

Abstract
We recently reported the anti-cancer and anti-cytomegalovirus (CMV) activity of artemisinin-derived trioxane diphenylphosphate dimer 838. To probe the relationship between chemical structure and anti-CMV and anti-cancer activities, we now report synthesis and evaluation of a series of eight new dimer phosphate ester analogs of 838. This series of novel molecules was screened against human foreskin fibroblasts (HFFs) infected with CMV and against the human Jurkat T cell acute lymphoblastic leukemia cell line. This SAR study confirms the very high anti-CMV and anti-cancer potencies of dimer diphenyl phosphate ester 838 without its being toxic to normal cells.
Introduction
Despite increased spending over the past two decades, the number of FDA approved drugs per dollar spent continues to decline.1 As such, focus has increasingly shifted to finding new uses for therapeutics already evaluated in humans in order to lower the cost of drug development.1, 2 In addition to this decline in success, malaria parasites and some cancer cell lines have developed resistance to several currently used drugs.3–5 Chemotherapy of cancer and other diseases, such as cytomegalovirus (CMV), is often associated with undesirable side effects. Therefore, the need for continued drug discovery and development is of paramount importance. The natural product artemisinin (1, Figure 1) is a well-known antimalarial drug that is the active ingredient of an ancient Chinese remedy discovered by the lab of Dr. Youyou Tu.6 The natural product has been functionalized to improve the pharmacokinetics of the molecule, and first generation derivatives such as dihydroartemisinin (DHA), artemether and sodium artesunate (2, 3, and 4, respectively, Figure 1) are currently part of artemisinin combination therapy (ACT), the first line of malaria chemotherapy.7 In addition to their potent and rapid antimalarial efficacy against all species of Plasmodium parasites, derivatives of artemisinin have shown also significant activity against a variety of other diseases, including helminth infections,8 CMV,9, 10 and many different cancer cell lines.10–16
Figure 1.

artemisinin and its first generation derivatives
A number of successful synthetic strategies have been employed to further improve the natural product or harness the activity of the essential peroxide linkage, ranging from semisynthetic17, 18 to fully synthetic analogs.19 It is important to note that many of the most potent artemisinin synthetic analogs that have been reported by our group and by others, against malaria and other diseases, are prepared by coupling two artemisinin units into one dimeric molecule.9, 11, 12, 20, 21 For example, we showed that artemisinin dimer primary alcohol 5 (Figure 2) had significantly greater anti-CMV activity than artemether (3) and artesunate (4)9 and that the deoxy version of dimer primary alcohol 5 (lacking the peroxide pharmacophore) was not active against CMV.10 Additionally, bis-benzylic alcohol dimer 6 (Figure 2) was >100 fold more active than DHA (2) against cervical cancer HeLa cells without showing toxicity to normal cells.11 Trioxane dimer diphenyl phosphate ester 838 (the name is derived from the compound’s molecular weight, Figure 2) has especially high potency and is more potent than the standard drug doxorubicin at inhibiting growth of prostate cancer LNCaP cells.12 We also recently demonstrated that dimer 838 has a selectivity index (anti-CMV/cytotoxicity) of approximately 1500, about 15 times higher than that of the standard anti-CMV drug ganciclovir (GCV).10 The high potency of this molecule spurred us to explore structure-activity relationships surrounding the diphenylphosphate moiety to probe what effect different substitutions might have on both the anti-CMV and anti-cancer activities. We show here the synthesis of this series of phosphate esters and their activities against CMV, a human leukemia cell line, and human normal cells.
Figure 2.

highly potent artemisinin-derived trioxane dimers
Chemistry
The syntheses of dimer primary alcohol 522 and dimer diphenylphosphate ester 83812 have been previously reported. The lactone moiety of artemisinin (1) is reduced by diisobutylaluminum hydride (DIBALH) and is subsequently trapped in situ with acetic anhydride (Ac2O) to provide exclusively α-dihydroartemisinin acetate (DHA-OAc, 7, Scheme 1). Sakurai bis-allylation using tin(IV) chloride (SnCl4) and bis-trimethylsilyl isobutylene linker 8 at −78 °C provides the isobutylene dimer 9 in excellent yield. The alkene is converted into primary alcohol 5 via hydroboration/oxidation, and coupling with diphenylphosphoryl chloride (10) affords the lead phosphate ester dimer 838. This lead compound 838 was partially hydrolyzed using sodium hydroxide (NaOH) to give the monophenylphosphate ester derivative 11 (Scheme 1). We wished to vary substitution patterns on the aromatic rings, requiring functionalized diphenylphosphoryl chlorides. Many of these were not commercially available and were therefore synthesized following literature procedure.23 Substituted phenols were reacted with phosphoryl chloride (POCl3, or thiophosphoryl chloride, PSCl3) in the presence of aluminum trichloride (AlCl3) to afford the desired phosphoryl chlorides 12–17 (Scheme 2a). Functionality was chosen to have both electron donating (12) and electron withdrawing (13) effects on the phosphate, and to incorporate a sterically crowded system (14). We also wished to explore the activity of thiophosphates (15 and 16) and rigidified systems (16 and 17). Finally, to test the effect of aromaticity on biological activity, a fully saturated phosphoryl chloride was prepared. Cyclohexanol (18) was treated with sodium hydride (NaH) and phosphorus trichloride (PCl3), yielding tricyclohexyl phosphite (19) which was subsequently reacted with triphosgene and dimethylimidazolidinone to provide the dicycohexylphosphoryl chloride 20 (Scheme 2b). Each synthesized phosphoryl chloride was then coupled to dimer primary alcohol 5, using the procedure shown for the preparation of 838 (Scheme 1), to provide the desired dimeric phosphate products.
