

Abstract
Human papillomaviruses (HPVs) are prevalent pathogens of mucosal and cutaneous epithelia. Productive infections of squamous epithelia lead to benign hyperproliferative warts, condylomata, or papillomas. Persistent infections of the anogenital mucosa by high-risk HPV genotypes 16 and 18 and closely related types can infrequently progress to high-grade intraepithelial neoplasias, carcinomas-in-situ, and invasive cancers in women and men. HPV-16 is also associated with a fraction of head and neck cancers. We discuss the interactions of the mucosotropic HPVs with the host regulatory proteins and pathways that lead to benign coexistence and enable HPV DNA amplification or, alternatively, to cancers that no longer support viral production.
Human papillomaviruses encode proteins to support viral replication. Some of these proteins inactivate and destabilize host tumor suppressor proteins and may lead to cancer in some cases.
Human papillomaviruses (HPVs) are ubiquitous small DNA viruses that comprise a family of more than 150 genotypes. Closely related types show a predilection for particular epithelial tissues and have similar pathogenic properties (de Villiers et al. 2004; Bravo et al. 2010). The mucosotropic HPVs can be sexually transmitted. Infections are frequently asymptomatic and are cleared by the immune systems. Active cases are manifested as hyperproliferative lesions but often regress into subclinical persistency within a year. Latent infections can reactivate following immunosuppression or upon undergoing repeated cycles of wounding and healing (Fig. 1). A small fraction of persistent infections by the high-risk (HR) HPV genotypes leads to cancers (reviewed by zur Hausen 2009). Worldwide, each year there are about 500,000 new cases of cervical cancer and 275,000 deaths. More than 99% of cervical cancers are caused by high-risk (HR) HPVs, and types 16 and 18 are responsible for about 70% of the cases (Walboomers et al. 1999). The HR HPVs are also responsible for a high percentage of cancers of other anogenital sites in men and women. Moreover, HPV-16 is associated with a fraction of the head and neck neoplasias, in particular tonsillar and oro-pharyngeal cancers (reviewed by Syrjänen 2010). In contrast, infections by the low-risk (LR) types 6 and 11 cause 90% of genital warts and essentially all laryngeal papillomas (recurrent respiratory papillomatosis or RRP) (reviewed by Derkay and Wiatrak 2008) but they rarely result in carcinomas. Juvenile-onset RRP results from transmission in utero or during passage through the birth canal of mothers with active condylomata, yet infections may become symptomatic years later. Recent studies indicate that HPVs can also replicate in trophoblasts and produce infectious particles and that HPVs can be detected in spontaneous aborted tissues (Gomez et al. 2008; You et al. 2008). Together, these observations add considerable medical significance to HPV infections during pregnancy.
Figure 1.

Natural history of HPV infections. Most infections are subclinical and are quickly cleared by host immune surveillance. Some infections become active but regress to a latent state or are cleared within a year. Immune suppression or cycles of wounding and healing, conditions that induce viral early gene expression in proliferating undifferentiated cells, may reactivate the infections. In tissues persistently infected by high-risk HPVs, repeated wounding and healing lead to recurring and extended inactivation of tumor suppressor proteins by the viral oncoproteins and hence excessive basal cell proliferation and progression to high-grade lesions. These cells may accumulate mutations and epigenetic changes, resulting over time in cell immortalization and transformation, integration of the viral DNA into host chromosomes (in most cases), and carcinomas.
There are no consistently effective drugs for treating HPV infections. Surgical removal or local ablation of lesions are the standards of care. The implementation of Pap smear screening in the developed countries over the past 60 years has enabled early detection of cytological changes caused by HPV infections of the cervix and has dramatically reduced morbidity and mortality. Nucleic acids-based HPV detection and genotyping have greatly improved screening sensitivity and specificity. However, in resource-poor countries where early detection and intervention are not generally available, cervical cancers remain a major malignancy among women.
How would a normally benign papillomavirus infection become cancerous? The answer resides in the virus–host interactions necessary to support the viral replicative cycle. Regardless of their oncogenic potential, the production of progeny virions occurs only in postmitotic, differentiated cells of squamous epithelia (reviewed by Chow et al. 2010). Thus, the viruses encode E7 and E6 proteins to reestablish a milieu conducive for viral DNA amplification. E7 does so by destabilizing p130, a member of the retinoblastoma (pRB) tumor suppressor protein family that controls cell cycle entry and progression (Litovchick et al. 2011, and references therein), whereas E6 inactivates the transcription-transactivation function of the tumor suppressor TP53 (Thomas and Chiang 2005), which functions to maintain genome stability by controlling critical cellular processes such as proliferation, DNA repair, senescence, and apoptosis. Unique to the HR HPVs, their E7 and E6 proteins also destabilize pRB and TP53, respectively (for reviews, see McLaughlin-Drubin and Münger 2009; Moody and Laimins 2010; Korzeniewski et al. 2011). This ability of the HR HPV oncoproteins to override the tumor suppressors then becomes a double-edged sword should they become up-regulated in undifferentiated cells as a result of multiple episodes of wounding and healing. Over time, critical host regulatory genes can incur mutations or become dysregulated because of epigenetic changes, resulting eventually in progression to oncogenic transformation. It is not understood why the HR HPVs acquired these unique properties, in view of the fact that progeny virions are no longer produced in cancers. In this chapter, we will discuss virus–host interactions, leading to divergent outcomes.
THE PERMISSIVE HOST TISSUES
Squamous epithelia turn over and are renewed every few weeks (for a review, Fuchs 1990). The basal reserve cells rarely divide except when replacing the cells immediately above, called parabasal transit amplifying (TA) cells. The TA cells have committed to differentiation but divide daily for up to 80 cell divisions whereupon they undergo terminal differentiation. With each asymmetrical cell division, one daughter cell remains in the TA layer while the other is pushed upward, exits the cell cycle, and differentiates into spinous cells that comprise the thickest strata of the epithelium. In the cutaneous skin, the upper spinous cells further differentiate into granulocytes. The superficial cells undergo programmed death and slough off as cornified envelopes.
Papillomavirions gain entry into basal cells through a wound or microlesion. The virus depends on the cells reentering into the cell cycle during wound healing to establish infection (Pyeon et al. 2009). Active HPV infections stimulate cell cycling and increase the thickness of the parabasal and spinous strata, resulting in warty growth. The viral genome is maintained as low copy number nuclear plasmids in lower strata in which viral DNA and RNA are present at or below the sensitivity of detection. Up-regulation of viral RNA transcription and DNA amplification occurs in a subset of mid- to upper-spinous cells. Progeny virus particles are produced in a small fraction of superficial cells that are shed during desquamation (Chow et al. 2010). This differentiation-dependent viral gene expression is controlled by transcription factors that bind to the viral enhancer and promoter sequences (reviewed by Bernard 2002) and by chromatin remodeling (Zhao et al. 1999; Kim et al. 2003; Wu et al. 2006; Wooldrige and Laimins 2008; Jha et al. 2010).
Three-dimensional organotypic culture systems were developed to achieve squamous differentiation, starting with native epithelial tissue fragments, with isolated primary human keratinocytes (PHKs) (reviewed by Fuchs 1990; Chow and Broker 1997), or with certain immortalized epithelial cell lines (Lambert et al. 2005). In these systems, epithelial cells are supported on a dermal equivalent consisting of rat tail type 1 collagen and fibroblasts and are held for 10 to 21 d at the air–medium interface, where the drying effects of the air help promote squamous differentiation. Using such “raft” cultures, the complete HPV reproductive program has been recapitulated.
VIRAL GENOME ORGANIZATION
The double-stranded circular DNA genome is approximately 7900 bp long. All genes are encoded along the same template strand (Fig. 2). Transcription regulatory elements and the origin of replication (ori) are situated in the upstream regulatory region (URR) (also called the long control region or LCR). Promoters are located in the URR and within the early regions. The E and L gene blocks are each followed by a polyadenylation site. The usage of different promoters and polyA sites as well as alternative mRNA splicing provides access to each of the ORFs and also enables the fusions of portions of different ORFs. Some messages are bicistronic (Wang et al. 2011; reviewed by Chow and Broker 2007). This organization is conserved among human and animal papillomaviruses except that many cutaneous HPVs do not have an obvious E5 ORF. The functions of the URR and proteins are summarized in Table 1. Briefly, the E proteins function directly or indirectly to support viral DNA maintenance, amplification (E1, E2, E8∧E2C, E1M∧E2C, E5, E6, and E7) and to modulate the host immune responses (E5, E6, and E7). The E1∧E4 fusion protein binds the cytokeratin network and may weaken the cellular envelopes and facilitate egress of virions from the shed squames (Bryan and Brown 2000; Khan et al. 2011). L1 and L2 are the major and minor capsid proteins necessary for encapsidating the viral genome. L2 also facilitates viral genome trafficking into the nucleus (Bienkowska-Haba et al. 2012). At high concentrations, L1 self-assembles into empty virus-like particles (VLPs). VLPs are the immunogens in type-restricted prophylactic vaccines that effectively prevent new infections (reviewed by Schiller and Lowy 2006).
