

Abstract
Tea polyphenols are of great benefit to the treatment of several neurodegenerative diseases. In order to explore the neuroprotective effects of tea polyphenols and their potential mechanisms, an established in vivo subarachnoid hemorrhage (SAH) model was used and alterations of mitochondrial function, ATP content, and cytochrome c (cyt c) in cerebral cortex were detected. This study showed that the alteration of mitochondrial membrane potential was an early event in SAH progression. The trend of ATP production was similar to that of mitochondrial membrane potential, indicating that the lower the mitochondrial membrane potential, lesser the ATP produced. Due to mitochondrial dysfunction, more cyt c was released in the SAH group. Interestingly, the preadministration of tea polyphenols significantly rescued the mitochondrial membrane potential to basal level, as well as the ATP content and the cyt c level in the brain cortex 12 h after SAH. After pretreatment with tea polyphenols, the neurological outcome was also improved. The results provide strong evidence that tea polyphenols enhance neuroprotective effects by inhibiting polarization of mitochondrial membrane potential, increasing ATP content, and blocking cyt c release.
1. Introduction
Polyphenols, the most abundant components in fruits, tea, wine, and vegetables, have attracted attention in recent years due to their healthy effects. It has long been known that tea polyphenols have neuroprotective effects in various pathological states of the nervous system, for example, by lowering the cognitive impairment in most neurodegenerative diseases and by reducing the risk of mortality after stroke [1–3]. Human epidemiological data shows that the risk of a fatal versus nonfatal stroke is significantly reduced by approximately 21% in people that drink 3 cups of tea per day when compared with nontea drinkers. In fact, high consumption of tea, particularly green tea, contributes to the lower rates of cardiovascular disease observed in the Asian population, especially in China, indicating that tea drinking may be a benefiting lifestyle. However, other authors indicate that tea consumption has no correlation with hemorrhagic stroke [4].
Stroke is a leading cause of morbidity and mortality worldwide. Subarachnoid hemorrhage (SAH) accounts for only 5%–10% of all strokes but is a major devastating subtype of stroke affecting 30,000 people in North America yearly [5]. SAH is caused by the rupture of a brain aneurysm and can be divided in two stages, the first 72 h correspond to the early brain injury (EBI) and the period after 72 h corresponds to the delayed vasospasm. About 21% of SAH survivors do not experience delayed vasospasm indicating that EBI may be an important stage predicting the outcome of SAH [6, 7]. In fact, all the factors related to the pathological mechanisms of EBI after SAH may eventually induce irreversible neuronal death, which is associated with neurological deficits and poor outcome [8–10].
A growing body of evidence from animal models and clinical studies indicate that mitochondrial dysfunction is a common event in brain injury, including in SAH [11]. Several lines of research suggest that mitochondrial dysfunction, induced by oxidative stress and inflammation, results in mitochondrial membrane potential (Δψ m) reduction, cytochrome c (cyt c) release, and then activation of mitochondrial-dependent cell death, providing evidence that mitochondrial impairment is deleterious [12, 13]. Therefore, mitochondrial function is a potential therapeutic target [14].
Recent results suggest that inhibition of a signal pathway may not be required for the treatment of SAH [15]. In such a scenario, tea polyphenols may be of special interest due to their multiple functions, such as antioxidants, antimutagenic, iron chelators, and in glutamate release [16]. However, tea polyphenols' effects in the time course after SAH are still unknown. The aim of this study was to investigate whether tea polyphenols have neurological and neurobehavioural effects or not, and the pathway involved in these effects in the in vivo SAH model. This study contributes to a better understanding of the role of tea polyphenols in SAH prevention, which are nontoxic and inexpensive dietary components.
2. Materials and Methods
2.1. Preparation Oxyhemoglobin (OxyHb)
Arterial blood collected from Kunming mouse with heparin was centrifuged at 2,500 ×g for 15 min. The supernatant was discarded. Erythrocytes were washed 3 times with saline solution, lysed with methylbenzene, and centrifuged at 15,000 ×g for 20 min; the supernatant was collected. Using filter membrane, the OxyHb solution was collected, adjusted to 3 μmol/L using measured optical density (OD) at 540 nm and 576 nm, and stored at −80°C [17].