Scheme 1.

synthesis of artemisinin dimer phosphates, reagents and conditions: (a) (i) DIBALH, CH2Cl2, −78 °C; (ii) DMAP, py, Ac2O, −78 °C to rt, 95%; (b) SnCl4, CH2Cl2, −78 °C, 85%; (c) (i) BH3:SMe2, THF, 0 °C; (ii) NaBO3:4H2O, 0 °C to rt, 85%; (d) diphenylphosphoryl chloride 10, DMAP, py, CH2Cl2, rt, 95%; (e) NaOH, DMF, 50 °C, 56%.
Scheme 2.


synthesis of phosphoryl chlorides, reagents and conditions: (a) AlCl3, PXCl3, 150 °C, microwave, 1 h 62–93% crude; (b) (i) NaH, THF, rt to 50 °C; (ii) PCl3; (c) dimethylimidazolinone, triphosgene, CH2Cl2, 85% over two steps.
Results and discussion
The series of trioxane phosphate dimers were screened in vitro against both CMV-infected cells and leukemia cells. For CMV, HFFs were infected and treated with the compounds in a dose-dependent manner, and at 72 hours post infection (hpi) luciferase activity was measured as an indication for late CMV gene expression.9
The anti-leukemia effect of these dimers against Jurkat cells (T-cell acute lymphoblastic leukemia cell line from American Type Cell Collection, VA) was assessed after 48 hours of drug treatment using the AlamarBlue assay (Invitrogen, CA).24–26 To measure toxicity to nonmalignant cells, peripheral blood mononuclear cells (PBMCs) were isolated from 4 consenting healthy volunteers and cultured with phytohemagglutinin (PHA, Sigma, MO; 10 µg/ml) in multiwell plates. After 24hrs, drug or vehicle was added for 48 additional hours before Alamar Blue assays were performed.27, 28
The results from these assays are summarized in Table 1. As previously determined, both artemether (3) and artesunate (4) showed weak activity against CMV.9 In sharp contrast, however, all of the trioxane dimer phosphates reported here were significantly more potent than artemether (3) and artesunate (4) against CMV replication, having IC50 activities in the low-nanomolar range. Lead compound 838 displayed the most potent activity, and dicyclohexylphosphate 851 also was very potent.
Table 1.
In vitro results from anti-CMV and anti-leukemia studies1
| Key Structural Unit | Trioxane Dimer | Anti-CMV EC50 (nM)2 |
Anti-leukemia IC50(nM)3 |
PHA-stimulated PBMC IC50(nM)4 |
TI5 | |
|---|---|---|---|---|---|---|
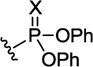 |
X = O | 838 | 39±3 | 100±9 | 1477±639 | 14.8 |
| X = S | 855 | 78±4 | 1750±132 | >400007 | >22.9 | |
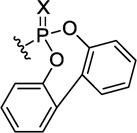 |
X = O | 836 | 72±4 | 220±16 | 2281±710 | 10.4 |
| X = S | 853 | 70±9 | 1933±278 | >400007 | >20.7 | |
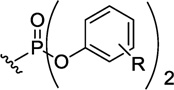 |
R = 4-CI | 907 | 66±4 | 152±7 | >400007 | >263 |
| R = 4-iPr | 923 | 300±2 | 511±63 | >400007 | >78.3 | |
| R = 2,6-diMe | 895 | 71±4 | 296±37 | >400007 | >135 | |
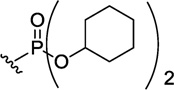 |
851 | 44±5 | 287±10 | 2031±2192 | 7.1 | |
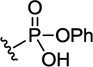 |
762 | >300 | 7436±536 | ~400008 | ~5.4 | |
| monomer | Artemether | 5300±27006 | >30000 | >1000007 | - | |
| monomer | Sodium Artesunate | 18500±52006 | 6887±770 | 27431±20993 | 4.0 | |
| Doxorubicin | - | 9.6±0.6 | 271±163 | 28.2 | ||
| Ganciclovir | 2700±52009 | - | - | - | ||
| Dimer alcohol 5 | 160±89 | - | - | - | ||
average of triplicate experiments;
activity determined in HFF cells;
activity determined in Jurkat T-ALL cells;
average of 4 experiments, each in triplicate;
therapeutic index, ratio of IC50 in PHA-stimulated PBMCs to the IC50 in Jurkat cells;
data were determined in reference 9;
50% growth inhibition was not achieved at the highest tested dose;
50% growth inhibition was achieved only at the highest tested dose,
data were determined in reference 9.