Figure 2.
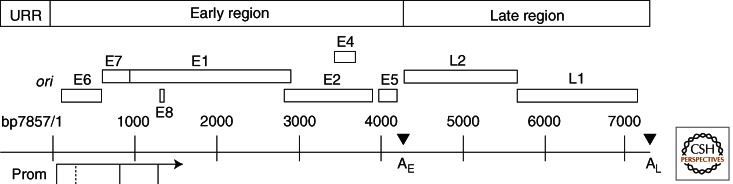
The genome organization of HPV-18. The upstream regulatory region (URR) contains transcription regulatory elements as well as the origin of DNA replication (ori), which overlaps the major promoter preceding E6. Major promoters (Prom) are indicated (solid vertical lines connected to an arrow, which denotes the direction of transcription). Open reading frames (ORFs) are depicted by open boxes. The early (E) and late (L) gene blocks are each followed by a polyadenylation site (AE and AL). This genome organization is largely conserved among HPVs. Some mucosotropic genotypes have one or two E5 ORFs whereas many cutaneous HPVs have no recognizable E5 ORF. The HR HPVs use the E6 promoter to express both E6 and E7 and possibly E1 and E2. Alternative E6 intragenic mRNA splicing allows efficient expression of the E7 protein. The LR HPV types use a separate promoter located in E6 (dotted vertical line) to express E7. Downstream ORFs are accessed by alternative RNA splicing. Transcripts from the promoter located within E7 encode E1, E2, E1M∧E2C (a fusion of the amino-terminal portion of E1 to the middle of E2), E1∧E4 (a fusion of the amino terminus of E1 to E4) and E5, as well as L2 and L1. The minor promoter embedded within E1 generates a short E8 exon, which is spliced to the middle of the E2 ORF, creating E8∧E2C. The expression of late genes from the promoter in E7 occurs only in the superficial squamous cells after viral DNA amplification and requires elongation of transcripts through AE to AL and stabilization of the late 3′ UTR. The functions of encoded proteins and regulatory elements are detailed in Table 1.
Table 1.
Summary of HPV protein functions and regulatory elements
| URR | Upstream regulatory region containing the replication ori, transcription promoters and regulatory protein binding sites. |
| E6 | Inactivation of TP53. HR HPV E6 also up-regulates telomerase and destabilizes TP53 and many PDZ domain-containing host proteins in a complex with E6AP and other ubiquitin ligases. |
| E7 | Destabilization of p130, leading to S phase-entry in differentiated and quiescent cells. HR HPV E7 also destabilizes pRB. |
| E1 | DNA helicase for viral DNA replication. Recruitment of cellular replication proteins to the origin. |
| E2 | Origin binding protein and recruitment of E1 to the ori. Interaction with mitotic apparatus or chromosomes to enable viral genome partitioning during mitosis. Negative regulation of the URR promoter. |
| E4 | Association with cytokeratins. It might weaken the cellular envelope, facilitating virion release. |
| E5 | Membrane protein, enhancing signal transduction of receptor tyrosine kinases. |
| L2 | Minor capsid protein, essential for DNA packaging. |
| L1 | Major capsid protein. It self-assembles into virus-like particles, antigens for prophylactic vaccines. |
| E5, E6, E7 | Down-regulation of host immune surveillance. |
| HR E6, E7 | Immortalization of primary human keratinocytes in vitro. Essential to maintain transformed cell phenotypes. |
THE FUNCTIONS OF E1 AND E2 PROTEINS IN VIRAL DNA REPLICATION
Replication requires the viral ori, the ori binding protein E2, the E1 replicative DNA helicase, and host replication proteins (reviewed by Chow and Broker 2006). The ori consists of a cluster of three E2 binding sites (BSs) flanking a series of overlapping E1 BSs. Three E2 dimers bind to the ori to form a toroidal ring, presumably partially denaturing the AT-rich E1 BSs in the negatively supercoiled DNA (Sim et al. 2008). E2 interacts with E1 and recruits it to ori. E1 assembles into a pair of hexameric rings (Liu et al. 1998; reviewed by Schuck and Stenlund 2005) while E2 is being released (Abbate et al. 2004). In the presence of topoisomerase I, RPA, and ATP, the E1 dihexamer is potent bidirectional helicase on a supercoiled plasmid (Lin et al. 2002). It is required throughout replication initiation and elongation (Liu et al. 1995), as it also recruits the host DNA polymerase α and the single-stranded DNA binding protein RPA to initiate and sustain replication.
Being a highly active DNA helicase with low sequence specificity, the E1 protein must be tightly controlled to avoid unintended DNA strand separation or damage (Fradet-Turcotte et al. 2011; Sakakibara et al. 2011), and this is achieved by a cell-cycle regulated E1 nuclear localization. Near the E1 protein amino terminus is a localization regulatory region (LRR), which controls reversible nucleo-cytoplasmic shuttling. Efficient nuclear entry of HPV-11 E1 requires a bipartite, positively charged nuclear localization sequence (NLS) as well as phosphorylation by mitogen-activated protein kinases (MAPKs), primarily ERK1/2, on serine residue(s) within the LRR (Yu et al. 2007). MAP kinase docking motifs are located in the carboxyl-terminal helicase domain. The default position of HPV-11 E1 in the cytoplasm is attributable to a dominant nuclear export sequence (NES) and the nuclear exportin Crm1. The NES is inactivated by cyclin-dependent kinases (cdks) that phosphorylate a serine residue within NES (Deng et al. 2004). Indeed, cyclin E/cdk2 is required for efficient amplification of HPV-11 ori plasmids (Ma et al. 1999; Lin et al. 2000). Similarly, cdk2 also regulates the nucleo-cytoplasmic location of HPV-31 E1 and is required for genome maintenance (Fradet-Turcotte et al. 2010). LRR sequences and the MAPK docking motifs are conserved among human and animal PVs. In light of the crucial role of MAP kinases, the ability of the HPV E5 protein to enhance signal transduction from receptor tyrosine kinases (Pedroza-Saavedra et al. 2010; Suprynowicz et al. 2010; Belleudi et al. 2011) that function upstream of MEK1/2 and ERK1/2 would predict that it plays an important role in viral DNA amplification, and indeed it does in organotypic cultures (J-H Yu, TR Broker, and LT Chow, unpubl.).
The E2 protein is additionally responsible for maintenance of the plasmid in dividing cells to ensure equitable partitioning by associating with the mitotic apparatus or with mitotic chromosomes (Van Tine et al. 2004a; Dao et al. 2006; Parish et al. 2006; reviewed by Kadaja et al. 2009). Two fusion proteins E1M∧E2C (Chiang et al. 1991, 1992) and E8∧E2C (Zobel et al. 2003; Lace et al. 2008), each with an alternative amino terminus from the full-length E2 while retaining the DNA binding and protein dimerization domain, negatively control viral DNA amplification. The E2BS are immediately adjacent to the URR promoter, which controls E6 and E7 expression, and binding of the E2 or the E2-related proteins suppresses the URR promoter activity, constituting a feedback regulation.
THE INDUCTION OF S-PHASE REENTRY IN THE DIFFERENTIATED KERATINOCYTES BY E7
The tumor suppressor protein that confers susceptibility to retinoblastomas (pRB) is the gatekeeper of the G1 to S-phase transition during the cell cycle. It is inactivated via sequential phosphorylation by cyclin D/cdk4 or cyclin D/cdk6 and by cyclin E/cdk2 to initiate transcription activation of a large suite of E2F-dependent genes, thus promoting cell cycle entry and progression (Sherr and Roberts 2004). The HR but not the LR HPV E7 proteins effectively destabilize the under-phosphorylated form of pRB (McLaughlin-Drubin and Münger 2009; Moody and Laimins 2010), bypassing the need for cyclin D/cdk4 or cyclin D/cdk6.