2.2. Animals
The animal use and care protocols were approved by Institutional Animal Care and Use Committee (IACUC) of Xinxiang Medical University. One hundred and eighty adult male Kunming mice weighing from 18 to 20 g were purchased from Xinxiang Medical University. All animals were required to undergo institutional quarantine for 7 days prior to use. The environment for animal housing was equipped with controlled temperature (22 ± 3°C), humidity (40%–70%), and a 12 h light/dark alternation. The mice were divided into three groups: sham group (n = 60), SAH group (n = 60), and preadministration tea polyphenols SAH group (n = 60). Subsequently, each group was subdivided into 3 h, 6 h, 12 h, 24 h, and 72 h subgroup (n = 12), respectively. SAH group was injected with OxyHb, and sham group was given isotonic saline for the same period. Pretreatment group was administered intragastrically with water containing tea polyphenols at a dose of 450 mg/kg/d for 7 days before OxyHb injection [18, 19].
2.3. Mouse SAH Model
SAH was performed using the model reported by Shi et al. [20]. Under general anesthesia with 1% pentobarbital (50 mg/kg, intraperitoneally), animals were placed in prone position. The posterior cervical muscles were dissected through a suboccipital midline skin incision and retracted laterally. The exposed transparent atlantooccipital membrane was penetrated by a 30 gauge needle. Under spontaneous breathing, a 23-gauge needle without point was inserted percutaneously into the skull at a controlling depth of 1.5 mm in the cross position at the sagittal suture 2 mm and sutura coronaria 1 mm. Then, 50 μL (150 μmol/L) of OxyHb was injected through this hole into subarachnoid space. After the neurological assessment, the animals were decapitated at different time points after SAH. Prior to decapitation, one part (n = 6) was perfused through the left cardiac ventricle with isotonic saline, followed by 4% paraformaldehyde in phosphate-buffered saline (PBS). The brain tissue was removed and fixed in 4% paraformaldehyde for 48 h and then embedded in paraffin. Sections of 4 mm thickness were cut using a microtome for histologic studies. The others (n = 6) were sacrificed with 0.9% saline solution perfusion through the left cardiac ventricle. The fresh brain was immediately removed and cortex sections selected and then stored at −80°C.
2.4. Mortality and Neurological Functions Assessment
Recently, the Garcia scoring system has been developed to evaluate the animal neurological behavior and function in a blinded fashion [21]. The results showed that the higher the neurological score, the better the outcome. Briefly, the neurobehavioral examination was performed at 6 h, 12 h, 24 h, and 72 h. An 18-point scoring system was used based on (1) spontaneous activity, (2) symmetry of limb movement, (3) climbing, (4) body proprioception, (5) movement of forelimbs, and (6) response to vibrissae touch (score scale: 0–3 each).
2.5. Lactate Dehydrogenase (LDH) Assay from Brain Cortex
The supernatant of all the samples was collected after homogenate and the LDH content was determined using an LDH assay kit according to the manufacturer's instructions (Nanjing Institute of Jiancheng Biological Engineering, China) [22]. LDH cytotoxicity was calculated using OD as LDH cytotoxicity (U/g protein) = (OD sample − OD blank)/(OD standard solution − OD blank standard solution) × standard solution concentration/sample protein concentration.
2.6. Isolation of Mitochondria from Cerebral Cortex
Intact mitochondria were isolated from fresh brain cortex layer using a tissue mitochondria isolation kit (Beyotime Institute of Biotechnology, China). In brief, after homogenization of 0.5 g cortical tissue in ice-cold MSH buffer (10 mM HEPES, pH 7.5, containing 200 mM mannitol, 70 mM sucrose, 1.0 mM EGTA, and 2.0 mg/mL serum albumin), the homogenate was centrifuged at 1,000 ×g at 4°C for 10 min. The collected supernatant was then centrifuged at 3,500 ×g at 4°C for 10 min to obtain a mitochondrial pellet [23].
2.7. Assay of Mitochondrial Membrane Potential
Changes in mitochondrial membrane potential (Δψ m) were measured using a JC-1 (5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanide iodide) staining (mitochondrial membrane potential assay kit, Beyotime Institute of Biotechnology, China) according to the manufacturer's instructions. Briefly, isolated mitochondria were suspended in 0.5 mL medium containing 5 mM JC-1. Samples were analyzed using an automatic microplate reader (Thermo Scientific, USA) at time scan method. The intensities of green (excitation/emission wavelength = 485/538 nm) and red (excitation/emission wavelength = 485/590 nm) fluorescence were analyzed in each sample and represented a surrogate marker of loss of mitochondrial Δψ m [24].