The anti-leukemic activities also were in the low-nanomolar range for most of this series of phosphates, and important information was gained from the SAR. First, partial hydrolysis of diphenylphosphate 838 into the monophenylphosphate ester 762 resulted in a 70-fold loss in potency (Table 1). This is likely due to the diminished ability of the more hydrophilic compound 762 to penetrate the leukemia cells. A second point of note is the replacement of the phosphoryl oxygen with sulfur. These compounds (853 and 855) showed ~9-fold and ~18-fold loss in potency, respectively, compared to their oxygenated counterparts. The aryl phosphate with one alkyl substituent (compound 923) was 5-fold less potent than dimer 838, while the aryl phosphate with two alkyl substituents (compound 895) had comparable potency to dimer 838. Rigidified phosphate ester 836 and saturated dicyclohexyl phosphate dimer 851 were about half as efficacious as dimer 838. Finally, artesunate was nearly 70-fold less efficacious than the lead compound 838, which was the most active dimer against leukemia, while artemether was essentially inactive.
The therapeutic index was determined for each compound by measuring the activity against phytohemagglutinin (PHA)-stimulated peripheral blood mononuclear cells (PBMCs) and determining the ratio of activities (PBMC IC50/Jurkat cell IC50). While lead compound 838 was the most active compound in this series against leukemia cells, it had a reduced therapeutic window (TI = 14.8) as compared to the standard drug doxorubicin (TI = 28.2, Table 1). Two other analogs (compounds 855 and 853) had comparable therapeutic indices (TI = 22.9 and 20.7, respectively), while isopropylphenyl dimer 923 had a noteworthy therapeutic index of 78.3. Importantly, 4-chloro-substituted dimer 907 was both active against leukemia cells (IC50 = 152 nM) and was essentially inactive against PHA-stimulated PBMCs (IC50 >40,000), providing a therapeutic index of >263, approximately 10-fold greater than that of doxorubicin.
Conclusion
It is imperative to continue the pursuit of new therapies, and it may be beneficial to find multiple uses for the same therapeutic. We have previously demonstrated that trioxane dimer diphenylphosphate ester 838 is highly potent against both cancer and CMV-infected cells.9, 10 Additionally, dimer 838 has a significant therapeutic window in each case.9, 10 Herein we report an SAR study surrounding this lead compound. The structure-activity relationships based on this study confirm and emphasize the very high antileukemic and anti-CMV activities of the lead compound 838 and its low toxicity to human normal cells. This study also identified bis(4-chlorophenyl)phosphate dimer 907 as a highly active compound against leukemia that is also non-toxic to normal human cells.
Experimental
All reactions were performed under argon in oven-dried or flame-dried glassware. Microwave reactions were performed in a Biotage Initiator microwave. Dichloromethane was dispensed from an LC Technology Solutions SPBT-1 bench top solvent purification system. All commercially available reagents were purchased from Sigma Aldrich and used as received. All experiments were monitored by analytical thin layer chromatography (TLC) performed on Silicycle silica gel 60 Ǻ glass supported plates with 0.25mm thickness. Flash chromatography was performed with EMD silica gel (40–63 µM). Yields are not optimized. Infrared (IR) spectra were recorded on a Perkin Elmer 1600 FT-IR spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400 MHz FT-NMR spectrometer (400 MHz for 1H, 100 MHz for 13C). The following abbreviations are used in the experimental section for the description of 1H NMR spectra: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad singlet (bs), doublet of doublets (dd), doublet of triplets (dt), and doublet of quartets (dq). Low resolution mass spectra (electrospray ionization) were acquired on an Agilent Technologies 6130 quadrupole spectrometer coupled to an Agilent Technologies 1200 series HPLC. High resolution mass spectrum-electron ionization sprary (HRMS-ESI) were obtained on an Agilent Technologies 1200 series Dual Absorbance Detector HPLC system equipped with a Phenomenex Luna 75×3mm, C18, 3 µm column at 45 °C (UV detection at 220nm, BW 8nm, and 254nm BW 8nm, flow rate: 0.8 mL/min (increasing), Injection volume: 1.0 µL, sample solvent: 100% Methanol, sample conc.: ~0.01 mg/mL, mobile phase A: Water with 0.1% acetic acid, mobile phase B: Acetonitrile with 0.1% acetic acid) coupled to a Agilent 6210 time-of-flight mass spectrometer (ion source: Duel ESI, min range: 115 m/z, max range: 1400 m/z, scan rate: 0.9 seconds, gas temp: 340°C, gas flow: 10 L/min, nebulizer: 50 PSI, ion polarity: positive, VCap: 3500 V, fragmentor: 175 V, skimmer1: 65 V, OctopoleRFPeak: 250 V, ref mass: enabled (Agilent P/N G1969-85001). Data were analyzed using Agilent Masshunter Workstation Data Acquisition (v B.02.00, Patch 1,2,3) and Agilent Masshunter Qualitative Analysis (v B.02.00, Build 2.0.197.7, Patch 3). List of abbreviations are as follows: dichloromethane – DCM; dimethylformamide – DMF; sat. aq. NaCl – brine; ethyl acetate – EtOAc.