However, in normal squamous epithelia, p130, rather than pRB, is principally responsible for maintaining quiescence of basal cells and the homeostasis of the postmitotic differentiated cells. In contrast, pRB is primarily detected in actively cycling basal and TA cells (Genovese et al. 2008). In organotypic cultures of PHKs acutely transduced with a retrovirus expressing E7 of HPV-6, -11, -16, -18, or the plantar wart virus HPV-1, suprabasal S-phase reentry is induced stochastically, and HR HPV E7 proteins are more effective than the nononcogenic HPV E7 (Banerjee et al. 2006; Genovese et al. 2008, 2011). This is because E7 proteins are able to destabilize p130 to different avidity (Zhang et al. 2006; Genovese et al. 2008, 2011).
The E7 protein binds to the pRB family of pocket proteins via the LxCxE motif in the conserved region 2 (CR2), which is also present in adenovirus E1A protein and SV40/PY large T-antigen. Phosphorylation of E7 by casein kinase II (CKII) on one or two serine residues that closely follow the LxCxE motif increases its affinity for the pocket proteins and is critical for efficient p130 destabilization and for S-phase reentry (Chien et al. 2000; Genovese et al. 2008). Remarkably, mutations of just three amino acid residues that are within or downstream of CR2 in the LR HPV-11 E7 can increase its affinity for the pocket proteins and its ability to induce S-phase reentry to the levels achieved by the HR HPV E7 proteins (Genovese et al. 2011).
THE PROPERTIES OF THE HR HPV E6 ONCOPROTEIN
The best-known property of the HR HPV E6 protein is TP53 degradation via the proteasome pathway. E6 and a host protein E6AP function as a ubiquitin ligase of p53. In complexes with E6AP or other ubiquitin ligases, the HR HPV E6 also destabilizes many PDZ domain-containing host proteins that regulate signal transduction, proliferation, cell–cell communication, cell polarity, and differentiation (reviewed by Thomas et al. 2008 and by Howie et al. 2009). Moreover, E6 activates transcription of the human telomerase catalytic subunit (hTERT) (Klingelhutz et al. 1996). In contrast, the LR HPV E6 proteins do not have these activities, although both the HR and LR HPV E6 proteins can inactivate the transcription activation activity of TP53 (Thomas and Chiang 2005). These major distinctions in E6 and E7 protein properties between the HR and LR HPV genotypes largely account for their differential oncogenic potentials in vivo. In vitro, only the HR HPV E6 and E7 can immortalize primary human keratinocytes (Halbert et al. 1992, and references therein).
Unique to the HR HPVs, the great majority of the E6 transcripts undergo intragenic or intergenic alternative splicing. The spliced mRNAs encode truncated E6* peptides that lack carboxy-terminal residues can still destabilize certain PDZ proteins but not TP53 (Pim et al. 2009). The intragenic mRNA splice is thought to facilitate translation of the downstream E7, E1, and E2 ORFs (Hubert and Laimins 2002; Tang et al. 2006). The reduction of HR HPV E6 protein by RNA splicing would also ensure the presence of a certain level of TP53 and other targeted host proteins needed to maintain host genome stability or for some unknown aspect of the viral life cycle. For the LR HPVs, a promoter located within the E6 gene is responsible for E7 transcription (DiLorenzo and Steinberg 1995) (Fig. 2), negating the need for intragenic RNA splicing.
RECAPITULATION OF THE VIRAL PRODUCTIVE PROGRAM IN ORGANOTYPIC CULTURES OF EPITHELIAL CELL LINES
The productive phase of HPV infections was initially recapitulated in organotypic raft cultures of explanted HPV-11 infected human epithelial tissue (Dollard et al. 1992) and in TPA-treated raft cultures of an HPV-31 plasmid-containing epithelial cell line derived from a dysplasia (Meyers et al. 1992). For genetic analyses in the context of an autonomously replicating viral genome in the raft cultures, HR HPV DNA is excised from recombinant plasmids and used to transfect and immortalize PHKs or, alternatively, to transfect an already immortalized epithelial cell line (reviewed by Lambert et al. 2005; Wilson and Laimins 2005). However, raft cultures of immortalized cells usually do not support a highly productive viral program, thus compromising the analyses. Moreover, many functionally significant HR HPV E6 mutants cannot be studied by using this approach because they cannot immortalize PHKs and are not stably maintained as plasmids in transfected cells (Park and Androphy 2002; Lee et al. 2007, and references therein).
PSEUDOVIRIONS
As an alternative to raft cultures, high titers of pseudovirions can be produced in human 293 TT cells (Buck et al. 2004; Pyeon et al. 2005). The cells are cotransfected with an L1 and L2 expression vector and a reporter plasmid or HPV DNA excised from a recombinant plasmid. Packaging of plasmid DNA of 8 kbp or shorter is not sequence-specific. However, pseudovirions have low infectivity in PHKs and have not been used to study the virus life cycle. In contrast, pseudovirions containing a reporter gene are used extensively to elucidate the initial virus–host cell interactions in the infection processes in cell lines and in a mouse genital tract model (Roberts et al. 2007; reviewed by Sapp and Bienkowska-Haba 2009; Schiller et al. 2010). They also have been highly instrumental in developing and validating prophylactic HPV vaccines and candidates (Jagu et al. 2009; Lin et al. 2010).
HPV PRODUCTION IN ORGANOTYPIC CULTURES OF PRIMARY HUMAN KERATINOCYTES
Robust production of infectious HPV-18 virus has recently been achieved in raft cultures of PHKs from neonatal foreskin (Chow et al. 2009; Wang et al. 2009a). In this system, HPV-18 genomic plasmids are efficiently generated in transfected PHKs via excision from supercoiled recombinant plasmids using Cre-LoxP site-specific recombination. 20%–30% of the cells survive the 4-day selection for the drug-resistance gene expressed from the residual vector, and there is no need for selection based on the immortalization functions of the HR HPVs. Within a week of transfection, the cells are used to develop raft cultures. The highly productive viral life cycle resembles the infected tissue xenografts in athymic mice (Stoler et al. 1990), as neither has the adaptive immune system to curb the viral activities. The abundant yield of virus particles recovered elicited productive infection of naïve PHKs in raft cultures.
INITIATION OF VIRAL DNA AMPLIFICATION IN G2 PHASE
The high HPV-18 productivity in PHK raft cultures affords an opportunity to analyze the viral life cycle in detail (Wang et al. 2009a). Unexpectedly, host DNA replication and viral DNA amplification are temporally separate events. In situ assays showed that host chromosomal DNA replicates first in the spinous cells, as denoted by BrdU incorporation and the nuclear S-phase cyclin A. In contrast, viral DNA amplification initiates during a protracted G2 phase, as revealed by elevated cytoplasmic G2 cyclin B1 (Fig. 3). Pulse-chase experiments have verified this time course. As the viral DNA amplifies, the E7 activity is reduced and eventually extinguished such that p130 reappears and cells exit the cell cycle. The infectious program then switches to the late phase in which capsid proteins are synthesized and progeny virions are assembled in the superficial keratinocytes and finally mature as these cells die.
Figure 3.

The initiation of viral DNA amplification in G2 phase cells. A formalin-fixed, paraffin-embedded section of an HPV-18 containing PHK raft culture held at the air–medium interface for 12 days was simultaneously probed for viral DNA (red) by in situ hybridization and cyclin B1 protein (green) with an antibody. Low viral DNA signal was detected in cells with cytoplasmic G2 cyclin B1. Cells with high viral DNA signal were negative for cyclin B1 (and cyclin A, not shown) as these cells have already exit the cell cycle. (Adapted from Wang et al. 2009a.)
Interestingly, E7 alone is responsible for the prolonged G2 phase by inducing or activating host proteins typically associated with the DNA damage response (DDR) (Banerjee et al. 2011). Briefly, cdc2 is elevated but inactivated following hyperphosphorylation by Wee1 and Myt1 kinases and is sequestered in the cytoplasm. Moreover, the cdc25 phosphatase, which activates cdc2, is itself phosphorylated and inactivated by activated ATM, Chk1, Chk2, or JNK, also accumulates in the cytoplasm (Fig. 4). The ATM DDR was also reported in cells harboring HPV-31 plasmids (Moody and Laimins 2009).
Figure 4.

