington's and Parkinson's diseases. In: Steiner H, Tseng KY, editors. Basal ganglia structure and function. San Diego: Elsevier; 2010. pp. 607–623. [Google Scholar]
- Chamberlain SR, Blackwell AD, Fineberg NA, Robbins TW, Sahakian BJ. The neuropsychology of obsessive compulsive disorder: the importance of failures in cognitive and behavioural inhibition as candidate endophenotypic markers. Neurosci Biobehav Rev. 2005;29:399–419. doi: 10.1016/j.neubiorev.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Charpier S, Mahon S, Deniau JM. In vivo induction of striatal long-term potentiation by low-frequency stimulation of the cerebral cortex. Neuroscience. 1999;91:1209–1222. doi: 10.1016/S0306-4522(98)00719-2. [DOI] [PubMed] [Google Scholar]
- Cornish JL, Duffy P, Kalivas PW. A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience. 1999;93:1359–1367. doi: 10.1016/S0306-4522(99)00214-6. [DOI] [PubMed] [Google Scholar]
- Cowan RL, Wilson CJ. Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J Neurophysiol. 1994;71:17–32. doi: 10.1152/jn.1994.71.1.17. [DOI] [PubMed] [Google Scholar]
- Darvas M, Palmiter RD. Restriction of dopamine signaling to the dorsolateral striatum is sufficient for many cognitive behaviors. Proc Natl Acad Sci U S A. 2009;106:14664–14669. doi: 10.1073/pnas.0907299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M, Palmiter RD. Contributions of striatal dopamine signaling to the modulation of cognitive flexibility. Biol Psychiatry. 2011;69:704–707. doi: 10.1016/j.biopsych.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M, Morsch M, Racz I, Ahmadi S, Swandulla D, Zimmer A. Modulation of the Ca2+ conductance of nicotinic acetylcholine receptors by Lypd6. Eur Neuropsychopharmacol. 2009;19:670–681. doi: 10.1016/j.euroneuro.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin B, Doudnikoff E, Gonon F, Bloch B. Differences in ultrastructural localization of dopaminergic D1 receptors between dorsal striatum and nucleus accumbens in the rat. Neurosci Lett. 2007;419:273–277. doi: 10.1016/j.neulet.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Etchepareborda MC, Mulas F. Cognitive flexibility, an additional symptom of attention deficit hyperactivity disorder. Is it a therapeutically predictive element? Rev Neurol. 2004;38:S97–S102. [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fellous JM, Rudolph M, Destexhe A, Sejnowski TJ. Synaptic background noise controls the input/output characteristics of single cells in an in vitro model of in vivo activity. Neuroscience. 2003;122:811–829. doi: 10.1016/j.neuroscience.2003.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Permenter LK, Lake R, Worley PF, Kalivas PW. Homer1 proteins and AMPA receptors modulate cocaine-induced behavioural plasticity. Eur J Neurosci. 2003;18:1645–1651. doi: 10.1046/j.1460-9568.2003.02880.x. [DOI] [PubMed] [Google Scholar]
- Gorelova N, Yang CR. The course of neural projection from the prefrontal cortex to the nucleus accumbens in the rat. Neuroscience. 1997;76:689–706. doi: 10.1016/S0306-4522(96)00380-6. [DOI] [PubMed] [Google Scholar]
- Hansen ST, Mark GP. The nicotinic acetylcholine receptor antagonist mecamylamine prevents escalation of cocaine self-administration in rats with extended daily access. Psychopharmacology (Berl) 2007;194:53–61. doi: 10.1007/s00213-007-0822-z. [DOI] [PubMed] [Google Scholar]
- Jog MS, Kubota Y, Connolly CI, Hillegaart V, Graybiel AM. Building neural representations of habits. Science. 1999;286:1745–1749. doi: 10.1126/science.286.5445.1745. [DOI] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, André VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, Bamford NS. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29:2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, McFarland K, Bowers S, Szumlinski K, Xi ZX, Baker D. Glutamate transmission and addiction to cocaine. Ann N Y Acad Sci. 2003;1003:169–175. doi: 10.1196/annals.1300.009. [DOI] [PubMed] [Google Scholar]
- Kasper EM, Larkman AU, Lübke J, Blakemore C. Pyramidal neurons in layer 5 of the rat visual cortex. II. Development of electrophysiological properties. J Comp Neurol. 1994;339:475–494. doi: 10.1002/cne.903390403. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Low MJ, Rubinstein M, Phillips TJ. Role of dopamine D1-like receptors in methamphetamine locomotor responses of D2 receptor knockout mice. Genes Brain Behav. 2008;7:568–577. doi: 10.1111/j.1601-183X.2008.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Glutamate and reinstatement. Curr Opin Pharmacol. 2009;9:59–64. doi: 10.1016/j.coph.