2.8. Assay for Cellular ATP Levels from Brain Cortex
After being thawed, samples were homogenized in boiling double distilled water in order to denature endogenous ATPase present in the tissue. The supernatant fraction of the homogenate was collected after centrifugation at 10,000 ×g for 10 min. ATP levels were measured using ATP colorimetric assay kit according to manufacturer's instructions (Nanjing Institute of Jiancheng Biological Engineering, China) [25].
2.9. Immunohistochemistry for Cyt c Antigen
Deparaffinized sections were treated with 0.3% hydrogen peroxide in methanol for 15 min at room temperature to block endogenous peroxidase activity. The sections were incubated in 0.01 M, pH 6.5 sodium citrate buffer for 10 min at 121°C, and cooled to room temperature. After being blocked with 10% normal goat serum for 1 h at room temperature, the slides were subsequently incubated overnight with anti-cyt c antibody (BA0781, Boster Bio-Engineering Limited Company, China) at a dilution of 1 : 100. After being extensively washed with PBS, the slides were incubated with Histostain-Plus kit (SP-9001, Zymed, USA). The sections were then counterstained with DAB (ZLI-9032, Zhongshan Golden Bridge Biotechnology Co., LTD, China). Quantitative evaluation was measured using IDA-2000 software (Beijing Konghai Technology Company, China). At least 10 visual fields were captured and more than 500 cells were counted [26, 27].
2.10. Statistical Analysis
The statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS Inc., Chicago, IL, USA) program. All data were reported as means ± SD of three independent experiments. The physiological variables were analyzed by one-way ANOVA followed by LSD multiple comparison post hoc analysis. The neurological scores were compared by Kruskal-Wallis nonparametric test followed by multiple comparison procedures by Duncan's method. For all comparisons, P < 0.05 was considered statistically significant.
3. Results
3.1. Mortality and Neurological Scores
Of a total of 180 mice, 8 (4.4%) died over the course of the experiment, 5 (8.3%) in the SAH group, 3 (5.0%) in the tea polyphenols pretreated animals, and 0 in the sham group. This suggests that tea polyphenols pretreatment reduced the mortality in consequence of SAH.
The neurological scores obtained for sham, SAH, and tea polyphenols + SAH groups are depicted in Figure 1. The neurological score observed for mice with SAH was significantly lower than that of the sham group from 6 h to 72 h after SAH (P < 0.01 versus sham), whereas the improved neurological scores were observed after 12 h of SAH in animals pretreated with tea polyphenols pretreatment group (P < 0.01 versus SAH), indicating that tea polyphenols rescued neuronal injury.
Figure 1.

Neurological deficits in OxyHb-induced SAH mice. Values are expressed as mean ± SD of triplicate samples. **P < 0.01 versus sham; ∆∆ P < 0.01 versus SAH.
3.2. The Neuroprotective Role of Tea Polyphenols in Early Brain Injury after SAH
LDH activity is the most widely used marker in cytotoxic studies. Using this assay, we detected a neuroprotective role for tea polyphenols in the EBI stage after SAH. LDH activity was stable in the cortical tissue of the sham group (Figure 2). After SAH, LDH levels were significantly increased in the cortex along with the SAH progress (P < 0.01 versus sham), but the peak level was observed 12 h after SAH, indicating that LDH activity might be time dependent. However, pretreatment with tea polyphenols led to a significant decrease in LDH activity compared to the SAH group (P < 0.01 versus SAH). A significant difference was observed between groups pretreated with tea polyphenols and sham (P < 0.01 versus sham), except at 12 h. LDH levels after pretreatment with tea polyphenols were variable compared to those detected in the sham group.
Figure 2.
