

Abstract
Hydrogen sulfide (H2S) has emerged as a critical mediator of multiple physiological processes in mammalian systems. The pathways involved in the production, consumption, and mechanism of action of H2S appear to be sensitive to alterations in the cellular redox state and O2 tension. Indeed, the catabolism of H2S through a putative oxidation pathway, the sulfide quinone oxido-reductase system, is highly dependent on O2 tension. Dysregulation of H2S homeostasis has also been implicated in numerous pathological conditions and diseases. In this review, the chemistry and the main physiological actions of H2S are presented. Some examples highlighting the cytoprotective actions of H2S within the context of cardiovascular disease are also reported. Elucidation of the redox biology of H2S will enable the development of new pharmacological agents based on this intriguing new redox cellular signal.
Abbreviations: ARE, Antioxidant response element; CO, Carbon monoxide; CBS, Cystathionine-β-synthase; CGL, Cystathionine-γ-lyase; CcO, Cytochrome c oxidase; GSH, Glutathione; HSP, Heat shock protein; H2S, Hydrogen sulfide; HIF, Hypoxic inducible factor; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; 3-MST, 3-mercaptopyruvate S-transferase; NO, Nitric oxide; NF-κB, Nuclear factor light chain enhancer of activated B cells; oxLDL, Oxidized low density lipoprotein; PAG, Propargylglycine; PGE2, Prostaglandin E2; NaHS, Sodium hydrosulfide; Na2S, Sodium sulfide; SQR, Sulfide quinone oxido-reductase; TNF-α, Tumor necrosis factor alpha; VEGF, Vascular endothelial growth factor; VSMC, Vascular smooth muscle cells
Keywords: Hydrogen sulfide, Redox biology, Oxygen, Oxidative stress, Mitochondria
Graphical abstract
Highlights
► Hydrogen sulfide (H2S) is an endogenously produced cell signaling molecule, which plays a key role in multiple physiological processes including vasodilation, angiogenesis, and inflammation. ► The production and consumption of H2S in vivo is redox sensitive. ► Dysregulation of H2S production and consumption contributes to disease pathogenesis. ► Cytoprotective properties of H2S may be due, in part, to restoration of cellular redox status and upregulation of antioxidant defenses.
1. Introduction
Hydrogen sulfide (H2S); a toxic gas, is endogenously produced, bioactive, and contributes to numerous physiological functions in mammalian systems. Studies support the possibility that H2S has therapeutic potential for treating multiple diseases including cardiovascular diseases. For example, experimental animal studies show that H2S may be effective in treating atherosclerosis and protecting against ischemia-reperfusion injury [1–3]. Interest in the cytoprotective actions of H2S has grown since the discovery that it can induce a hypometabolic state characterized by decreased O2 consumption, heart rate, and body temperature in non-hibernating rodents [4]. Although not discussed in this review, H2S-dependent hypometabolism is an O2-dependent phenomenon [5]. The proposed mitochondrial and signaling actions of H2S make this molecule an attractive intervention for preventing and treating diseases and trauma-associated injuries. In this review article, we provide an overview of H2S redox biology as it relates to the biological and pharmacological actions of this interesting new signaling molecule in mammalian systems.
2. Historical benefits of H2S
The ancient Greeks, Egyptians, and Romans regularly bathed in natural sulfur springs as treatments for disease [6]. Depending on the microbiota and oxygen content, sulfur springs typically contain H2S concentrations ranging from 1 to 500 μM [7] with anti-inflammatory, anti-bacterial, vasodilatory, and anti-fungal properties attributed to the sulfur-containing water [8]. Epidemiological studies report that a diet rich in organosulfur species is associated with longevity and decreased morbidity [9]. Members of the Allium genus (garlic and onions), which contain organosulfur compounds have a well-documented history of health benefits [10]. Indeed, garlic-derived compounds such as diallyl trisulfide release H2S in the presence of cellular reductants like glutathione (GSH) [11]. Populations that consume garlic regularly have low blood pressure, low cholesterol, and less vascular disease [12]. While administration of exogenous sulfur-containing compounds shows strong promise as therapies, H2S is also endogenously produced in many different human tissues.
3. Endogenous production of H2S
In the early 1990s, it was discovered that H2S is enzymatically produced by two cytosolic enzymes; cystathionine β-synthase (CBS) and cystathionine γ-lyase (CGL) [13,14]. Seminal work of Abe and Kimura showed, for the first time, that H2S enhances long-term potentiation in the hippocampus [15]. Specifically, they demonstrated that H2S was produced by CBS and that exogenous H2S enhanced NMDA receptor-mediated responses. Since then many studies have shown that CBS and CGL are expressed in human tissues with H2S contributing to physiological and pathophysiological processes (Table 1). In addition to CBS and CGL, there are other enzymes that produce H2S with several utilizing cysteine as a substrate. The enzyme 3-mercaptopyruvate S-transferase (3-MST) is found in mitochondria and cytosol and produces H2S [16]. Several H2S producing enzymes are pyridoxal-5′-phosphate (PLP) dependent enzymes [17]. Moreover, other sulfur-containing amino acids, such as cystine and homocysteine, can be metabolized to generate H2S. The enzymatic mechanisms of H2S production are shown in Fig. 1. Many of these enzymes participate in the cellular sulfur cycle and have multiple enzymatic activities, including H2S generation. Because the concentration of reduced sulfur species has an effect on many cellular processes [18], the activity of these enzymes is tightly regulated. Much of this regulation is linked to substrate availability [19]. Moreover, these are redox-sensitive enzymes, which exhibit increased activity under oxidative conditions [20]. Considering that H2S; a reductant, is a product of these enzymes, it is conceivable that enzymatic activity may also be subject to negative feedback regulation. Finally, work by Wang et al. suggests that under certain conditions, such as oxidative stress, H2S-producing enzymes translocate from the cytosol to mitochondria [21]. This dynamic regulation bolsters the argument that H2S may function as a redox signaling molecule.
Table 1.