General procedure for the synthesis of diaryl chlorophosate esters 12–17
To a 0.5 – 2 mL microwave vial were added 4.46 mmol of substituted phenol (1.0 eq), 2.23 mmol of phosphorus oxychloride (POCl3, 0.5 eq), and 0.446 mmol of aluminum chloride (AlCl3, 0.1 eq). The reaction mixture was heated in the microwave at 150 °C for 1 h. Upon completion, the mixture was diluted with CH2Cl2, filtered through a pad of celite, and purified directly on silica. Gradient elution (5–15% ethyl acetate in hexanes) afforded the desired products as colorless amorphous solids or oils.
Synthesis of tricyclohexyl phosphite (19)
Cyclohexanol (18, 2.26 g, 22.57 mmol, 3.1 eq) and THF (40 mL) were added to a round bottom flask. Sodium hydride (NaH, 542 mg, 22.57 mmol, 3.1 eq) was added and the reaction mixture was heated to 50 °C for 3 h. At that time, phosphorus trichloride (PCl3, 0.635 mL, 7.28 mmol, 0.32 eq) was added dropwise over several minutes. The heat was turned off, allowing the reaction to slowly cool to rt, and the reaction mixture was stirred at rt overnight. The reaction was deemed complete by TLC, and the solvent was removed under reduced pressure and the residue was dissolved in hexanes (50 mL). The solution was filtered through celite and the solvent was again removed under reduced pressure. The resulting crude oil was taken on to the next step without further purification.
Synthesis of dicyclohexyl phosphorochloridate (20)
Tricyclohexyl phosphite 19 (2.3g, 7.0 mmol, 1.0 eq) and 1,3-dimethylimidazolidin-2-one (24 mg, 0.21 mmol, 0.13 eq) and dichloromethane (30 mL) were added to a round bottom flask, and the stirring solution was cooled to 0 °C in an ice bath. A separate solution of triphosgene (706 mg, 2.38 mmol, 0.34 eq) in dichloromethane (5 mL) was prepared and added dropwise to the original stirring solution over 10 mins. The reaction mixture was stirred at 0 °C for 30 mins, the ice bath was removed, and the mixture was heated to 35 °C for 3 h. The reaction was deemed complete by TLC, and the solvent was removed under reduced pressure. The residue was distilled in a Buchi Kugelrohr, affording a colorless oil: (1.68 g, 5.98 mmol, 85%).
General procedure for the synthesis of dimer phosphate esters
Primary alcohol 18 (1.0 eq) was dissolved in dichloromethane (2 mL) under argon at room temperature. To the stirring solution were added pyridine (5.0 eq), dimethylaminopyridine (DMAP, 1.0 eq) and the appropriate phosphoryl chloride (5.0 eq) in that order. The reaction mixture was allowed to stir at rt for 3 hr, at which point the reaction was deemed complete by analytical TLC. The reaction mixture was diluted with dicholoromethane and washed consecutively with 10% citric acid and brine. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified directly on silica gel. Gradient elution (10 – 40% ethyl acetate in hexanes) afforded the desired products as a colorless, amorphous solid.
Synthesis of dimer diphenylthiophosphate ester 855
The title compound was prepared as described in the general procedure, using diphenylthiophosphoryl chloride (15): yield (7.5 mg, 0.009 mmol, 27%), 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.17 (m, 12 H), 5.33 (s, 1 H), 5.30 (s, 1 H), 4.55 – 4.37 (m, 3 H), 4.27 – 4.24 (m, 1 H), 2.73 – 2.65 (m, 1 H), 2.59 – 2.52 (m, 1 H), 2.37 – 2.28 (m, 3 H), 2.04 – 1.98 (m, 3 H), 1.94 – 1.82 (m, 3 H), 1.79 – 1.63 (m, 6 H), 1.52 – 1.35 (m, 12 H, including singlets at 1.40 and 1.38), 1.31 – 1.23 (m, 4 H), 1.00 – 0.82 (m, 12 H); 13C NMR (100 MHz, CDCl3) δ 150.7, 129.7, 125.1, 120.2, 120.1, 103.1, 102.4, 89.9, 89.0, 81.2, 81.0, 74.2, 72.1, 70.8, 52.4, 52.1, 44.5, 44.2, 38.8, 38.0, 36.6, 36.5, 35.0, 34.5, 30.0, 30.0, 29.9, 29.4, 26.1, 25.9, 24.9, 24.9, 24.7, 20.2, 20.1, 13.2, 12.6; ESI – HRMS for C46H63NaO11PS (M + Na)+ calc. = 877.3721, found = 877.3741; [α]D 23 = 62 (c = 0.39, CHCl3); FT-IR (cm−1) 2919, 2861, 1589, 1485, 1455, 1376, 1290, 1225, 1194, 1110, 1009, 943, 760.