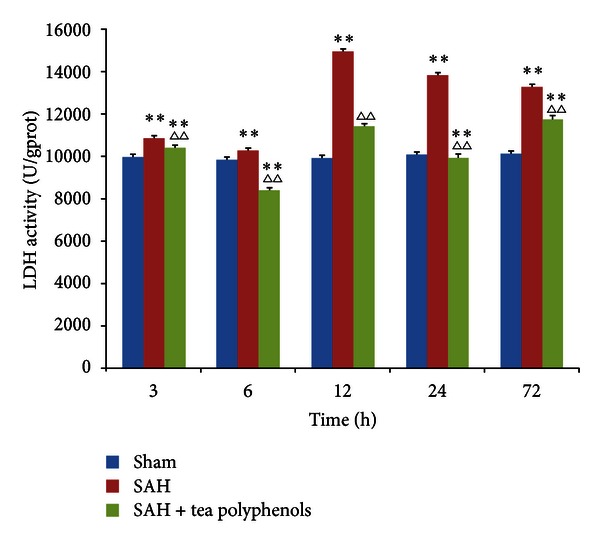
Effect of tea polyphenols on LDH activity in OxyHb-induced SAH. Data are expressed as the mean ± SD of three independent experiments. **P < 0.01 versus sham; ∆∆ P < 0.01 versus SAH.
3.3. The Preventive Effect of Tea Polyphenols on Mitochondrial Depolarization in the Cortex after SAH
Mitochondrial membrane potential (Δψ m), a widely recognized biomarker of mitochondrial function, can be measured using a cationic lipophilic dye, JC-1. OxyHb caused significant mitochondrial membrane depolarization in the SAH group, which was expressed as an increase in JC-1 green/red fluorescence ratios (P < 0.01 versus sham) (Figure 3). However, the preadministration of tea polyphenols prevented the loss of mitochondrial membrane potential (P < 0.05 versus sham); 12 h after SAH, no significant difference was observed compared with the sham group (P > 0.05 versus sham). The decrease of mitochondrial membrane potential in SAH group was greatly alleviated by tea polyphenols pretreatment with a time-dependent effect (P < 0.05 versus SAH).
Figure 3.
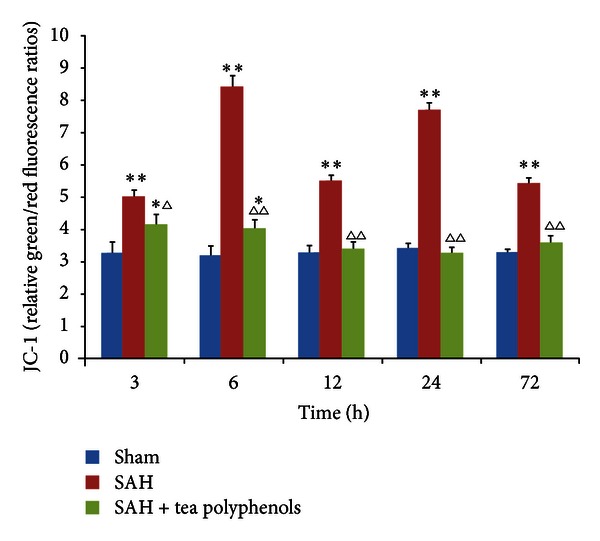
Effect of tea polyphenols on the mitochondrial membrane potential. Data are expressed as the mean ± SD of three independent experiments. *P < 0.05 versus sham; **P < 0.01 versus sham; ∆ P < 0.05 versus SAH; ∆∆ P < 0.01 versus SAH.
3.4. Tea Polyphenols Increasing ATP Content in the Development of SAH
After SAH, ATP content was determined using a validated ATP detection assay in the cortex (Figure 4). Within the cortical region, the steady-state ATP level was stable in the sham group at different time points. However, ATP levels were dramatically altered both in the SAH and the tea polyphenols pretreatment groups with a distinct pattern. Interestingly, the ATP content increased rapidly, showing the highest values 3 h after SAH both in the SAH and the tea polyphenols pretreatment groups (P < 0.01 versus sham). After 3 h of SAH, ATP levels dramatically declined and the lowest levels were observed at 3 days after SAH, suggesting a time-dependent ATP depletion (P < 0.01 versus sham). In the tea polyphenols treatment group, a fluctuation in the ATP level was observed. ATP levels were gradually reduced from 6 h to 24 h, whereas a significant increase in ATP content was observed 3 days after SAH (P > 0.05 versus sham, P < 0.01 versus SAH). In conclusion, tea polyphenols significantly increased ATP content after SAH.