Biological and therapeutic actions of H2S are O2-dependent.
| Biological context | Low O2 | High O2 |
|---|---|---|
| Inflammation | Anti-inflammatory: IL-10; ↓ IL-6, ICAM | Pro-inflammatory: ↑ NF-κB, TNF-α |
| Vasoactivity | Vasodilatory: ↑ KATP channel conductance | Vasoconstrictive |
| Angiogenesis | Pro-angiogenic: VEGF, Hif1-α | No effect |
| Respiratory inhibition | Higher [H2S] results in more inhibition | O2 acts as an H2S antagonist: ↑SOx |
| Ischemia-reperfusion | Ischemic tissue has higher [H2S]: mito KATP, ARE genes | Unknown |
Fig. 1.
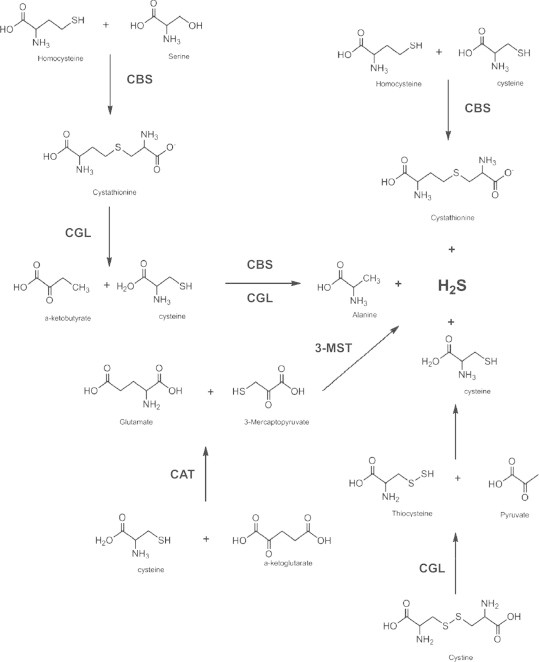
The enzymatic production of H2S. The two primary enzymes responsible for H2S production, cystathionine-γ-lyase (CGL) and cystathionine-β-synthase (CBS), are found in the cytosol. CBS catalyzes the first step in H2S production through the transsulfuration of homocysteine to cystathionine. CGL in an elimination reaction catalyzes the formation of cysteine and α-ketobutyrate. Cysteine is the substrate from which H2S is directly produced either through elimination (CGL) or β-replacement (CBS). Cysteine amino transferase (CAT) catalyzes the formation of 3-mercaptopyruvate, a substrate for the mitochondrial enzyme 3-mercaptopyruvate-S-transferase (3-MST). 3-MST can directly produce H2S, albeit at lower levels than CBS and CGL, in mitochondria.
4. Catabolism of H2S
Several regulated and unregulated non-enzymatic processes participate in H2S catabolism. These pathways maintain in vivo H2S concentrations, most likely, in the nM to low μM range. H2S can react with heme proteins in mitochondria and therefore H2S can function as a mitochondrial respiratory toxicant [22,23]. Fatal industrial accidents have been documented in individuals exposed to high concentrations of H2S gas (e.g., >1000 pm) [24]. Therefore, the toxicological profile of H2S has been well-studied and documented [24]. The mechanism of toxicity is through the binding of H2S to cytochrome c oxidase (CcO) mediating respiratory inhibition [22]. However, this interaction is complex and poorly understood because H2S can act as both an inhibitor and an electron donor for CcO [25]. H2S binds to the oxidized states of the heme a-a3 binuclear center, resulting in the reduction of the heme molecules [26]. Excess H2S can also reduce CuB [27]. While the stoichiometry may vary, Cooper and Brown reported that 3 molecules of H2S bind per inhibited CcO [28]. In this inhibitory reaction, H2S is oxidized to sulfane sulfur and this is coupled to consumption of molecular O2 [28]. Unlike nitric oxide (•NO), the inhibition of CcO by H2S is noncompetitive with O2 [27,29]. In addition, H2S can also directly reduce the electron carrier cytochrome c producing the one electron oxidation product, the thiyl radical (•SH) [28].
Rhodanese, a mitochondrial sulfur transferase enzyme, catalyzes the oxidation of H2S [30]. It is one part of three enzymatic activities characterized as a major pathway for H2S catabolism. This pathway consists of a sulfide quinone oxido-reductase (SQR), a sulfur dioxygenase, and the sulfur transferase enzyme rhodanese (Figs. 2 and 3). H2S reduces the external disulfide on the SQR to form a thiol (RSH) and a perthiol (RSSH). This two electron oxidation of H2S reduces the FAD prosthetic group, which uses ubiquinone (Q) as an electron acceptor [31] The second sulfur atom on the perthiol is a reactive sulfane (S0), which is oxidized by a sulfur dioxygenase enzyme encoded by the gene ETHE1, consuming O2 and H2O to form sulfite (SO3−2). While the protein responsible for this enzymatic activity is not known, the ETHE1 gene encoding the protein has been identified. Mutations in this gene cause a buildup of H2S leading to ethylmalonic encephalopathy [32,33]. Rhodanese then transfers a sulfane sulfur to sulfite to form thiosulfate (S2O3−2) [34]. This proposed oxidation pathway, in close proximity to CcO, functions as a major clearance pathway of cellular H2S. H2S can also be oxidized by non-mitochondrial heme proteins such as hemoglobin (Hb) and myoglobin [35]. H2S will reduce the ferric iron in met-Hb, restoring the oxygen binding abilities of the protein [36]. At high concentrations of H2S, sulf-Hb can also be formed from oxy-Hb [37]. While displaying very weak affinity for O2, sulf-Hb can still deliver O2, albeit with no cooperativity [38]. As a result, the bioavailability of H2S, whether in the context of steady state in vivo concentrations or exogenously administered, is dictated by the O2 concentration. Therefore, O2 can be considered an H2S antagonist, accelerating its oxidation and attenuating its biological actions. The effect of O2 on H2S concentration is both direct and indirect. The spontaneous reaction of H2S with O2, while slow, can cause an appreciable decrease in the H2S concentration. Thus, tissues with relatively high O2 concentrations (e.g., alveolar epithelium) may have less H2S compared to tissues that are in a lower O2 environment (e.g., centrilobular region of liver). This has implications in pathological states of hypoxia such as ischemia-reperfusion, where the availability, and thus the signaling effects of H2S may be augmented. Furthermore, O2 concentration can indirectly affect H2S concentration through changes in the redox state of heme proteins. Proteins such as Hb will react with H2S at different rates depending on the redox status of the hemes. For example, H2S will react more rapidly with met-Hb (Fe+3) than with deoxy-Hb (Fe+2) [36].