The activities of E7 in differentiated keratinocytes. The expression of E7 in the differentiated keratinocytes destabilizes p130, leading to S-phase reentry. Wee1 and Myt 1 kinases are also activated, hyperphosphorylating and inactivating cdc2. Moreover, E7 further induces a DNA damage repair (DDR)-like response in that ATM, Chk1, Chk2, and JNK are each activated, resulting in the inactivation of the cdc25C phosphatase, disabling it from removing the inhibitory phosphates from cdc2. The inactive cdc2 and cyclin B1 accumulate in the cytoplasm in a prolonged G2 phase during which viral DNA amplifies (see Fig. 3). However, an antibody to γH2AX detected only sporadic signals (LT Chow and TR Broker, unpubl.), inconsistent with widespread DNA damage.
HPV-16 E7 also activates the Fanconi anemia (FA) pathway involved in DNA repair (Spardy et al. 2007). FA patients with a mutated component gene are prone to developing squamous cell carcinomas at multiple body sites. In transgenic mice in which HPV-16 E7 expression was targeted to the basal cells, FancD2 knockout increased the frequency of head and neck cancers relative to the control mice with wild-type FancD2 (Park et al. 2010). In organotypic cultures of HPV-16 E6/E7 immortalized keratinocytes, knockdown of a FA component leads to hyperplasia, consistent with a role of the FA pathway in HPV pathogenesis (Hoskins et al. 2009).
THE ROLE OF E6 IN THE PRODUCTIVE LIFE CYCLE
In PHK raft cultures, the expression of the HR HPV E6 alone does not promote S-phase reentry in differentiated cells, whereas expression of E7 alone induces S-phase reentry and TP53 stabilization, via phosphorylation by kinases activated by E7 just described. When E6 and E7 are coexpressed, TP53 is abolished (Jian et al. 1998), consistent with known biochemical properties of the HR HPV E6 protein. The Cre-loxP excisional recombination system to generate HPV-18 whole genomic plasmids enabled the examination of an HPV-18 E6*I mutant not previously possible. In this mutant, the E6 gene is deleted of the E6*I intron coding sequence and hence only expresses the truncated E6*I peptide because of premature translation determination after the splice. In PHK raft cultures, the mutant DNA amplification is greatly reduced and L1 is not expressed. Notably, elevated TP53 accumulates for prolonged periods of time in numerous basal and suprabasal cells without inducing apoptosis (Wang et al. 2009a). The E6 mutant could be partially complemented in trans by a retrovirus delivering HPV-18 URR-E6, increasing viral DNA amplification and L1 expression, whereas TP53 protein was no longer detected (EY Kho, HK Wang, NS Banerjee, TR Broker, and LT Chow, unpubl.). These observations support the hypothesis that TP53 is an E6 target during the viral life cycle. Ectopic TP53 inhibits transient viral ori-dependent replication in transfected cells, mediated by TP53 interacting with the E2 protein (Lepik et al. 1998; Massimi et al. 1999). Detailed examination of missense E6 mutants is in progress to verify this conclusion and to identify additional E6 targets critical for the productive cycle.
MODULATION OF THE PRODUCTIVE PROGRAM BY p21CIP1 AND p27KIP1
Although the HPV E7 protein reestablishes an S-phase potential in practically all the differentiated cells, as suggested by the general loss of p130 and the widespread induction of the proliferating cell nuclear antigen (PCNA), only a fraction of these cells reenters S phase. This outcome appears to be determined by the levels of p27kip1, an inhibitor of cdks (CKI) (Fig. 2) (Noya et al. 2001; Chien et al. 2002). Briefly, in suprabasal cells of normal PHK raft cultures, p27kip1 is variably but stably expressed, whereas the related p21cip1 is constitutively expressed, but is quickly degraded by proteasomes. When E7 was expressed, suprabasal cells that reentered S phase were negative for p27kip1, p21cip1, and cyclin E, whereas cells that did not reenter S phase were positive for all three. We suggest that, if the differentiated cells contain low levels of p27kip1, E7 expression induces S-phase genes, including cyclin E and cdk2, and p27kip1 is phosphorylated and degraded, allowing the cells to enter into S phase. The normal feedback regulation then prevents further expression of cyclin E. If, however, the p27kip1 protein is already high, it would bind to and inhibit cyclin E/cdk2, preventing S-phase entry. Cyclin E continues to be expressed and is then costabilized in an inactive complex with p21cip1/cdk2, unable to reenter S phase. This reciprocal relationship between p21cip1 stabilization and HPV-11 DNA amplification was observed in RRP patient specimens (Schmidt-Grimminger et al. 1998; Jian et al. 1999). Thus, the pair of CKIs appears to serve as a defense against viral DNA amplification in a fraction of the infected cells.
Nonetheless, in a study of benign genital lesions, this relationship was not consistently observed in cells with high HR or LR HPV DNA (Zehbe et al. 1999). Similarly, in the highly productive HPV-18 PHK raft cultures, some cells with amplified viral DNA were positive for p21cip1 (AA Duffy, HK Wang, TR Broker, and LT Chow, unpubl.). One interpretation would have CKI accumulation occurring following viral DNA amplification, as it did following host DNA replication in E7-transduced raft cultures (Chien et al. 2002). Alternatively, HR but not the LR HPV E7 can overcome the inhibitory effects of CKI (Jones et al. 1997; Helt et al. 2002; Shin et al. 2009). This issue of host defense deserves further investigation.
ATTRIBUTES OF HR HPV IN CANCERS AND MODEL SYSTEMS
The trade-off for restricting the HPV productive phase to postmitotic differentiated cells is the occasional development of cancers that no longer support progeny virus production. Cancers caused by HR HPVs have several attributes. First, they harbor wild-type TP53 and pRB but are phenotypically negative for the encoded proteins. TP53 mutations are rare in cervical cancers. Thus, cancers that are frequently associated with mutated TP53 (and therefore with more stable and elevated TP53) are not likely to be caused by HR HPVs. Second, HPV DNA is often, but not always, integrated into host chromosomes. Third, the HR HPV E6 and E7 mRNAs are consistently expressed at elevated levels. Experimentally, reenabling the pRB or TP53 pathways in cancer cell lines by repressing the viral E7 or E6 expression leads to senescence or apoptosis, underscoring the addiction to the viral oncoproteins for sustaining the transformed phenotypes (DeFilippis et al. 2003; Wells et al. 2003).
The initiation of viral carcinogenesis is attributable to repeated dysregulation of the viral oncogenes in the basal reserve or stem cells when the tissue is subjected to cycles of wounding and healing, events known to activate viral early genes. Diminished immune capability is also a contributing factor. The loss of pRB and TP53 promotes excessive cell cycling while increasing the probability of the cell acquiring mutations and incurring chromosome instability (Fig. 1). Detailed mechanisms have been expertly reviewed recently (Howie et al. 2009; Moody and Laimins 2010; Korzeniewski et al. 2011) and will not be repeated here.
HPV-associated cancer development usually takes years or decades until the reserve or stem cells have accumulated combinations of genetic and epigenetic changes to become transformed. Gain or loss of host chromosomal loci has been reported (Wilting et al. 2009, and references therein). Epigenetic changes in certain host genes increase with lesion severity (Wilting et al. 2010; Hesselink et al. 2011; van der Meide et al. 2011). The reprogramming of p16INK4a and Hox has been attributed to viral oncoproteins (McLaughlin-Drubin et al. 2011). In fact, the accumulation of p16INK4a is one of the validated biomarkers for HPV-induced high-grade dysplasias and cancers (Roelens et al. 2012) along with S-phase biomarkers such as Ki-67, MCM7, and PCNA in the suprabasal cells of a squamous epithelium. Furthermore, HPV oncoproteins also affect the expression of oncogenic or tumor suppressive miRNAs (Martinez et al. 2008; Wang et al. 2008, 2009b; Wald et al. 2011). These viral activities very likely contribute to the productive life cycle or to viral carcinogenesis. It remains to be seen whether some of these events might be potential biomarkers predictive for risk of progression to cancers.
HR HPV E6/E7 immortalized cells are excellent models to characterize molecular and phenotypic changes associated with progression of lesions in vivo. In organotypic cultures, the immortalized cells resemble various grades of dysplasias. Upon extended cell passage, the raft cultures show increasingly dysplastic histology, consistent with additional mutations in host genes (Hurlin et al. 1991; Merrick et al. 1992; Steenbergen et al. 1998). For example, immortalized cells that acquire anchorage-independent growth have up-regulated PIK3CA, which is upstream in the AKT pathway. Indeed, up-regulated PIK3CA is consistently observed in cervical cancers (Henken et al. 2011, and references therein).