Figure 4.
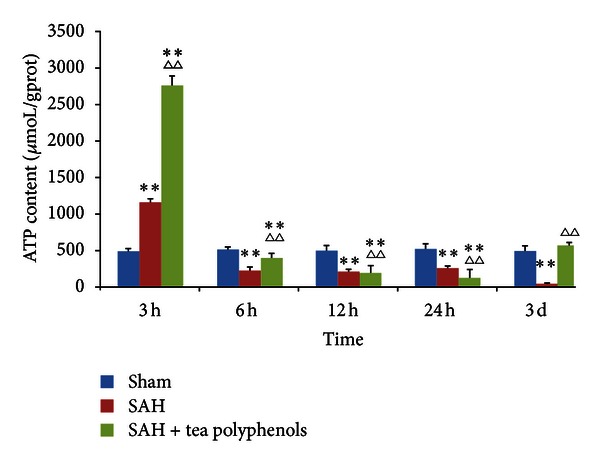
Alteration of ATP content after SAH. All the experiments were performed as described in Section 2. Data are expressed as the mean ± SD of three independent experiments. **P < 0.01 versus sham; ∆∆ P < 0.01 versus SAH.
3.5. Effect of Tea Polyphenols on the Cyt c in EBI after SAH
Considering that cytoplastic cyt c level is increased along with depolarization of mitochondrial membrane potential, cyt c content was measured in the different experimental models using immunohistology. As shown in Figure 5(a), a moderate and stable immunoreactivity was observed in the sham group, whereas robust cyt c levels were observed at all time points after SAH. However, after pre-administration of tea polyphenols, cyt c levels were gradually decreased to basal level. The cyt c levels were significantly increased in the SAH group compared to the sham group (P < 0.01 versus sham) (Figure 5(b)). Interestingly, cyt c values after pre-administration of tea polyphenols were higher than that of sham, but lower than that of SAH group. Significant difference was observed from 3 h to 24 h of sham (P < 0.01 versus sham), as well as 3 h, 24 h and 3 days of SAH (P < 0.05 versus SAH), indicating that tea polyphenols can block cyt c release after SAH.
Figure 5.
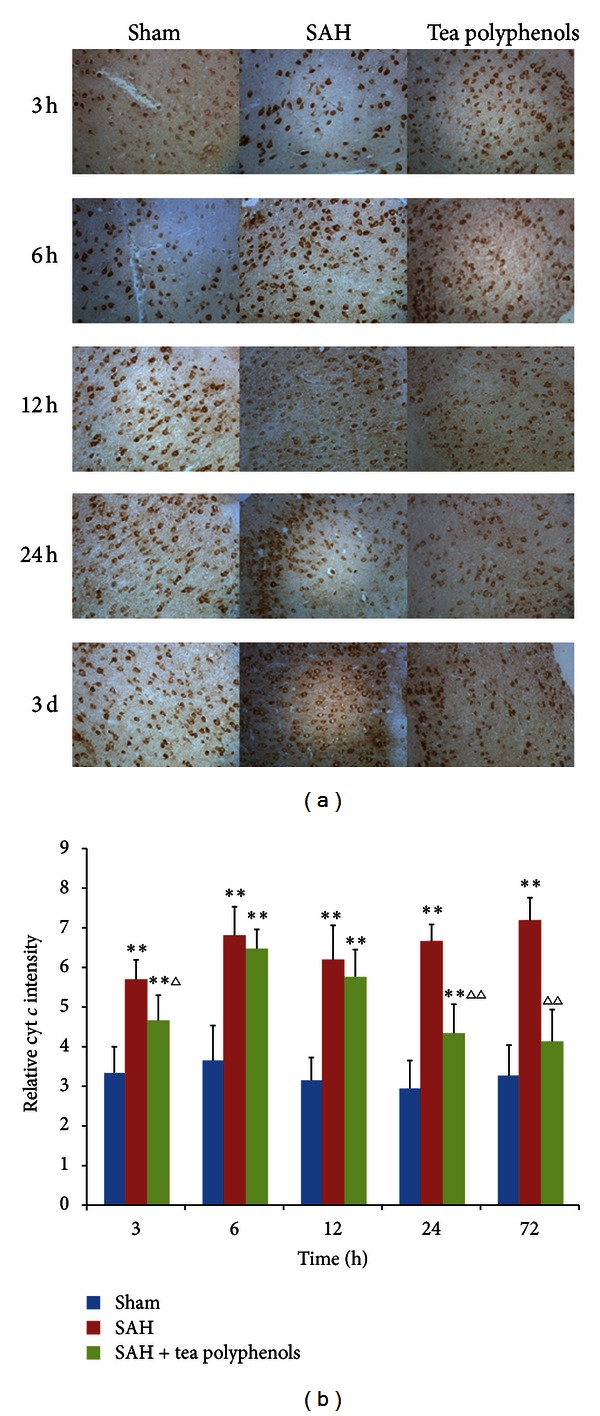
Tea polyphenols prevented cyt c release. (a) Identification of cyt c by immunohistochemical assay (×400). (b) Quantification of cyt c level by densitometry. At least 10 visual fields were captured and more than 500 cells were counted. The quantification represents means and standard deviations of results from three independent experiments. **P < 0.01 versus sham; ∆ P < 0.05 versus SAH; ∆∆ P < 0.01 versus SAH.