Synthesis of dimer biphenylphosphate ester 836
The title compound was prepared as described in the general procedure, using biphenylphosphoryl chloride (17): yield (7.0 mg, 0.008 mmol, 34%), 1H NMR (400 MHz, CDCl3) δ 7.55 – 7.26 (m, 8 H), 5.29 (s, 1 H), 5.28 (s, 1 H), 4.62 – 4.46 (m, 2 H), 4.39 – 4.35 (m, 1 H), 4.24 – 4.19 (m, 1 H), 2.73 – 2.65 (m, 1 H), 2.60 – 2.52 (m, 1 H), 2.37 – 2.26 (m, 3 H), 2.08 – 1.97 (m, 2 H), 1.92 – 1.80 (m, 3 H), 1.79 – 1.70 (m, 2 H), 1.68 – 1.55 (m, 5 H), 1.50 – 1.35 (m, 12 H, including singlets at 1.40 and 1.36), 1.31 – 1.27 (m, 6 H), 1.00 – 0.92 (m, 6 H), 0.89 – 0.80 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 134.4, 129.9, 129.6, 129.5, 105.3, 102.8, 89.3, 88.5, 81.2, 81.1, 74.1, 72.0, 52.5, 52.2, 43.6, 43.4, 37.4, 37.3, 34.5, 31.0, 26.1, 26.0, 24.8, 20.6, 20.2, 20.1, 17.1, 13.5; ESI - HRMS for C46H61NaO12P (M + Na)+ calc. = 859.3793, found = 859.3814; [α]D 23 = 73 (c = 0.42, CHCl3); FT-IR (cm−1) 2930, 2927, 1582, 1489, 1376, 1175, 1130, 1009, 965, 941, 878, 852, 752.
Synthesis of dimer biphenylthiophosphate ester 853
The title compound was prepared as described in the general procedure, using biphenylthiophosphoryl chloride (16): yield (5.5 mg, 0.006 mmol, 26%), 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.31 (m, 8 H), 5.30 (s, 1 H), 5.28 (s, 1 H), 4.57 – 4.54 (m, 2 H), 4.41 – 4.38 (m, 1 H), 4.24 – 4.20 (m, 1 H), 2.71 – 2.65 (m, 1 H), 2.59 – 2.51 (m, 1 H), 2.37 – 2.28 (m, 2 H), 2.07 – 1.98 (m, 4 H), 1.96 – 1.81 (m, 4 H), 1.79 – 1.60 (m, 4 H), 1.60 – 1.42 (m, 12 H, including singlets at 1.40 and 1.39), 1.35 – 1.24 (m, 6 H), 1.00 – 0.92 (m, 6 H), 0.88 – 0.80 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 134.4, 131.6, 131.9, 129.6, 103.3, 102.8, 89.3, 81.2, 81.1, 73.1, 71.0, 55.5, 52.2, 44.6, 44.2, 38.4, 37.4, 34.5, 30.4, 26.1, 26.0, 24.8, 20.6, 20.2, 17.1; ESI – HRMS for C46H62O11PS (M + H)+ calc. = 853.3745, found = 853.3747; [α]D 23 = 124 (c = 0.40, CHCl3); FT-IR (cm−1) 2930, 2924, 2915, 1582, 1481, 1376, 1279, 1189, 1130, 1009, 965, 941, 878, 852, 752.
Synthesis of dimer bis-(4-chlorophenyl)phosphate ester 907
The title compound was prepared as described in the general procedure, using bis(4-chlorophenyl)phosphoryl chloride (13): yield (21.3 mg, 0.023 mmol, 71%); 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.65 Hz, 4 H), 7.15 – 7.21 (m, 4 H), 5.28 (s, 1 H), 5.25 (s, 1 H), 4.45 – 4.54 (m, 2 H), 4.41 (dd, J = 9.28, 6.76 Hz, 1 H), 4.26 (dd, J = 9.38, 6.54 Hz, 1 H), 2.57 – 2.71 (m, 1 H), 2.39 – 2.51 (m, 2 H), 2.29 (t, J = 13.80 Hz, 4 H), 1.96 – 2.04 (m, 2 H), 1.82 – 1.94 (m, 2 H), 1.70 – 1.80 (m, 4 H), 1.55 – 1.68 (m, 4 H), 1.39 – 1.50 (m, 4 H), 1.36 (d, J = 10.74 Hz, 6 H), 1.16– 1.31 (m, 8 H), 0.93 (dd, J = 9.54, 4.93 Hz, 6 H), 0.80 (t, J = 8.27 Hz, 4 H); 13C NMR (100 MHz, CDCl3) δ 149.2, 149.1, 130.7, 129.8, 121.6, 121.5, 121.5, 103.1, 102.8, 89.7, 89.0, 81.2, 77.4, 77.1, 76.8, 73.5, 72.3, 72.2, 70.2, 52.3, 52.1, 44.4, 44.0, 37.5, 37.4, 36.7, 36.6, 35.4, 35.4, 34.5, 34.4, 30.6, 30.5, 30.3, 29.7, 26.1, 26.1, 24.9, 24.9, 24.8, 24.7, 20.2, 20.1, 13.0, 12.5; ESI-HRMS for C46H61NaO12P (M + Na)+ calc. = 929.3170, found = 929.3186; [α]D25 +60.09 (c = 0.27, CHCl3); FT-IR (cm−1) 2923, 2918, 1580, 1486, 1375, 1301, 1197, 1092, 1009, 949, 878, 834, 754.