Fig. 2.
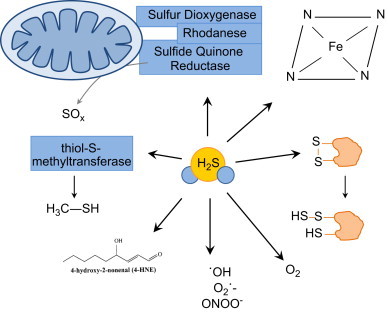
Proposed pathways of H2S removal in mammalian cells. The physiological steady-state concentration of H2S in vivo is believed to be maintained in the sub-micromolar range. This steady state concentration is established by the production pathways shown in Fig. 1 and the proposed consumption pathways shown within this figure. H2S will react non-enzymatically with many biomolecules such as reactive oxygen and nitrogen species, electrophilic lipids like 4-hydroxy-2-nonenal, free heme, and disulfide bonds to form a thiol and perthiol. The catabolism of H2S can also be catalyzed enzymatically by the sulfide quinone oxido-reductase system (SQR) comprised by sulfur dioxygenase, rhodanese, and sulfur quinone reductase.
Fig. 3.
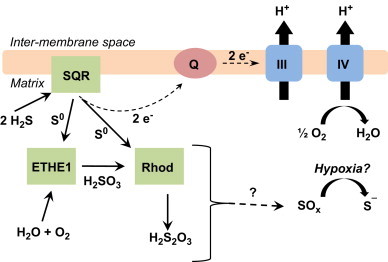
The oxidation of H2S by the sulfide quinone oxido-reductase system in mitochondria. H2S reduces the disulfide composed of the vicinal thiols on the sulfide quinone reductase (SQR) forming a thiol and a perthiol. The second sulfur atom on the perthiol, the sulfane sulfur (S0), is the substrate for both the sulfur transferase enzyme, rhodanese, and the sulfur dioxygenase enzyme encoded by the gene ETHE1. Rhodanese catalyzes the formation of thiosulfate (S2O3−2) from sulfite (SO3−2) and S0. ETHE1 catalyzes the formation of SO3−2. The reduced SQR can then transfer electrons into the ubiquinone (Q) pool, thus coupling the oxidation of H2S to electron transfer, H+ pumping, and ultimately ATP synthesis.
Because H2S is a nucleophile, it can also react with electrophilic lipids [39]; and the thiolate anion, HS−, can also reduce disulfide bonds (Fig. 2) [40]. Indeed, the exfoliation of skin cells in hot sulfur springs is due to H2S reducing the structural disulfide bonds of cellular junctions in keratinocytes [8]. While this can be harmful at high concentrations, the reduction of external disulfide bonds by H2S may, in some instances, reverse a deleterious post-translational protein modification. Although still contentious, the S-sulfhydration of cysteine residues may represent an important sink for free H2S [41]. In theory, H2S can also reduce higher thiol oxidation states such as S-nitrosothiols and sulfenic acids [42]. H2S can also be methylated by the cytosolic enzyme thiol-S-methyltransferase to form methane thiol. As with virtually all molecules, H2S can react with other free radical species, as well as, a number of non-radical reactive oxygen (ROS) and nitrogen (RNS) species (Fig. 2) [43]. Many of the oxidized sulfur species as well as sulfur-centered radicals formed are less reactive than their oxygen-containing counterparts [44]. One of the most important oxidants responsible for the catabolism of H2S is O2. In the presence of molecular O2 and redox active metals, H2S will spontaneously oxidize [45]. In an oxygenated biological medium, metalloproteins catalyze H2S oxidation. This makes O2 tension a critical methodological consideration when conducting biologically relevant experiments.
5. Measurement of H2S in mammalian samples
As the field of H2S continues to grow, accurate measurement of H2S in biological samples is critical for proper understanding of its biochemistry and identifying its key physiological roles. Early studies reported endogenous H2S concentrations in mammalian tissues to be approximately 160 μM [46]. However, when considering that the Ki of CcO for H2S is 0.2 μM [28] it is unlikely that free H2S exists at high μM concentrations. Older methods relied on inducing large pH shifts during sample preparation followed by capturing the released sulfide anions with metals such as silver or zinc [47] and then measuring these complexes by spectrophotometry or chromatography [46]. One problem with these methods is that they liberate multiple acid-labile sulfur species, including those in Fe–S centers, giving artificially high values. Recently, the mono-bromobimane assay has been refined to exclude sulfur species other than free H2S [47]. As techniques improve, the reported concentrations of H2S in mammalian samples have decreased from the 100–200 μM range to less than 500 nM [48]. Other techniques, including polarography, have measured H2S concentrations in mouse blood in the sub-μM range (Stein et al., unpublished data). These lower values are more plausible when considering the inhibitory potential of H2S for heme-containing proteins, in addition to, parallels between H2S and other signaling molecules such as •NO and carbon monoxide (CO). It is also important to note that whole tissue or cell lysate measurements of H2S overlook compartmental differences in H2S concentrations and thus likely underestimate the effective localized concentrations of H2S in vivo. Because the technology required to accurately measure H2S on the sub-cellular level is unavailable, it is important to recognize these limitations when considering physiologically relevant levels and reactions of H2S.
6. Biological roles of H2S
Like •NO and CO, H2S is produced in many different cell types and can easily diffuse without the need for transporters. Therefore, it is not surprising that H2S has diverse biological actions (Fig. 4). Important factors that determine the biological actions of H2S include, but are not limited to, differences in the solubility of H2S in aqueous vs. lipid phases, proximity of the target to H2S-detoxifying enzymes, heme redox state, and inter- and intracellular differences in O2 tension.
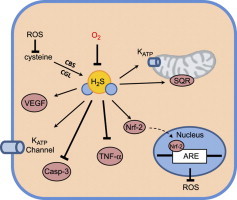
Fig. 4.