Transgenic mice models for cervical cancer, head and neck cancer, and anal cancers have been established by targeting the expression of HPV-16 E7, E6, or E5 alone or in combination to basal cells of the epithelia. Long-term exposure to estrogen is required. These studies have verified the importance of destabilization of pRB, TP53, and PDZ domain proteins in cancer development (Chung et al. 2010; Jabbar et al. 2010; Maufort et al. 2010; Stelzer et al. 2010, and references therein).
INTEGRATION OF HPV DNA IN CERVICAL CANCERS AND CLONAL SELECTION OF CANCER CELLS
In cervical cancers and cell lines derived therefrom, as well as in many HPV DNA-immortalized cell lines, viral DNA integration and clonal expansion appear to be important early events (Klaes et al. 1999; Park et al. 2003; Hopman et al. 2004; Melsheimer et al. 2004; Pett et al. 2004; Vinokurova et al. 2008). The HPV DNA is integrated as single copies or as tandem repetitions, and it along with flanking chromosomal DNA can be translocated to multiple sites (Macville et al. 1999). Integration appears to favor the open chromatin of expressed host genes (Kraus et al. 2008). The viral DNA breakage and integration site invariably occurs within the E1 or E2 genes. There is no sequence specificity. One predominant E6/E7-host chimeric transcript is derived from the viral copy located at the downstream integration junction, whereas all upstream copies are silenced by DNA methylation, resulting in the loss of expression from all other viral genes (Wentzensen et al. 2002; Van Tine et al. 2004b). Consequently, viral DNA methylation is high in carcinomas and cell lines thereof and low in benign lesions (Kalantari et al. 2004; Brandsma et al. 2009; Ding et al. 2009; Vinokurova and von Knebel Doeberitz 2011). The host 3′ untranslated region (UTR) and polyA site in the chimeric E6/E7 transcript may confer increased stability (Fig. 5). Moreover, the loss of E2 expression eliminates the negative feedback regulation of the URR promoter, elevating the expression of E6 and E7 (Broker et al. 1989). Collectively, these mechanisms would provide the cells with a growth advantage (Jeon and Lambert 1995; Jeon et al. 1995).
Figure 5.
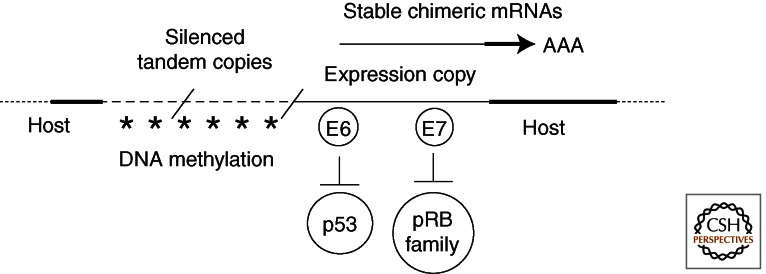
Epigenetic regulation of HPV transcription in a clonally selected cancer cell with tandemly integrated viral DNA. Thick dotted lines represent host sequences. Thin dashed lines represent HPV genomes integrated in tandem arrays. The predominant viral transcripts are chimeric. They originate from the URR promoter of the downstream integration junction copy, which is disrupted in E1 or E2. The E6/E7 transcripts have 3′ UTR and polyA site sequence derived from the host. All upstream viral DNA copies are silenced by DNA methylation (Wentzensen et al. 2002; Van Tine et al. 2004b) such that other viral genes are no longer expressed.
THE POTENTIAL ROLES OF HPV DNA REPLICATION, INTEGRATION, AND E1 AND E2 PROTEINS IN ONCOGENESIS
What leads to the integration of the viral DNA is a matter of conjecture and ongoing research. Nonhomologous recombination between a broken viral DNA and the host chromosome would result in integration. As discussed, E2 proteins of mucosotropic HPVs associate with mitotic apparatus or mitotic chromosomes. The tight association of E2 to ori in the integrated HPV genomes might then act as a viro-centromere (Van Tine et al. 2004a), which, in combination with the host chromosome centromere, could establish a multicentric chromosome, thereafter initiating rounds of the breakage-fusion-bridge cycle (McClintock 1951). In cells with integrated viral genomes, continued synthesis of E1 and E2 proteins would also lead to repeated reinitiation of replication from the viral origin to form “onion-skin” replication bubbles, making the host chromosome highly vulnerable to breakage. Newly replicated viral DNA fragments could integrate into additional chromosomal sites, increasing host genome instability (reviewed by Kadaja et al. 2009). To avoid mitotic catastrophe, the expression of E2 protein from intact copies of the gene must cease via epigenetic silencing in the tumor cell that eventually emerges. The absence of E2 and E2-related proteins would deregulate the viral oncogene expression, further driving the process of oncogenesis, as already discussed.
DOWN-REGULATION OF HOST IMMUNE SURVEILLANCE BY HPV ONCOPROTEINS
Virus production is usually sporadic and low, unless the individual is compromised in their immune systems such as during pregnancy, in patients who are taking immunosuppressive drugs, in cancer patients undergoing chemotherapy and radiation, and in patients with HIV-AIDS. Minimizing gene expression in cycling cells and delaying elevated protein expression to the mid to upper differentiated strata are viral strategies to avoid detection by the host immune systems. In addition, HPVs have developed elaborate schemes to undermine host immune surveillance (reviewed by Bhat et al. 2011). The E6 and E7 proteins affect both innate and adaptive immunity whereas the E5 protein down-regulates class I and II HLA molecules. Collectively, these strategies allow HPVs and host cells to establish a benign co-existence. In the long run, the virus spreads and casts a wider net than otherwise possible. In cancers, the HR HPV E6 and E7 genes are expressed at elevated levels. Thus, the ability to impair the host immune responses exacerbates viral carcinogenesis and additionally presents a challenge to the development of therapeutic vaccines that target the viral oncoproteins (reviewed by Su et al. 2010).
CONCLUDING REMARKS
The inability to propagate HPVs in conventional cell cultures has been a blessing in disguise. It forced investigators to develop model systems that recapitulate the spectrum of patient lesions. The viral oncoproteins have been invaluable tools to probe the functions of the two major host tumor suppressor proteins, pRB and TP53. The HPV system continues to provide fertile opportunities for investigations of chromosome dynamics, cellular and molecular responses to DNA damage, signal transduction, host immune response, and cancer biology.
Within 30 years since the cloning of the HR HPV genomes from cervical cancers, intensive basic research made tremendous progress in prevention and diagnosis. The development and licensing of highly effective prophylactic vaccines against the oncogenic HPV-16 and HPV-18 (Gardasil, Cervarix), as well as against nononcogenic HPV-6 and HPV-11 (Gardasil), is the biggest recent advancement that impacts on the public health (Schiller and Lowy 2006). Less expensive and cross-reactive vaccines are being developed (Jagu et al. 2009; Lin et al. 2010). Probe technologies for HPV genotypes and RNA transcripts are leading the transition from cytology to molecular-based screening. Identification of protein biomarkers for staging of HPV lesions is ongoing and will be important for guiding optimal treatments. Looking ahead, a thorough investigation of the functions of the viral proteins during productive infections and elucidation of the mechanisms of their regulation remain as high priorities. These studies will lead to the identification of therapeutic targets for pharmacologic treatment of active papillomavirus infections that, despite concerted efforts at prevention, will continue to occur globally for decades to come.
ACKNOWLEDGMENTS
The authors are supported by USPHS/NIH/National Cancer Institute grants CA36200, CA83679, and CA107338 and gratefully acknowledge the invaluable contributions of our students, fellows, and staff members as well as many collaborators. We thank Carolyn S. Ashworth, MD and the nurses in the Newborn Nursery at UAB Hospital for collecting foreskins from elective circumcision that are critical for our research. We regret a less than comprehensive description and citation of the numerous relevant publications by colleagues because of page limits.