Synthesis of dimer bis-(4-isopropylphenyl)phosphate ester 923
The title compound was prepared as described in the general procedure, using bis(4-isopropylphenyl)phosphoryl chloride (12): yield (8.6 mg, 0.009 mmol, 56%), 1H NMR (400 MHz, CDCl3) δ 7.16 - 7.09 (m, 8 H), 5.33 (s, 1 H), 5.28 (s, 1 H), 4.56 – 4.99 (m, 1 H), 4.84 – 4.37 (m, 2 H), 4.27 – 4.20 (m, 1 H), 2.91 – 2.83 (m, 2 H), 2.71 – 2.64 (m, 1 H), 2.54 – 2.48 (m, 1 H), 2.33 – 2.26 (m, 3 H), 2.03 – 1.95 (m, 2 H), 1.92 – 1.81 (m, 4 H), 1.78 – 1. 70 (m, 4 H), 1.68 – 1.55 (m, 4 H), 1.54 – 1.43 (m, 4 H), 1.42 – 1.35 (m, 9 H), 1.29 – 1.20 (m, 15 H), 0.99 – 0.89 (m, 6 H), 0.85 – 0.78 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 149.2, 149.1, 130.7, 129.8, 121.6, 121.5, 121.5, 103.1, 102.8, 89.7, 89.0, 81.2, 77.4, 77.1, 76.8, 73.5, 72.3, 72.2, 70.2, 52.3, 52.1, 52.1, 44.4, 37.5, 37.4, 36.7, 36.6, 35.4, 34.5, 34.4, 30.9, 30.3, 29.7, 26.3, 26.1, 24.9, 24.9, 24.8, 24.7, 20.2, 20.1, 13.0, 12.5; ESI – HRMS for C52H76O12P (M + H)+ calc. = 923.5069, found = 923.5036; [α]D23 = 57 (c = 0.14, CHCl3); FT-IR (cm−1) 2923, 2918, 1580, 1486, 1375, 1301, 1197, 1092, 1009, 949, 878, 834, 754.
Synthesis of dimer bis-(2,6-dimethylphenyl)phosphate ester 895
The title compound was prepared as described in the general procedure, using bis(2,6-dimethylphenyl)phosphoryl chloride (14): yield (18.0 mg, 0.02 mmol, 61%); 1H NMR (400 MHz, CDCl3) δ 7.07 – 6.91 (m, 6 H), 5.29 (s, 1 H), 5.25 (s, 1 H), 4.53 - 4.44 (m, 1 H), 4.42 - 4.30 (m, 2 H), 4.19 - 4.11 (m, 1 H), 2.72 - 2.62 (m, 1 H), 2.56 - 2.46 (m, 1 H), 2.34 (s, 6 H), 2.32 (s, 6 H), 2.29 - 2.18 (m, 2 H), 2.02 – 1.93 (m, 2 H), 1.92 - 1.79 (m, 5 H), 1.72 (d, J = 15.54 Hz, 4 H), 1.66 - 1.49 (m, 6 H), 1.36 (d, J = 8.46 Hz, 8 H), 1.34 - 1.16 (m, 8 H), 0.94 (d, J = 6.00 Hz, 3 H), 0.88 (d, J = 4.67 Hz, 3 H), 0.79 (t, J = 6.41 Hz, 4 H); 13C NMR (100 MHz, CDCl3) δ 157.7, 148.5, 148.4, 130.4, 129.0, 125.1, 103.2, 102.8, 89.4, 88.6, 81.2, 81.1, 74.0, 72.1, 72.0, 70.9, 52.5, 52.2, 44.6, 44.2, 37.5, 37.3, 36.7, 36.6, 35.4, 35.4 34.6, 34.5, 30.5, 30.4, 30.2, 29.2, 26.1, 26.1, 24.9, 24.7, 20.2, 20.1, 17.2, 13.3, 12.7; ESI-HRMS for C50H71NaO12P (M + Na)+ calc. = 917.4575, found = 917.4589; [α]D25 +60.33 (c = 0.69, CHCl3); FT-IR (cm−1) 2937, 2925, 1585, 1471, 1376, 1281, 1159, 1092, 1009, 953, 878, 753.