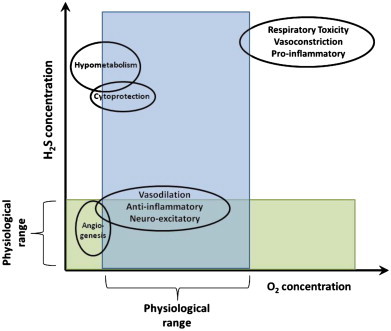
Interaction of O2 and H2S on physiological outcomes. H2S plays a role in many physiological processes. Additionally, it is capable of acting as a therapeutic agent. The concentration of H2S, whether endogenously produced or exogenously administered, will dictate the outcome. This can be beneficial at low and intermediate concentrations or harmful at high concentrations. High O2 can reverse many of the beneficial roles of H2S seen at lower O2 concentrations, resulting in, for example, vasoconstriction rather than vasodilation. Additionally, under hypoxic and normoxic conditions, H2S promotes angiogenesis. However, at higher concentrations of both O2 and H2S, an inhibition of cellular proliferation is seen. H2S has a narrow therapeutic window within which it is cytoprotective. At high concentrations it can be pro-apoptotic and pro-inflammatory. Finally, the larger doses of H2S necessary to induce a hypometabolic effect, can, if pushed further, result in cardiac and respiratory toxicity.
7. Vasoactivity
One of the first physiological roles that prompted investigators to regard H2S as the “third gaseous signaling molecule” was vasodilation. In 2001, Zhao et al. showed that H2S decreased blood pressure in rats in vivo and caused vascular smooth muscle cell (VSMC) relaxation in vitro [49]. H2S-mediated vasodilation has also been shown in the ileum of the gastrointestinal tract and the vas deferens [50]. Additionally, others have shown that transgenic mice deficient in CBS are chronically hypertensive [51]. H2S is thought to induce vasodilation by increasing the conductance of adenosine triphosphate (ATP) sensitive potassium channels (KATP). Furthermore, the specific molecular targets of H2S were shown to be cysteine 6 and 26 of the extracellular portion of the rvSUR1 subunit of the KATP channel complex [52]. These vicinal thiols form a disulfide bond, which H2S reduces, increasing channel conductance. However, the vasodilatory effect of H2S is highly O2-dependent as supra-physiological levels of O2 (200 μM) cause H2S-induced vasoconstriction [53]. Recently, conflicting reports have emerged showing that the contribution of the KATP channels to H2S-induced vasodilation is minimal and that vasodilation is due to metabolic inhibition (i.e., decrease in ATP), intracellular pH changes, and modulation of Cl−/HCO−3 channels [54]. There are several methodological differences including O2 tension, pre-contraction agent, vessel type, and the type of sulfide-based chemical used, which may account for discrepancies among experimental studies. There are also indications that H2S may act through •NO to stimulate vasodilation. Studies suggest that H2S liberates •NO from S-nitrosothiols [55]. Others show that endothelial denudation and nitric oxide synthase (NOS) inhibitors shift the dose–response curve to the right [56]. However, H2S increases eNOS phosphorylation and subsequent •NO production in an Akt-dependent manner [57]. While the mechanism(s) responsible for vasodilation remain unclear, the role of H2S as a vasodilator is accepted and there is great interest in employing H2S-releasing agents as therapies to treat hypertension.
8. Angiogenesis
H2S can cause cell proliferation and migration [58,59]; however, there appears to be a narrow concentration range of the proliferative effect, below which no effect is seen and above which there is anti-proliferation and H2S cytotoxicity [60]. In cell culture experiments, low micromolar concentrations of H2S increase endothelial cell number, proto-vessel formation, and cell migration [58]. Chicken chorioallantoic membranes, an in vivo model of angiogenesis, display increased branching and lengthening of blood vessels in response to 48 h incubation with H2S [59]. Additionally, aortic tissue isolated from transgenic mice lacking CSE, the primary H2S-producing enzyme in the endothelium, exhibit marked decreases in angiogenesis [59].
The mechanism of H2S-induced angiogenesis operates through several pathways, including activation of ATP-sensitive potassium (KATP) channels [49]. Papapetropoulos et al. showed that treatment of endothelial cells with the KATP channel inhibitor glibenclamide reduced cell migration, which was accompanied by decreased H2S-induced p38 and heat shock protein 27 (Hsp27) phosphorylation [59]. Additionally, H2S can stimulate angiogenesis through phosphatidylinositol 3-kinase (PI3K) and Akt activation [61]. H2S can also activate hypoxia inducible factor-1α (HIF-1α) and thus increase expression of vascular endothelial growth factor (VEGF) [62]. Conversely, VEGF-stimulated angiogenesis is suppressed in CSE knockout mice [58].
Endogenous H2S production is known to be upregulated during wound healing [63]. Topically applied H2S accelerates wound closure and healing [59]. Angiogenesis is very important in both acute and chronic ischemia as poorly vascularized tissue will lose function and possibly become necrotic. In models of chronic hind limb ischemia, sodium hydrosulfide (NaHS) increased capillary formation and blood flow [64]. Similar results were found in chronically ischemic hearts with improvements in cardiac function following H2S treatment [65]. These studies indicate that endogenous H2S is crucial in physiological angiogenesis and that those capabilities can be employed in disease treatment.