4. Discussion
Mitochondria contribute to many cellular events involving intracellular calcium homeostasis, reduction-oxidation potential, cell cycle regulation, and synaptic plasticity [28]. There is a current awareness that mitochondria are highly likely subjected to insults; therefore, mitochondrial dysfunction may act as one of the main trigger events in central nervous system disease, such as Parkinson's disease, Alzheimer's disease, and ischemic stroke, leading to cell death, and ultimately diseased brain [29–31]. The depolarization of the mitochondrial membrane potential (Δψ m) is a common event in mitochondrial dysfunction through which apoptosis, necrosis, and autophagy can be driven [32, 33]. Our results show a significant decrease in Δψ m in the SAH group compared with sham group. Several studies have shown that tea polyphenols significantly prevent cell swelling and the decline in the Δψ m [34]. In line with these reports, we observed that in the pretreated tea polyphenols group, the Δψ m gradually increased to basal level 12 h after SAH indicating that one mechanism by which tea polyphenols exert their protective effects is possibly by inhibition of the depolarization of the inner membrane potential. Due to lack of glycolytic capacity, more mitochondria are required to produce the necessary energy in the brain than that in other organs [35]. In pathological conditions, loss of Δψ m leads to the mitochondrial permeability transition (MPT) pore opening and to osmotic swelling of the mitochondrial matrix and to defective oxidative phosphorylation, thus impairing ATP synthesis. After SAH, the ATP levels were decreased compared to sham controls but could be restored by tea polyphenols pretreatment, indicating that tea polyphenols may block mitochondrial dysfunction followed by increased ATP content, eventually leading to neuronal cell survival.
Since the brain is highly sensitive to changes in mitochondrial respiration due to its higher consumption of oxygen and fewer free radicals scavenger ability, changes in Δψ m may also ultimately lead to apoptosis through downregulation of antiapoptotic proteins, as well as activation of proapoptotic pathways [36]. More specifically, mitochondrial dysfunction would inhibit cytochrome c oxidase activity and lead to release of cyt c into the cytosol, which initiates the caspase cascade. There is no doubt that mitochondrial respiratory dysfunctions leading to low Δψ m might result in the release of pro-apoptotic proteins, such as the apoptosis inducing factor (AIF) and cyt c [37]. The latter activates caspase-9, thus activating caspase-3-dependent apoptotic pathway. In addition, in low Δψ m conditions, PINK1 accumulates on the surface of the mitochondria and recruits Parkin, an ubiquitin ligase, which ubiquitinylates Bcl-2, further inducing cyt c related and apoptosis [38, 39]. It has been shown that increased cyt c mediates DNA fragmentation and apoptosis in mouse brains in subarachnoid hemolysate [40]. In other words, with SAH progression, neuronal cells enter the apoptotic pathway in the cortex. However, tea polyphenols can significantly inhibit cyt c release by blocking mitochondrial dysfunction at the EBI stage after SAH. This finding is supported by previous results that showed that the number of apoptotic cells was reduced after using green tea extract for pre-treatment of ischemia in gerbils [41].