Synthesis of dimer dicyclohexylphosphate ester 851
The title compound was prepared as described in the general procedure, using dicyclohexylphosphoryl chloride (20): yield (7.1 mg, 0.008 mmol, 25%); 1H NMR (400 MHz, CDCl3) δ 5.30 (s, 2 H), 4.63 - 4.53 (m, 1 H), 4.50 - 4.34 (m, 2 H), 4.30 (d, J = 4.86 Hz, 2 H), 4.24 (dd, J = 9.76, 6.16 Hz, 1 H), 2.74 - 2.64 (m, 1 H), 2.63 - 2.54 (m, 1 H), 2.38 - 2.25 (m, 2 H), 2.25 - 2.18 (m, 1 H), 2.04 – 1.97 (m, 3 H), 1.95 - 1.85 (m, 7 H), 1.82 - 1.69 (m, 8 H), 1.68 - 1.59 (m, 6 H), 1.58 - 1.47 (m, 6 H), 1.47 - 1.38 (m, 10 H), 1.37 - 1.29 (m, 6 H), 1.28 - 1.19 (m, 6 H), 0.95 (d, J = 6.06 Hz, 6 H), 0.85 (t, J = 7.14 Hz, 4 H); 13C NMR (100 MHz, CDCl3) δ 154.8, 103.2, 102.9, 89.37, 81.1, 81.1, 73.8, 71.4, 52.5, 52.2, 44.6, 44.2, 37.4, 36.6, 34.5, 34.4, 31.6, 30.6, 30.3, 29.7, 26.1, 25.3, 25.0, 24.7, 23.8, 23.6, 20.2, 13.2, 12.8; ESI – HRMS for C46H75NaO12P (M + Na)+ calc. = 873.4888, found = 873.4890; [α]D23 = 49.95 (c = 0.355, CHCl3); FT-IR (cm−1) 2936, 2925, 1737, 1451, 1376, 1254, 1105, 1038, 1008, 963, 880.
Synthesis of monophenylphosphate ester 762
To a 1 dram vial were added 83812 (62.0 mg, 0.074 mmol) and DMF (1 mL), followed by 1N NaOH (89 µL, 0.089 mmol, 1.2 eq). The reaction mixture was heated to 50 °C for 3 h. The reaction was deemed complete via analysis on LCMS, and the reaction mixture was by direct injection onto reverse phase chromatography system. The fractions containing the desired product were lyophilized overnight, providing the title compound as a colorless, amorphous solid: yield (31.5 mg, 0.041 mmol, 56%); 1H NMR (400 MHz, CDCl3) δ 11.22 (brs, 1 H), 7.47 - 7.02 (m, 5 H), 5.46 - 5.24 (m, 2 H), 4.41 - 4.22 (m, 2 H), 4.21 - 4.06 (m, 2 H), 2.83 - 2.65 (m, 1 H), 2.65 - 2.50 (m, 1 H), 2.29 (t, J = 11.87 Hz, 2 H), 2.21 - 2.09 (m, 1 H), 2.08 – 1.95 (m, 2 H), 1.93 - 1.81 (m, 2 H), 1.75 (d, J = 12.13 Hz, 2 H), 1.69 - 1.50 (m, 6 H), 1.37 (d, J = 8.65 Hz, 12 H), 1.25 (t, J = 7.07 Hz, 5 H), 0.92 (dd, J = 13.93, 5.34 Hz, 6 H), 0.88 - 0.75 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 129.5, 124.7, 120.2, 103.5, 89.4, 88.8 81.1, 81.1, 74.7, 71.9, 69.7, 52.4, 52.2, 44.6, 44.2, 37.4, 37.1, 36.5, 34.4, 30.2, 29.5, 25.8, 24.8, 20.2, 20.1, 14.2, 13.3, 12.7; ESI-HRMS for C40H60O12P (M + H)+ calc. = 763.3840, found = 763.3835; [α]D25 +70.38 (c = 1.315, CHCl3); FT-IR (cm−1) 2952, 2876, 1739, 1593, 1491, 1453, 1377, 1208, 1159, 1103, 1012, 938, 876, 824, 757, 690.