9. Inflammation
Several studies report that H2S is a mediator of inflammation [66–69], while others report that H2S ameliorates inflammatory sequelae [70–72]. These studies are likely in conflict because different models of inflammation, H2S production inhibitors, H2S “donors”, and different O2 tensions were used for experiments. Using NaHS, several groups have shown that H2S augments neutrophil migration and adhesion, nociception, and increases endotoxemia [66,67,73]. This is likely because upon application NaHS “releases” all its H2S instantly, which may be pro-inflammatory in some cases. This is in contrast to the much slower kinetics and magnitude of endogenous H2S production. Moreover, a burst of H2S may alter •NO homeostasis as well, decreasing its bioavailability through direct reactions and through inhibition of NOS [74]. Regarding endogenous production, some groups report that CSE is upregulated during lipopolysaccharide (LPS)-induced endotoxemia and that pharmacological inhibition of CSE attenuated inflammation [68,69]. Whiteman et al. highlighted differences between using rapid release of high concentrations of H2S vs. using slow-releasing donor compounds, which more closely mimic endogenous production. They showed that a slow-releasing H2S donor dose-dependently inhibited LPS-induced inflammation by decreasing the pro-inflammatory mediators IL-1β, IL-6, TNF-α, and PGE2 [74]. Additionally, they showed an increase in the anti-inflammatory cytokine IL-10 in an NFκB-dependent mechanism [75]. This report is similar to others that show H2S-dependent inhibition of •NO and TNF-α through p38 inhibition; a known downstream target of H2S [76]. In contrast to [68], which used a non-specific PLP-dependent enzyme inhibitor, propargylglycine (PAG), others have used the CSE inhibitor β-cyanoalanine [70]. Treatment with β-cyanoalanine increased leukocyte adhesion to the endothelium, indicating some off-target effects of PAG and that endogenous H2S is anti-inflammatory [70]. Like vasodilation, the anti-inflammatory effects of H2S appear to be KATP channel-dependent with glibenclamide reversing the inhibitory effects of H2S on leukocyte adhesion [71]. Although there are conflicting reports regarding the role of H2S in inflammation, common themes have emerged that shed light on the reasons for these disparate results. H2S donor compounds with slow release kinetics that mimic endogenous production appear to ameliorate inflammation. This is contrasted against the apparent pro-inflammatory potential of low purity sulfide salts, which immediately dissociate in solution to rapidly deliver large doses of H2S. As with vasoactivity, the role of H2S in inflammation is redox sensitive and this becomes important when impure sulfide salts are used in experiments. In addition to sulfide, these salts contain oxidized sulfur species such as sulfates, sulfenic acid, and sulfonates. In contrast to H2S, these species can have proinflammatory properties [75]. Additionally, experiments conducted at supra-physiological O2 tensions may yield misleading results, as high O2 concentrations increase production of proinflammatory oxidized sulfur species [53,75]. In order to move H2S from the “bench to the bedside”, comprehensive pharmacological studies are needed, which focus on routes of administration, type of donor compounds used, kinetics and magnitude of H2S release, timing of the intervention, physiologically and pathologically relevant O2 tensions, and the type of inflammatory condition being addressed.
10. Hydrogen sulfide and cytoprotection
As mentioned in earlier sections, the therapeutic window for effective H2S treatment is likely very narrow because H2S is a potent inhibitor of mitochondrial respiration. Therefore, several pharmacological factors including dose, route of administration, and timing of H2S exposure must be carefully considered when using H2S as a therapy. In the following paragraphs, select examples from the literature are presented showing cytoprotective effects of H2S and other sulfide-based compounds in models of cardiovascular disease.
11. Cardiovascular disease
Studies show decreased plasma levels of H2S in atherosclerosis models [51,77]. Furthermore, endogenous production of H2S in sclerotic aortic tissues is impaired due to decreased expression of CGL in VSMCs [78]. Conversely, in cases of H2S over-production (e.g., Trisomy 21) progression of atherosclerosis is slower [79]. H2S may slow disease progression of atherosclerosis by inhibiting several elements of foam cell formation. Macrophages incubated with oxidized lipoprotein (oxLDL) show decreased intracellular lipid accumulation when treated with H2S, whereas inhibiting endogenous H2S production exacerbates lipid accumulation [80]. Expression of scavenger receptors responsible for oxLDL accumulation was also decreased with H2S treatment [80]. Using apolipoprotein E knockout mice, H2S decreases expression of adhesion molecules, thus preventing the recruitment of macrophages into the vascular intima, which is accompanied by a reduction in pro-inflammatory cytokines [2]. In atherosclerotic plaques, VSMCs proliferate and assume a fibrogenic phenotype, contributing to vessel occlusion. H2S, at high concentrations, prevents VSMC proliferation [81]. Together, these results support the concept that sulfide-based therapies attenuate some of the early metabolic changes that contribute to cardiovascular disease. What is not known is whether H2S is efficacious in end stage cardiovascular disease.
Considerable work has also focused on the impact sulfide-based therapies have in ameliorating cardiac ischemia-reperfusion injury. Lefer and colleagues have shown that pretreatment with sodium sulfide (Na2S) before cardiac ischemia-reperfusion reduces infarct size and improves overall cardiac function [3,82,83]. There are several proposed mechanisms underlying this protective effect. First, H2S up-regulates anti-oxidant response element (ARE) genes by inducing the translocation of Nrf-2 to the nucleus [84]. This increases GSH and expression of antioxidant proteins such as heme oxygenase-1, glutathione S-transferase, and thioredoxin-1 [84]. Second, H2S acts through Akt-dependent pro-survival pathways to prevent caspase 3 activation and association of pro-apoptotic proteins to mitochondria that are necessary for mitochondrial permeability pore formation and cytochrome c release [85]. The effects of H2S on the mitochondrion are likely central to overall cardiomyocyte protection [86], which may also involve activation of KATP channels [87]. While some groups attribute the cytoprotection to mitochondrial KATP channels, others using organelle-specific KATP channel blockers show that cytoprotection is due to effects on sarcolemmal channels [88].
Interestingly, daily H2S administration for several months prevented perivascular fibrosis and subsequent arteriole occlusion in spontaneously hypertensive rats. [89], suggesting that H2S administration may prevent pathogenic vascular remodeling. H2S prevents thickening of damaged arterial intima and medial thickening of intramyocardial coronary arterioles [90]. Finally, H2S induces neovascularization in models of chronic ischemia [64]. This molecular action of H2S may be highly important following acute cardiac ischemia, as maintenance of a highly vascularized myocardium is required for proper organ function. These select findings, when taken together, demonstrate the promise of H2S as a treatment for chronic cardiovascular diseases, as well as, in the emergency room for patients suffering from cardiac arrest.
12. Summary
The actions of H2S described in this review highlight its ability to function in redox biology and cell signaling. With a pKa of 6.9, both protonated and anion forms of H2S are present at physiological pH. This allows for the free diffusion of H2S to all cellular compartments. Because H2S-producing enzymes play a diverse role in the larger sulfur cycle of the cell, the production of H2S is dependent on cellular redox status. During conditions of oxidative stress the amount of reduced substrates like cysteine available for H2S production may be limited. Moreover, steady state H2S concentrations will be determined by H2S consumption pathways that are redox sensitive as well. Importantly, O2 being an antagonist of H2S will also accelerate H2S oxidation and inhalation of 100% O2 is, in fact, a treatment for H2S gas inhalation. Also highlighted herein, is the impact of O2 tension on the biological actions and outcomes of H2S. For example, H2S-mediated vasodilation is observed at physiological O2 concentrations, whereas at vasoconstriction occurs at hyperoxia (Fig. 4). These same patterns can also be extended to other biological processes such as inflammation and angiogenesis (Fig. 4). Furthermore, many of the cytoprotective actions of H2S are triggered by and act to attenuate oxidative stress as exemplified by the ability of H2S to induce the expression of antioxidant enzymes during myocardial ischemia-reperfusion. In conclusion, investigation of the redox biology of H2S will not only increase understanding of its role in human physiology, but its potential therapeutic role in pathologies where oxidative stress is central to the disease process.