Footnotes
Editors: Stephen D. Bell, Marcel Méchali, and Melvin L. DePamphilis
Additional Perspectives on DNA Replication available at www.cshperspectives.org
REFERENCES
- Abbate EA, Berger JM, Botchan MR 2004. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev 18: 1981–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee NS, Genovese N, Noya F, Chien W-M, Broker TR, Chow LT 2006. Inducible E7 proteins of high-risk and low-risk HPVs promote S phase re-entry in post-mitotic differentiated keratinocytes in organotypic cultures. J Virol 80: 6517–6524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee NS, Wang H-K, Broker TR, Chow LT 2011. HPV E7 protein induces prolonged G2 following S-phase reentry in differentiated human keratinocytes. J Biol Chem 286: 11473–11482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belleudi F, Leone L, Purpura V, Cannella F, Scrofani C, Torrisi MR 2011. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 30: 4963–4876 [DOI] [PubMed] [Google Scholar]
- Bernard H-U 2002. Gene expression of genital human papillomaviruses and considerations on potential antiviral approaches. Antiviral Ther 7: 219–237 [PubMed] [Google Scholar]
- Bhat P, Mattarollo SR, Gosmann C, Frazer IH, Leggatt GR 2011. Regulation of immune responses to HPV infection and during HPV-directed immunotherapy. Immunol Rev 239: 85–98 [DOI] [PubMed] [Google Scholar]
- Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86: 9875–9887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK 3rd, Martel M, Harigopal M, Schofield K, Neapolitano M 2009. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology 389: 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo IG, de Sanjose S, Gottschling M 2010. The clinical importance of understanding the evolution of papillomaviruses. Trends Microbiol 18: 432–438 [DOI] [PubMed] [Google Scholar]
- Broker TR, Chow LT, Chin MT, Rhodes CR, Wolinsky SM, Whitbeck A, Stoler MH 1989. A molecular portrait of human papillomavirus carcinogenesis. Cancer Cells 7: 197–208 [Google Scholar]
- Bryan JT, Brown DR 2000. Association of the human papillomavirus type 11 E1∧E4 protein with cornified cell envelopes derived from infected genital epithelium. Virology 277: 262–269 [DOI] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, Schiller JT 2004. Efficient intracellular assembly of papillomaviral vectors. J Virol 78: 751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-M, Broker TR, Chow LT 1991. An E1M∧E2C fusion protein encoded by human papillomavirus type 11 is a sequence-specific transcription repressor. J Virol 65: 3317–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C-M, Dong G, Broker TR, Chow LT 1992. Control of human papillomavirus type 11 origin replication by the E2 family of transcriptional regulatory proteins. J Virol 66: 5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien W-M, Parker JN, Schmidt-Grimminger D-C, Broker TR, Chow LT 2000. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S phase entry. Cell Growth Diff 11: 425–435 [PubMed] [Google Scholar]
- Chien W-M, Noya F, Benedict-Hamilton HM, Broker TR, Chow LT 2002. Alternative fates of keratinocytes transduced by human papillomavirus type 18 E7 during squamous differentiation. J Virol 76: 2964–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LT, Broker TR 1997. In vitro experimental systems for HPV: Epithelial raft cultures for viral reproduction and pathogenesis and for genetic analyses of viral proteins and regulatory sequences. Clin Dermatol 15: 217–227 [DOI] [PubMed] [Google Scholar]
- Chow LT, Broker TR 2006. Mechanisms and regulation of papillomavirus DNA replication. In Recent advances in papillomavirus research (ed. Campo MS), pp. 53–71 Caister Academic, Norfolk, UK [Google Scholar]
- Chow LT, Broker TR 2007. Human papillomavirus RNA transcription. In Papillomaviruses (ed. Garcea RL, DiMaio D), pp. 109–144 Springer, New York [Google Scholar]
- Chow LT, Duffy AA, Wang H-K, Broker TR 2009. A highly efficient system to produce infectious human papillomavirus: Elucidation of natural virus-host interactions. Cell Cycle 8: 1319–1323 [DOI] [PubMed] [Google Scholar]
- Chow LT, Broker TR, Steinberg BM 2010. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 118: 422–449 [DOI] [PubMed] [Google Scholar]
- Chung SH, Franceschi S, Lambert PF 2010. Estrogen and ERα: Culprits in cervical cancer? Trends Endocrinol Metab 21: 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao LD, Duffy AA, Van Tine BA, Wu S-Y, Chiang C-M, Broker TR, Chow LT 2006. Dynamic localization of the HPV-11 origin binding protein E2 during mitosis while in association with the spindle apparatus. J Virol 80: 4792–4800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis RA, Goodwin EC, Wu L, DiMaio D 2003. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 77: 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Lin BY, Jin G, Wheeler C, Ma T, Harper JW, Broker TR, Chow LT 2004. Cyclin/CDK regulates the nucleo-cytoplasmic localization of the human papillomavirus E1 DNA helicase. J Virol 78: 13954–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkay CS, Wiatrak B 2008. Recurrent respiratory papillomatosis: A review. Laryngoscope 118: 1236–1247 [DOI] [PubMed] [Google Scholar]
- de Villiers E-M, Fauquet C, Broker TR, Bernard H-U, zur Hausen H 2004. Classification of papillomaviruses. Virology 324: 17–27 [DOI] [PubMed] [Google Scholar]
- DiLorenzo TP, Steinberg BM 1995. Differential regulation of human papillomavirus type 6 and 11 early promoters in cultured cells derived from laryngeal papillomas. J Virol 69: 6865–6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding DC, Chiang MH, Lai HC, Hsiung CA, Hsieh CY, Chu TY 2009. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur J Obstet Gynecol Reprod Biol 147: 215–220 [DOI] [PubMed] [Google Scholar]
- Dollard SC, Wilson JL, Demeter LM, Bonnez W, Reichman RC, Broker TR, Chow LT 1992. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. Genes Dev. 6: 1131–1142 [DOI] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Moody C, Laimins LA, Archambault J 2010. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J Virol 84: 11747–11760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol 85: 8996–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E 1990. Epidermal differentiation: The bare essentials. J Cell Biol 111: 2807–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese NJ, Banerjee NS, Broker TR, Chow LT 2008. Casein kinase II motif-dependent phosphorylation of HPV E7 protein promotes p130 degradation and S-phase induction in differentiated human keratinocytes. J Virol 82: 4862–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese NJ, Broker TR, Chow LT 2011. Non-conserved lysine residues attenuate the biological function of the low-risk human papillomavirus E7 protein. J. Virol. 85: 5546–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LM, Ma Y, Ho C, McGrath CM, Nelson DB, Parry S 2008. Placental infection with human papillomavirus is associated with spontaneous preterm delivery. Hum Reprod 23: 709–715 [DOI] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA 1992. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol 66: 2125–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helt AM, Funk JO, Galloway DA 2002. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol 76: 10559–10568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henken FE, Banerjee NS, Snijders PJF, Meijer CJLM, De-Castro Arce J, Rösl F, Broker TR, Chow LT, Steenbergen RDM 2011. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Mol Cancer 10: 10.1186/1476-4598-10-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselink AT, Heideman DAM, Steenbergen RDM, Coupé VM, Overmeer RM, Rijkaart D, Berkhof J, Meijer CJLM, Snijders PJF 2011. Combined promoter methylation analysis of CADM1 and MAL: An objective triage tool for high-risk human papillomavirus DNA-positive women. Clin Cancer Res 17: 2459–2465 [DOI] [PubMed] [Google Scholar]
- Hopman AHN, Smedts F, Dignef W, Ummelen M, Sonke G, Mravunac M, Vooijs GP, Speel EJ, Ramaekers FC 2004. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J Pathol 202: 23–33 [DOI] [PubMed] [Google Scholar]
- Hoskins EE, Morris TA, Higginbotham JM, Spardy N, Cha E, Kelly P, Williams DA, Wikenheiser-Brokamp KA, Duensing S, Wells SI 2009. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 28: 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Katzenellenbogen RA, Galloway DA 2009. Papillomavirus E6 proteins. Virology 384: 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert WG, Laimins LA 2002. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J Virol 76: 2263–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Kaur P, Smith PP, Perez-Reyes N, Blanton RA, McDougall JK 1991. Progression of human papillomavirus type 18-immortalized human keratinocytes to a malignant phenotype. Proc Natl Acad Sci 88: 570–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbar S, Strati K, Shin MK, Pitot HC, Lambert PF 2010. Human papillomavirus type 16 E6 and E7 oncoproteins act synergistically to cause head and neck cancer in mice. Virology 407: 60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagu S, Karanam B, Gambhira R, Chivukula SV, Chaganti RJ, Lowy DR, Schiller JT, Roden RB 2009. Concatenated multitype L2 fusion proteins as candidate prophylactic pan-human papillomavirus vaccines. J Natl Cancer Inst 101: 782–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Lambert PF 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl Acad Sci USA 92: 1654–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Allen-Hoffmann BL, Lambert PF 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 69: 2989–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, Dutta A 2010. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60 dependent transcriptional regulation and apoptotic pathway. Mol Cell 38: 700–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Y, Schmidt-Grimminger D-C, Chien W-M, Wu X, Broker TR, Chow LT 1998. Post-transcriptional induction of p21cip1 protein by HPV E7 in differentiated epithelial cells inhibits reactivated unscheduled DNA synthesis. Oncogene 17: 2027–2038 [DOI] [PubMed] [Google Scholar]