Acknowledgements
Financial support is acknowledged from the following agencies: NIH grant R37 AI34885 (GHP), NIH grant R01 AI093701 (RAB), National Foundation for Cancer Research (CIC), NIH grant PO1 CA70970 (CIC), the Maryland Stem Cell Research Foundation/TEDCO 2010-MSCRFII-0065 (CIC), the Curing Kids Cancer Foundation (XC) and the William Lawrence & Blanche Hughes Foundation (XC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scannell JW, Blanckley A, Boldon H, Warrington B. Nat. Rev. Drug. Discov. 2012;11:191. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 2.Aubé J. Med. Chem. Lett. 2012;3:442. doi: 10.1021/ml300114c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foth BJ. Nat. Rev. Microbiol. 2012;10:524. doi: 10.1038/nrmicro2847. [DOI] [PubMed] [Google Scholar]
- 4.Al-Lazikani B, Banerji U, Workman P. Nat. Biotech. 2012;30:1. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues AS, Dinis J, Gromicho M, Martins C, Laires A, Rueff J. Curr. Pharm. Biotechno. 2012;13:651. doi: 10.2174/138920112799857549. [DOI] [PubMed] [Google Scholar]
- 6.Miller LH, Su X-z. Cell. 2011;146:855. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mutabingwa TK. Acta Tropica. 2005;95:305. doi: 10.1016/j.actatropica.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Keiser J, Utzinger J. Curr. Opin. Infect. Dis. 2007;20:605. doi: 10.1097/QCO.0b013e3282f19ec4. [DOI] [PubMed] [Google Scholar]
- 9.Arav-Boger R, He R, Chiou C-J, Liu J, Woodard L, Rosenthal A, Jones-Brando L, Forman M, Posner G. PLoS One. 2010;5:e10370. doi: 10.1371/journal.pone.0010370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He R, Mott BT, Rosenthal AS, Genna DT, Posner GH, Arav-Boger R. PLoS One. 2011;6:e24334. doi: 10.1371/journal.pone.0024334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paik I-H, Xie S, Shapiro TA, Labonte T, Sarjeant AAN, Baege AC, Posner GH. J. Med. Chem. 2006;49:2731. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 12.Alagbala AA, McRiner AJ, Borstnik K, Labonte T, Chang W, D’Angelo JG, Posner GH, Foster BA. J. Med. Chem. 2006;49:7836. doi: 10.1021/jm060803i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou J, Wang D, Zhang R, Wang H. Clin. Cancer Res. 2008;14:5519. doi: 10.1158/1078-0432.CCR-08-0197. [DOI] [PubMed] [Google Scholar]
- 14.Rosenthal AS, Chen X, Liu JO, West DC, Hergenrother PJ, Shapiro TA, Posner GH. J. Med. Chem. 2009;52:1198. doi: 10.1021/jm801484v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crespo-Ortiz MP, Wei MQ. J. Biomed. Biotechnol. 2012 doi: 10.1155/2012/247597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai HC, Singh NP, Sasaki T. Invest. New. Drugs. 2012 doi: 10.1007/s10637-012-9873-z. [DOI] [PubMed] [Google Scholar]
- 17.Bégué J-P, Bonnet-Delpon D. ChemMedChem. 2007;2:608. doi: 10.1002/cmdc.200600156. [DOI] [PubMed] [Google Scholar]
- 18.Haynes RK, Fugmann B, Stetter J, Rieckmann K, Heilmann H-D, Chan H-W, Cheung M-K, Lam W-L, Wong H-N, Croft SL, Vivas L, Rattray L, Stewart L, Peters W, Robinson BL, Edstein MD, Kotecka B, Kyle DE, Beckermann B, Gerisch M, Radtke M, Schmuck G, Steinke W, Wollborn U, Schmeer K, Römer A. Angew. Chem. Int. Ed. 2006;45:2082. doi: 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- 19.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Tomas JS, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Nature. 2004;430:900. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 20.Slack RD, Jacobine AM, Posner GH. MedChemComm. 2012;3:281. [Google Scholar]
- 21.Chaturvedi D, Goswami A, Saikia PP, Barua NC. Chem. Soc. Rev. 2010;39:435. doi: 10.1039/b816679j. [DOI] [PubMed] [Google Scholar]
- 22.Posner GH, McRiner AJ, Paik I-H, Sur S, Borstnik K, Xie S, Shapiro TA, Alagbala A, Foster B. J. Med. Chem. 2004;47:1299. doi: 10.1021/jm0303711. [DOI] [PubMed] [Google Scholar]
- 23.Dhawan B, Redmore DJ. J. Org. Chem. 1986;51:179. [Google Scholar]
- 24.Ahmed SA, Gogal RM, Jr, Walsh JE. J. Immunol. Methods. 1994;170:211. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 25.de Fries R, Mitsuhashi M. J. Clin. Lab. Anal. 1995;9:89. doi: 10.1002/jcla.1860090203. [DOI] [PubMed] [Google Scholar]
- 26.Yan C, Huang D, Zhang Y. Exp. Toxicol. Pathol. 2011;63:413. doi: 10.1016/j.etp.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 27.Chang S, Griesgraber GW, Southern PJ, Wagner CR. J. Med. Chem. 2001;44:223. doi: 10.1021/jm000260r. [DOI] [PubMed] [Google Scholar]
- 28.Opsenica D, Pocsfalvi G, Juranic Z, Tinant B, Declerq JP, Kyle DE, Milhous WK. J. Med. Chem. 2000;43:3274. doi: 10.1021/jm000952f. [DOI] [PubMed] [Google Scholar]