Authors contributions
AS and SMB wrote the manuscript.
Acknowledgments
This work was supported by the NIH Grants T32 HL007918 (AS) and R01 HL092857 (SMB).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Jeney V., Komodi E., Nagy E., Zarjou A., Vercellotti G.M., Eaton J.W., Balla G., Balla J. Supression of hemin-mediated oxidation of low-density lipoprotein and subsequent endothelial reactions by hydrogen sulfide (H(2)S) Free Radical Biology & Medicine. 2009;46:616–623. doi: 10.1016/j.freeradbiomed.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Zhao X., Jin H., Wei H., Li W., Bu D., Tang X., Ren Y., Tang C., Du J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;29:173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 3.King A.L., Lefer D.J. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Experimental Physiology. 2011;96:840–846. doi: 10.1113/expphysiol.2011.059725. [DOI] [PubMed] [Google Scholar]
- 4.Blackstone E., Morrison M., Roth M.B. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 5.Stein A., Mao Z., Morrison J.P., Fanucchi M.V., Postlethwait E.M., Patel R.P., Kraus D.W., Doeller J.E., Bailey S.M. Metabolic and cardiac signaling effects of inhaled hydrogen sulfide and low oxygen in male rats. Journal of Applied Physiology. 2012;112:1659–1669. doi: 10.1152/japplphysiol.01598.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moss G.A. Water and health: a forgotten connection? Perspectives in Public Health. 2010;130:227–232. doi: 10.1177/1757913910379192. [DOI] [PubMed] [Google Scholar]
- 7.Reigstad L.J., Jorgensen S.L., Lauritzen S.E., Schleper C., Urich T. Sulfur-oxidizing chemolithotrophic proteobacteria dominate the microbiota in high arctic thermal springs on Svalbard. Astrobiology. 2011;11:665–678. doi: 10.1089/ast.2010.0551. [DOI] [PubMed] [Google Scholar]
- 8.Matz H., Orion E., Wolf R. Balneotherapy in dermatology. Dermatologic Therapy. 2003;16:132–140. doi: 10.1046/j.1529-8019.2003.01622.x. [DOI] [PubMed] [Google Scholar]
- 9.Mulrow C., Lawrence V., Ackermann R., Gilbert Ramirez G., Morbidoni L., Aguilar C., Arterburn J., Block E., Chiquette E., Gardener C., Harris M., Heidenreich P., Mullins D., Richardson M., Russell N., Vickers A., Young V. Garlic: effects on cardiovascular risks and disease, protective effects against cancer, and clinical adverse effects. Evidence Report—Technology Assessment (Summary) 2000;20:1–4. [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes J., Anderson L.A., Phillipson J.D. 3rd ed. Pharmaceutical Press; London; Grayslake, IL: 2007. Herbal medicines. [Google Scholar]
- 11.Benavides G.A., Squadrito G.L., Mills R.W., Patel H.D., Isbell T.S., Patel R.P., Darley-Usmar V.M., Doeller J.E., Kraus D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee S.K., Maulik S.K. Effect of garlic on cardiovascular disorders: a review. Nutrition Journal. 2002;1:4. doi: 10.1186/1475-2891-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stipanuk M.H. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annual Review of Nutrition. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 14.Stipanuk M.H., Beck P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. The Biochemical Journal. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe K., Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. The Journal of Neuroscience. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudek M., Frendo J., Koj A. Subcellular compartmentation of rhodanses and 3-mercaptopyruvate sulphurtransferase in the liver of some vertebrate species. Comparative Biochemistry and Physiology. 1979;65B:383–386. [Google Scholar]
- 17.Li L., Rose P., Moore P.K. Hydrogen sulfide and cell signaling. Annual Review of Pharmacology and Toxicology. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 18.Kabil O., Banerjee R. Redox biochemistry of hydrogen sulfide. The Journal of Biological Chemistry. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stipanuk M.H., Dominy J.E., Jr., Lee J.I., Coloso R.M. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. The Journal of Nutrition. 2006;136:1652S–1659S. doi: 10.1093/jn/136.6.1652S. [DOI] [PubMed] [Google Scholar]
- 20.Taoka S., Ohja S., Shan X., Kruger W.D., Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine beta-synthase activity. The Journal of Biological Chemistry. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 21.Fu M., Zhang W., Wu L., Yang G., Li H., Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholls P. Inhibition of cytochrome c oxidase by sulphide. Biochemical Society Transactions. 1975;3:316–319. doi: 10.1042/bst0030316. [DOI] [PubMed] [Google Scholar]
- 23.Bouillaud F., Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxidants & Redox Signaling. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 24.Chou, S. Syracuse research corporation, and United States. Agency for toxic substances and disease registry. Toxicological profile for hydrogen sulfide: update, U.S. Dept. of Health and Human Services Public Health Service Agency for Toxic Substances and Disease Registry [Atlanta, Ga.]; 2006.