- Jian Y, Van Tine BA, Chien W-M, Shaw GM, Broker TR, Chow LT 1999. Concordant induction of cyclin E and p21cip1 in differentiated keratinocytes by the HPV E7 protein inhibits cellular and viral DNA synthesis. Cell Growth Diff 10: 101–111 [PubMed] [Google Scholar]
- Jones DL, Alani RM, Münger K 1997. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1–mediated inhibition of cdk2. Genes Dev 11: 2101–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaja M, Silla T, Ustav E, Ustav M 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384: 360–368 [DOI] [PubMed] [Google Scholar]
- Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ, Bernard H-U 2004. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol 78: 12762–12772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Davy CE, McIntosh PB, Jackson DJ, Hinz S, Wang Q, Doorbar J 2011. Role of calpain in the formation of human papillomavirus type 16 E1∧E4 amyloid fibers and reorganization of the keratin network. J Virol 85: 9984–9997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF 2003. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J Virol 77: 12450–12459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaes R, Woerner SM, Ridder R, Wentzensen N, Dürst M, Schneider A, Lotz B, Melsheimer P, von Knebel Doeberitz M 1999. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 59: 6132–6136 [PubMed] [Google Scholar]
- Klingelhut AJ, Foster SA, McDougall JK 1996. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 380: 79–82 [DOI] [PubMed] [Google Scholar]
- Korzeniewski N, Spardy N, Duensing A, Duensing S 2011. Genomic instability and cancer: Lessons learned from human papillomaviruses. Cancer Lett 305: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus I, Driesch C, Vinokurova S, Hovig E, Schneider A, von Knebel Doeberitz M, Dürst M 2008. The majority of viral-cellular fusion transcripts in cervical carcinomas cotranscribe cellular sequences of known or predicted genes. Cancer Res 68: 2514–2522 [DOI] [PubMed] [Google Scholar]
- Lace MJ, Anson JR, Thomas GS, Turek LP, Haugen TH 2008. The E8∧E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J Virol 82: 10841–10853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Ozbun MA, Collins A, Holmgren S, Lee D, Nakahara T 2005. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol Med 119: 141–155 [DOI] [PubMed] [Google Scholar]
- Lee C, Wooldridge TR, Laimins LA 2007. Analysis of the roles of E6 binding to E6TP1 and nuclear localization in the human papillomavirus type 31 life cycle. Virology 358: 201–210 [DOI] [PubMed] [Google Scholar]
- Lepik D, Ilves I, Kristjuhan A, Maimets T, Ustav M 1998. p53 protein is a suppressor of papillomavirus DNA amplificational replication. J Virol 72: 6822–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin BY, Ma T, Liu J-S, Kuo S-R, Jin G, Broker TR, Harper JW, Chow LT 2000. HeLa cells are phenotypically limiting in cyclin E/CDK2 for efficient human papillomavirus DNA replication. J Biol Chem 275: 6167–6174 [DOI] [PubMed] [Google Scholar]
- Lin BY, Makhov AM, Griffith JD, Broker TR, Chow LT 2002. Human chaperone proteins abrogate the human papillomavirus E2 protein inhibition of DNA unwinding by the replicative helicase. Mol Cell Biol 22: 6591–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Doolan K, Hung CF, Wu T.-C 2010. Perspectives for preventive and therapeutic HPV vaccines. J Formos Med Assoc 109: 4–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA 2011. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 25: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J-S, Kuo S-R, Broker TR, Chow LT 1995. The functions of human papillomavirus type 11 E1, E2, and E2C proteins in cell-free DNA replication. J Biol Chem 270: 27283–27291 [DOI] [PubMed] [Google Scholar]
- Liu J-S, Kuo S-R, Makhov AM, Cyr DM, Griffith JD, Broker TR, Chow LT 1998. Human Hsp70 and Hsp40 chaperone proteins facilitate HPV-11 E1 protein binding to the origin and stimulate cell-free replication. J Biol Chem 273: 30704–30712 [DOI] [PubMed] [Google Scholar]
- Ma T, Zou N, Lin BY, Chow LT, Harper JW 1999. Interaction between cyclin-dependent kinases and human papillomavirus replication initiation protein E1 is required for efficient viral replication. Proc Natl Acad Sci 96: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macville M, Schröck E, Padilla-Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T 1999. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res 59: 141–150 [PubMed] [Google Scholar]
- Martinez I, Gardiner AS, Board KF, Monzon FA, Edwards RP, Khan SA 2008. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 27: 2575–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimi P, Pim D, Bertoli C, Bouvard V, Banks L 1999. Interaction between the HPV-16 E2 transcriptional activator and p53. Oncogene 18: 7748–7754 [DOI] [PubMed] [Google Scholar]
- Maufort JP, Shai A, Pitot HC, Lambert PF 2010. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res 70: 2924–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B 1951. Chromosome organization and genic expression. Cold Spring Harb Symp Quant Biol 16: 13–47 [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Münger K 2009. Oncogenic activities of human papillomaviruses. Virus Res 143: 195–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Crum CP, Münger K 2011. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci 108: 2130–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melsheimer P, Vinokurova S, Wentzensen N, Bastert G, von Knebel Doeberitz M 2004. DNA aneuploidy and integration of human papillomavirus type 16 E6/E7 oncogenes in intraepithelial neoplasia and invasive squamous cell carcinoma of the cervix uteri. Clin Cancer Res 10: 3059–3063 [DOI] [PubMed] [Google Scholar]
- Merrick DT, Blanton RA, Gown AM, McDougall JK 1992. Altered expression of proliferation and differentiation markers in human papillomavirus 16 and 18 immortalized epithelial cells grown in organotypic culture. Am J Pathol 140: 167–177 [PMC free article] [PubMed] [Google Scholar]
- Meyers C, Frattini MG, Hudson JB, Laimins LA 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257: 971–973 [DOI] [PubMed] [Google Scholar]
- Moody CA, Laimins LA 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5: e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA 2010. Human papillomavirus oncoproteins: Pathways to transformation. Nat Rev Cancer 10: 550–560 [DOI] [PubMed] [Google Scholar]
- Noya F, Chien W-M, Broker TR, Chow LT 2001. p21cip1 degradation in differentiated keratinocytes is abrogated by co-stabilization with cyclin E induced by HPV E7. J Virol 75: 6121–6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish JL, Bean AM, Park RB, Androphy EJ 2006. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol Cell 24: 867–876 [DOI] [PubMed] [Google Scholar]
- Park RB, Androphy EJ 2002. Genetic analysis of high-risk E6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J Virol 76: 11359–11364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TW, Riethdorf S, Schulz G, Riethdorf L, Wright T, Loning T 2003. Clonal expansion and HPV-induced immortalization are early molecular alterations in cervical carcinogenesis. Anticancer Res 23: 155–160 [PubMed] [Google Scholar]
- Park JW, Pitot HC, Strati K, Spardy N, Duensing S, Grompe M, Lambert PF 2010. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res 70: 9959–9968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroza-Saavedra A, Lam EW, Esquivel-Guadarrama F, Gutierrez-Xicotencatl L 2010. The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 400: 44–52 [DOI] [PubMed] [Google Scholar]
- Pett MR, Alazawi WO, Roberts I, Dowen S, Smith DI, Stanley MA, Coleman N 2004. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res 64: 1359–1368 [DOI] [PubMed] [Google Scholar]
- Pim D, Tomaic V, Banks L 2009. The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J Virol 83: 9863–9874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyeon D, Lambert PF, Ahlquist P 2005. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc Natl Acad Sci 102: 9311–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5: e1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JN, Buck CB, Thompson CD, Kines R, Bernardo M, Choyke PL, Lowy DR, Schiller JT 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 13: 857–861 [DOI] [PubMed] [Google Scholar]