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozell B, Meshul K. Alterations in nerve terminal glutamate immunoreactivity in the nucleus accumbens and ventral tegmental area following single and repeated doses of cocaine. Psychopharmacology (Berl) 2003;165:337–345. doi: 10.1007/s00213-002-1296-7. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PF, Christensen CH, Hazelwood LA, Dobi A, Bock R, Sibley DR, Mateo Y, Alvarez VA. Dopamine D2 receptor overexpression alters behavior and physiology in Drd2-EGFP mice. J Neurosci. 2011;31:126–132. doi: 10.1523/JNEUROSCI.4287-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribara H. Dopamine D1 receptor antagonist SCH 23390 retards methamphetamine sensitization in both combined administration and early posttreatment schedules in mice. Pharmacol Biochem Behav. 1995;52:759–763. doi: 10.1016/0091-3057(95)00173-T. [DOI] [PubMed] [Google Scholar]
- Lee K, Dixon AK, Freeman TC, Richardson PJ. Identification of an ATP-sensitive potassium channel current in rat striatal cholinergic interneurones. J Physiol. 1998;510:441–453. doi: 10.1111/j.1469-7793.1998.441bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu XT, Berney TG, Vartanian AJ, Stine CD, Wolf ME, White FJ. Both glutamate receptor antagonists and prefrontal cortex lesions prevent induction of cocaine sensitization and associated neuroadaptations. Synapse. 1999;34:169–180. doi: 10.1002/(SICI)1098-2396(19991201)34:3<169::AID-SYN1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Memo M, Missale C, Carruba MO, Spano PF. Pharmacology and biochemistry of dopamine receptors in the central nervous system and peripheral tissue. J Neural Transm Suppl. 1986;22:19–32. [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/S0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Hang GB, Grueter BA, Pascoli V, Lüscher C, Malenka RC, Kreitzer AC. A comparison of striatal-dependent behaviors in wild-type and hemizygous Drd1a and Drd2 BAC transgenic mice. J Neurosci. 2012;32:9119–9123. doi: 10.1523/JNEUROSCI.0224-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Pakkanen JS, Jokitalo E, Tuominen RK. Up-regulation of beta2 and alpha7 subunit containing nicotinic acetylcholine receptors in mouse striatum at cellular level. Eur J Neurosci. 2005;21:2681–2691. doi: 10.1111/j.1460-9568.2005.04105.x. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin J. The mouse brain in stereotaxic coordinates. Ed 2. London: Academic; 2005. [Google Scholar]
- Pennartz CM, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino LJ, Smith HR, Nader MA, Beveridge TJ. The effects of cocaine: a shifting target over the course of addiction. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1593–1600. doi: 10.1016/j.pnpbp.2007.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Sanz E, Wang W, Storey GP, Guler AD, Wanat MJ, Roller BA, La Torre A, Amieux PS, McKnight GS, Bamford NS, Palmiter RD. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat Neurosci. 2012 doi: 10.1038/nn.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Kalivas PW. Using glutamate homeostasis as a target for treating addictive disorders. Behav Pharmacol. 2010;21:514–522. doi: 10.1097/FBP.0b013e32833d41b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Camp DM. Long-lasting effects of escalating doses of d-amphetamine on brain monoamines, amphetamine-induced stereotyped behavior and spontaneous nocturnal locomotion. Pharmacol Biochem Behav. 1987;26:821–827. doi: 10.1016/0091-3057(87)90616-2. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Takahashi Y, Roesch MR, Schoenbaum G. Neural substrates of cognitive inflexibility after chronic cocaine exposure. Neuropharmacology. 2009;56:63–72. doi: 10.1016/j.neuropharm.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Kincaid AE, Wilson CJ. Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol. 1997;77:1697–1715. doi: 10.1152/jn.1997.77.4.1697. [DOI] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kloot W. The regulation of quantal size. Prog Neurobiol. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-W. [DOI] [PubMed] [Google Scholar]
- Wang W, Nitulescu I, Lewis JS, Lemos JC, Bamford IJ, Posielski NM, Storey GP, Phillips PE, Bamford NS. Over-inhibition of corticostriatal activity following prenatal cocaine exposure. Ann Neurol. 2013;73:355–369. doi: 10.1002/ana.23805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, Bamford NS. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol. 2012;590:3743–3769. doi: 10.1113/jphysiol.2012.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. J Neurosci. 1996;16:2592–2604. doi: 10.1523/JNEUROSCI.16-08-02592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/S0896-6273(00)80402-X. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci. 2004;19:181–189. doi: 10.1111/j.1460-9568.2004.03095.x. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]