- 25.Nicholls P., Kim J.K. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Canadian Journal of Biochemistry. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 26.Nicholls P., Kim J.K. Oxidation of sulphide by cytochrome aa3. Biochimica et Biophysica Acta. 1981;637:312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- 27.Hill B.C., Woon T.C., Nicholls P., Peterson J., Greenwood C., Thomson A.J. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. The Biochemical Journal. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper C.E., Brown G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. Journal of Bioenergetics and Biomembranes. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 29.Wever R., van G.B., Dervartanian D.V. Biochemical and biophysical studies on cytochrome c oxidase. XX. Reaction with sulphide. Biochimica et Biophysica Acta. 1975;387:189–193. doi: 10.1016/0005-2728(75)90102-4. [DOI] [PubMed] [Google Scholar]
- 30.Wilson K., Mudra M., Furne J., Levitt M. Differentiation of the roles of sulfide oxidase and rhodanese in the detoxification of sulfide by the colonic mucosa. Digestive Diseases and Sciences. 2008;53:277–283. doi: 10.1007/s10620-007-9854-9. [DOI] [PubMed] [Google Scholar]
- 31.Jackson M.R., Melideo S.L., Jorns M.S. Human sulfide: quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry. 2012;51:6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- 32.Tiranti V., D'Adamo P., Briem E., Ferrari G., Mineri R., Lamantea E., Mandel H., Balestri P., Garcia-Silva M.T., Vollmer B., Rinaldo P., Hahn S.H., Leonard J., Rahman S., Dionisi-Vici C., Garavaglia B., Gasparini P., Zeviani M. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. American Journal of Human Genetics. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M.D., Prelle A., Fagiolari G., Rimoldi M., Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nature Medicine. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 34.Hildebrandt T.M., Grieshaber M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. The FEBS Journal. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 35.Berzofsky J.A., Peisach J., Blumberg W.E. Sulfheme proteins. I. Optical and magnetic properties of sulfmyoglobin and its derivatives. The Journal of Biological Chemistry. 1971;246:3367–3377. [PubMed] [Google Scholar]
- 36.Assendelft O.W. v. Spectrophotometry of haemoglobin derivatives. C.C. Thomas; Springfield, Ill: 1970. [Google Scholar]
- 37.Carrico R.J., Blumberg W.E., Peisach J. The reversible binding of oxygen to sulfhemoglobin. The Journal of Biological Chemistry. 1978;253:7212–7215. [PubMed] [Google Scholar]
- 38.Pietri R., Roman-Morales E., Lopez-Garriga J. Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxidants & Redox Signaling. 2011;15:393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schreier S.M., Muellner M.K., Steinkellner H., Hermann M., Esterbauer H., Exner M., Gmeiner B.M., Kapiotis S., Laggner H. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotoxicity Research. 2010;17:249–256. doi: 10.1007/s12640-009-9099-9. [DOI] [PubMed] [Google Scholar]
- 40.Toohey J.I. Sulphane sulphur in biological systems: a possible regulatory role. The Biochemical Journal. 1989;264:625–632. doi: 10.1042/bj2640625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mustafa A.K., Gadalla M.M., Sen N., Kim S., Mu W., Gazi S.K., Barrow R.K., Yang G., Wang R., Snyder S.H. H2S signals through protein S-sulfhydration. Science Signaling. 2009;2(ra72):1–8. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacob C., Anwar A., Burkholz T. Perspective on recent developments on sulfur-containing agents and hydrogen sulfide signaling. Planta Medica. 2008;74:1580–1592. doi: 10.1055/s-0028-1088299. [DOI] [PubMed] [Google Scholar]
- 43.Carballal S., Trujillo M., Cuevasanta E., Bartesaghi S., Moller M.N., Folkes L.K., Garcia-Bereguiain M.A., Gutierrez-Merino C., Wardman P., Denicola A., Radi R., Alvarez B. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radical Biology & Medicine. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 44.Iciek M., Wlodek L. Biosynthesis and biological properties of compounds containing highly reactive, reduced sulfane sulfur. Polish Journal of Pharmacology. 2001;53:215–225. [PubMed] [Google Scholar]
- 45.Chen K.Y., Morris J.C. Kinetics of oxidation of aqueous sulfide by O2. Environmental Sciences and Technology. 1972;6:529–537. [Google Scholar]
- 46.Ubuka T. Assay methods and biological roles of labile sulfur in animal tissues. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2002;781:227–249. doi: 10.1016/s1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 47.Shen X., Pattillo C.B., Pardue S., Bir S.C., Wang R., Kevil C.G. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radical Biology & Medicine. 2011;50:1021–1031. doi: 10.1016/j.freeradbiomed.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen X., Peter E.A., Bir S., Wang R., Kevil C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radical Biology & Medicine. 2012;52:2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao W., Zhang J., Lu Y., Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. The EMBO Journal. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teague B., Asiedu S., Moore P.K. The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. British Journal of Pharmacology. 2002;137:139–145. doi: 10.1038/sj.bjp.0704858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A.K., Mu W., Zhang S., Snyder S.H., Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang B., Tang G., Cao K., Wu L., Wang R. Molecular mechanism for H(2)S-induced activation of K(ATP) channels. Antioxidants & Redox Signaling. 2010;12:1167–1178. doi: 10.1089/ars.2009.2894. [DOI] [PubMed] [Google Scholar]
- 53.Koenitzer J.R., Isbell T.S., Patel H.D., Benavides G.A., Dickinson D.A., Patel R.P., Darley-Usmar V.M., Lancaster J.R., Jr., Doeller J.E., Kraus D.W. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. American Journal of Physiology Heart and Circulatory Physiology. 2007;292:H1953–H1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- 54.Kiss L., Deitch E.A., Szabo C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sciences. 2008;83:589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whiteman M., Li L., Kostetski I., Chu S.H., Siau J.L., Bhatia M., Moore P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochemical and Biophysical Research Communications. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 56.Zhao W., Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. American Journal of Physiology Heart and Circulatory Physiology. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- 57.Predmore B.L., Julian D., Cardounel A.J. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism. Frontiers in Physiology. 2011;2:104. doi: 10.3389/fphys.2011.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szabo C., Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. British Journal of Pharmacology. 