- Roelens J, Reuschenbach M, von Knebel Doeberitz M, Wentzensen N, Bergeron C, Arbyn M 2012. p16(INK4a) immunocytochemistry versus human papillomavirus testing for triage of women with minor cytologic abnormalities: A systematic review and meta-analysis. Cancer Cytopathol 10.1002/cncy.21205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara N, Mitra R, McBride AA 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85: 8981–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp M, Bienkowska-Haba M 2009. Viral entry mechanisms: Human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J 276: 7206–7216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller JT, Lowy DR 2006. Prospects for cervical cancer prevention by human papillomavirus vaccination. Cancer Res 66: 10229–10232 [DOI] [PubMed] [Google Scholar]
- Schiller JT, Day PM, Kines RC 2010. Current understanding of the mechanism of HPV infection. Gynecol Oncol 118: S12–S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Grimminger D-C, Wu X, Jian Y, Broker TR, Chow LT 1998. Post-transcriptional induction of p21cip1 in condylomata and dysplasias is inversely related to human papillomavirus activities. Am J Pathol 152: 1015–1024 [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Stenlund A 2005. Assembly of a double hexameric helicase. Mol Cell 20: 377–389 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM 2004. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18: 2699–2711 [DOI] [PubMed] [Google Scholar]
- Shin MK, Balsitis S, Brake T, Lambert PF 2009. Human papillomavirus E7 oncoprotein overrides the tumor suppressor activity of p21Cip1 in cervical carcinogenesis. Cancer Res 69: 5656–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim J, Ozgur S, Lin BY, Yu JH, Broker TR, Chow LT, Griffith JD 2008. Remodeling of the human papillomavirus type 11 replication origin into discrete nucleoprotein particles and looped structures by the E2 protein. J Mol Biol 375: 1165–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S 2007. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol 81: 13265–13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenbergen RDM, Parker JN, Isern S, Walboomers JMM, Meijer CJLM, Snijders PJF, Broker TR, Chow LT 1998. Viral E6/E7 transcription in the basal layers of organotypic cultures without apparent p21cip1 expression precedes immortalization of keratinocytes by transfected HPV16 and HPV18 DNAs. J Virol 72: 749–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer MK, Pitot HC, Liem A, Schweizer J, Mahoney C, Lambert PF 2010. A mouse model for human anal cancer. Cancer Prev Res (Phila) 3: 1534–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler MH, Whitbeck A, Wolinsky SM, Broker TR, Chow LT, Howett MK, Kreider J 1990. Infectious cycle of human papillomavirus type 11 in human foreskin xenografts in nude mice. J Virol 64: 3310–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JH, Wu A, Scotney E, Ma B, Monie A, Hung CF, Wu TC 2010. Immunotherapy for cervical cancer: Research status and clinical potential. BioDrugs 4: 109–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprynowicz FA, Krawczyk E, Hebert JD, Sudarshan SR, Simic V, Kamonjoh CM, Schlegel R 2010. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J Virol 84: 10619–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrjänen S 2010. The role of human papillomavirus infection in head and neck cancers. Ann Oncol 21: vii243–245 [DOI] [PubMed] [Google Scholar]
- Tang S, Tao M, McCoy JP Jr, Zheng ZM 2006. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol 80: 4249–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang C-M 2005. E6 oncoprotein represses p53–dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell 17: 251–264 [DOI] [PubMed] [Google Scholar]
- Thomas M, Narayan N, Pim D, Tomaić V, Massimi P, Nagasaka K, Kranjec C, Gammoh N, Banks L 2008. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27: 7018–7030 [DOI] [PubMed] [Google Scholar]
- Ueda Y, Enomoto T, Miyatake T, Ozaki K, Yoshizaki T, Kanao H, Ueno Y, Nakashima R, Shroyer KR, Murata Y 2004. Monoclonal expansion with integration of high-risk type human papillomaviruses is an initial step for cervical carcinogenesis: Association of clonal status and human papillomavirus infection with clinical outcome in cervical intraepithelial neoplasia. Lab Invest 83: 1517–1527 [DOI] [PubMed] [Google Scholar]
- van der Meide WF, Snellenberg S, Meijer CJLM, Baalbergen A, Helmerhorst TJM, van der Sluis WB, Snijders PJF, Steenbergen RDM 2011. Promoter methylation analysis of WNT/β-catenin signaling pathway regulators to detect adenocarcinoma or its precursor lesion of the cervix. Gynecol Oncol 123: 116–122 [DOI] [PubMed] [Google Scholar]
- Van Tine BA, Dao LD, Wu S-Y, Sonbuchner TM, Lin BY, Zou N, Chiang C-M, Broker TR, Chow LT 2004a. HPV origin binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc Natl Acad Sci 101: 4030–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tine BA, Kappes JC, Banerjee NS, Knops J, Lai L, Steenbergen RDM, Meijer CLJM, Snijders PJF, Chatis P, Broker TR, et al. 2004b. Clonal selection for transcriptionally active viral oncogenes during progression to cancer by DNA methylation-mediated silencing. J Virol 78: 11172–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinokurova S, von Knebel Doeberitz M 2011. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS ONE 6: e24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, Kisseljov F, Dürst M, Schneider A, von Knebel Doeberitz M 2008. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res 68: 307–313 [DOI] [PubMed] [Google Scholar]
- Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJF, Peto J, Meijer CJLM, Muñoz N 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. Pathology 189: 12–19 [DOI] [PubMed] [Google Scholar]
- Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA 2011. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck 33: 504–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM 2008. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE 3: e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-K, Duffy AA, Broker TR, Chow LT 2009a. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev 23: 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang H-K, McCoy JP, Banerjee NS, Rader JS, Broker TR, Meyers C, Chow LT, Zheng ZM 2009b. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 15: 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Meyers C, Wang H-K, Chow LT, Zheng ZM 2011. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J Virol 85: 8080–8092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SI, Aronow BJ, Wise TM, Williams SS, Couget JA, Howley PM 2003. Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc. Natl Acad Sci 100: 7093–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen N, Ridder R, Klaes R, Vinokurova S, Schaefer U, von Knebel Doeberitz M 2002. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 21: 419–426 [DOI] [PubMed] [Google Scholar]
- Wilson R, Laimins LA 2005. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med 119: 157–169 [DOI] [PubMed] [Google Scholar]
- Wilting SM, Smeets SJ, Snijders PJF, van Wieringen WN, van de Wiel MA, Meijer GA, Ylstra B, Leemans CR, Meijer CJLM, Brakenhoff RH, et al. 2009. Genomic profiling identifies common HPV-associated chromosomal alterations in squamous cell carcinomas of cervix and head and neck. BMC Med Genomics 2: 10.1186/1755-8794-2-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting SM, van Boerdonk RAA, Henken FE, Meijer CJLM, Diosdado B, Meijer GA, le Sage C, Agami R, Snijders PJF, Steenbergen RDM 2010. Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol Cancer 9: 10.1186/1476-4598-9-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge TR, Laimins LA 2008. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology 374: 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang C-M 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20: 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Liu Y, Agrawal N, Prasad CK, Edwards JL, Osborne AF, Korourian S, Lowery CL, Hermonat PL 2008. Multiple human papillomavirus types replicate in 3A trophoblasts. Placenta 29: 30–38 [DOI] [PubMed] [Google Scholar]
- Yu J-H, Lin BY, Deng W, Broker TR, Chow LT 2007. Mitogen-activated protein kinases activate the nuclear localization sequence of the human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J Virol 81: 5066–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehbe I, Ratsch A, Alunni-Fabbroni M, Burzlaff A, Bakos E, Dürst M, Wilander E, Tommasino M 1999. Overriding of cyclin-dependent kinase inhibitors by high and low risk human papillomavirus types: Evidence for an in vivo role in cervical lesions. Oncogene 18: 2201–2211 [DOI] [PubMed] [Google Scholar]
- Zhang B, Chen W, Roman A 2006. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci 103: 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Noya F, Chen WY, Townes TM, Chow LT, Broker TR 1999. Trichostatin A up-regulates HPV-11 URR-E6 promoter activity in undifferentiated primary human keratinocytes. J Virol 73: 5026–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel T, Iftner T, Stubenrauch F 2003. The papillomavirus E8-E2C protein represses DNA replication from extrachromosomal origins. Mol Cell Biol 23: 8352–8362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H 2009. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 384: 260–265 [DOI] [PubMed] [Google Scholar]