2010;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papapetropoulos A., Pyriochou A., Altaany Z., Yang G., Marazioti A., Zhou Z., Jeschke M.G., Branski L.K., Herndon D.N., Wang R., Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai W.J., Wang M.J., Moore P.K., Jin H.M., Yao T., Zhu Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovascular Research. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 61.Szabo C., Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. British Journal of Pharmacology. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kai S., Tanaka T., Daijo H., Harada H., Kishimoto S., Suzuki K., Takabuchi S., Takenaga K., Fukuda K., Hirota K. Hydrogen sulfide inhibits hypoxia—but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxidants & Redox Signaling. 2012;16:203–216. doi: 10.1089/ars.2011.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wallace J.L., Dicay M., McKnight W., Martin G.R. Hydrogen sulfide enhances ulcer healing in rats. The FASEB Journal. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- 64.Wang M.J., Cai W.J., Li N., Ding Y.J., Chen Y., Zhu Y.C. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxidants & Redox Signaling. 2010;12:1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- 65.Qipshidze N., Metreveli N., Mishra P.K., Lominadze D., Tyagi S.C. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. International Journal of Biological Sciences. 2012;8:430–441. doi: 10.7150/ijbs.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H., Zhi L., Moochhala S.M., Moore P.K., Bhatia M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. Journal of Leukocyte Biology. 2007;82:894–905. doi: 10.1189/jlb.0407237. [DOI] [PubMed] [Google Scholar]
- 67.Dal-Secco D., Cunha T.M., Freitas A., Alves-Filho J.C., Souto F.O., Fukada S.Y., Grespan R., Alencar N.M., Neto A.F., Rossi M.A., Ferreira S.H., Hothersall J.S., Cunha F.Q. Hydrogen sulfide augments neutrophil migration through enhancement of adhesion molecule expression and prevention of CXCR2 internalization: role of ATP-sensitive potassium channels. Journal of Immunology. 2008;181:4287–4298. doi: 10.4049/jimmunol.181.6.4287. [DOI] [PubMed] [Google Scholar]
- 68.Collin M., Anuar F.B., Murch O., Bhatia M., Moore P.K., Thiemermann C. Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. British Journal of Pharmacology. 2005;146:498–505. doi: 10.1038/sj.bjp.0706367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L., Bhatia M., Zhu Y.Z., Zhu Y.C., Ramnath R.D., Wang Z.J., Anuar F.B., Whiteman M., Salto-Tellez M., Moore P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. The FASEB Journal. 2005;19:1196–1198. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 70.Wallace J.L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends in Pharmacological Sciences. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 71.Fiorucci S., Antonelli E., Distrutti E., Rizzo G., Mencarelli A., Orlandi S., Zanardo R., Renga B., Di Sante M., Morelli A., Cirino G., Wallace J.L. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 72.Li L., Rossoni G., Sparatore A., Lee L.C., Del Soldato P., Moore P.K. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radical Biology & Medicine. 2007;42:706–719. doi: 10.1016/j.freeradbiomed.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 73.Kawabata A., Ishiki T., Nagasawa K., Yoshida S., Maeda Y., Takahashi T., Sekiguchi F., Wada T., Ichida S., Nishikawa H. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 74.Li L., Salto-Tellez M., Tan C.H., Whiteman M., Moore P.K. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radical Biology & Medicine. 2009;47:103–113. doi: 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 75.Whiteman M., Li L., Rose P., Tan C.H., Parkinson D.B., Moore P.K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxidants & Redox Signaling. 2010;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu L.F., Wong P.T., Moore P.K., Bian J.S. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. Journal of Neurochemistry. 2007;100:1121–1128. doi: 10.1111/j.1471-4159.2006.04283.x. [DOI] [PubMed] [Google Scholar]
- 77.Geng B., Chang L., Pan C., Qi Y., Zhao J., Pang Y., Du J., Tang C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochemical and Biophysical Research Communications. 2004;318:756–763. doi: 10.1016/j.bbrc.2004.04.094. [DOI] [PubMed] [Google Scholar]
- 78.Jiang H.L., Wu H.C., Li Z.L., Geng B., Tang C.S. Changes of the new gaseous transmitter H2S in patients with coronary heart disease. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:951–954. [PubMed] [Google Scholar]
- 79.Kamoun P., Belardinelli M.C., Chabli A., Lallouchi K., Chadefaux-Vekemans B. Endogenous hydrogen sulfide overproduction in Down syndrome. American Journal of Medical Genetics Part A. 2003;116A:310–311. doi: 10.1002/ajmg.a.10847. [DOI] [PubMed] [Google Scholar]
- 80.Zhao Z.Z., Wang Z., Li G.H., Wang R., Tan J.M., Cao X., Suo R., Jiang Z.S. Hydrogen sulfide inhibits macrophage-derived foam cell formation. Experimental Biology and Medicine (Maywood) 2011;236:169–176. doi: 10.1258/ebm.2010.010308. [DOI] [PubMed] [Google Scholar]
- 81.Yang G., Sun X., Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. The FASEB Journal. 2004;18:1782–1784. doi: 10.1096/fj.04-2279fje. [DOI] [PubMed] [Google Scholar]
- 82.Predmore B.L., Lefer D.J. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. Journal of Cardiovascular Translational Research. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- 83.Lavu M., Bhushan S., Lefer D.J. Hydrogen sulfide-mediated cardioprotection: mechanisms and therapeutic potential. Clinical Science (London) 2011;120:219–229. doi: 10.1042/CS20100462. [DOI] [PubMed] [Google Scholar]
- 84.Calvert J.W., Jha S., Gundewar S., Elrod J.W., Ramachandran A., Pattillo C.B., Kevil C.G., Lefer D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circulation Research. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Calvert J.W., Elston M., Nicholson C.K., Gundewar S., Jha S., Elrod J.W., Ramachandran A., Lefer D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elrod J.W., Calvert J.W., Morrison J., Doeller J.E., Kraus D.W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C.W., Lefer D.J. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maack C., Dabew E.R., Hohl M., Schafers H.J., Bohm M. Endogenous activation of mitochondrial KATP channels protects human failing myocardium from hydroxyl radical-induced stunning. Circulation Research. 2009;105:811–817. doi: 10.1161/CIRCRESAHA.109.206359. [DOI] [PubMed] [Google Scholar]
- 88.Pan T.T., Feng Z.N., Lee S.W., Moore P.K., Bian J.S. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 2006;40:119–130. doi: 10.1016/j.yjmcc.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 89.Shi Y.X., Chen Y., Zhu Y.Z., Huang G.Y., Moore P.K., Huang S.H., Yao T., Zhu Y.C. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. American Journal of Physiology Heart and Circulatory Physiology. 2007;293:H2093–H2100. doi: 10.1152/ajpheart.00088.2007. [DOI] [PubMed] [Google Scholar]
- 90.Elsey D.J., Fowkes R.C., Baxter G.F. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S) Cell Biochemistry and Function. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]