5. Conclusion
In the present study, we found that neuroprotective effects of tea polyphenols may rely on their mitochondrial protection behavior. Our studies showed that tea polyphenols pretreatment reduces mitochondrial dysfunction markers and increases neurological scores after SAH [42, 43]. This suggests that dietary supplementation with tea polyphenols could be a potential candidate for prevention of SAH. As tea polyphenols represent a class of natural, dietary components, further research will be necessary to better identify which polyphenols play the roles in SAH, and which signaling pathways are involved in these neuroprotective effects.
Conflict of Interests
The authors have declared no conflict of interests.
Acknowledgments
This research was supported by the National Nature Science Foundation of China (no. 31200519), the Program for New Century Excellent Talents in University (NCET), the Program for Science and Technology Innovation Talents in Universities of Henan Province (HASTIT), and the Science and Technology Key Projects of Henan Province (no. 122102110116).
References
- 1.Weinreb O, Mandel S, Amit T, Youdim MBH. Neurological mechanisms of green tea polyphenols in Alzheimer’s and Parkinson’s diseases. Journal of Nutritional Biochemistry. 2004;15(9):506–516. doi: 10.1016/j.jnutbio.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Sutherland BA, Rahman RMA, Appleton I. Mechanisms of action of green tea catechins, with a focus on ischemia-induced neurodegeneration. Journal of Nutritional Biochemistry. 2006;17(5):291–306. doi: 10.1016/j.jnutbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Kuriyama S, Hozawa A, Ohmori K, et al. Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project. American Journal of Clinical Nutrition. 2006;83(2):355–361. doi: 10.1093/ajcn/83.2.355. [DOI] [PubMed] [Google Scholar]
- 4.Sumpio BE, Cordova AC, Berke-Schlessel DW, Qin F, Chen QH. Green tea, the “Asian Paradox,” and cardiovascular disease. Journal of the American College of Surgeons. 2006;202(5):813–825. doi: 10.1016/j.jamcollsurg.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Bederson JB, Connolly ES, Jr., Batjer HH, et al. Guidelines for the management of aneurismal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the stroke council, American Heart Association. Stroke. 2009;40(3):994–1025. doi: 10.1161/STROKEAHA.108.191395. [DOI] [PubMed] [Google Scholar]
- 6.Alaraj A, Charbel FT, Amin-Hanjani S. Peri-operative measures for treatment and prevention of cerebral vasospasm following subarachnoid hemorrhage. Neurological Research. 2009;31(6):651–659. doi: 10.1179/174313209X382395. [DOI] [PubMed] [Google Scholar]
- 7.Cahill WJ, Calvert JH, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2006;26(11):1341–1353. doi: 10.1038/sj.jcbfm.9600283. [DOI] [PubMed] [Google Scholar]
- 8.Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. Journal of Neuroscience Research. 2007;85(7):1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- 9.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35(10):2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 10.Cahill J, Calvert JW, Marcantonio S, Zhang JH. p53 may play an orchestrating role in apoptotic cell death after experimental subarachnoid hemorrhage. Neurosurgery. 2007;60(3):531–545. doi: 10.1227/01.NEU.0000249287.99878.9B. [DOI] [PubMed] [Google Scholar]
- 11.Fiskum G, Rosenthal RE, Vereczki V, et al. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. Journal of Bioenergetics and Biomembranes. 2004;36(4):347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- 12.Marchi S, Giorgi C, Suski JM. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of Signal Transduction. 2012;2012:17 pages. doi: 10.1155/2012/329635.329635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mechanisms of Ageing and Development. 2010;131(7-8):536–543. doi: 10.1016/j.mad.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Current Alzheimer Research. 2011;8(4):393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage research: from delayed vasospasm to early brain injury. Molecular Neurobiology. 2011;43(1):27–40. doi: 10.1007/s12035-010-8155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Critical Reviews in Food Science and Nutrition. 2003;43(1):89–143. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki T, Kasuya H, Onda H, et al. Role of p38 mitogen-activated protein kinase on cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2004;35(6):1466–1470. doi: 10.1161/01.STR.0000127425.47266.20. [DOI] [PubMed] [Google Scholar]
- 18.Scolaro B, Delwing-De Lima D, Da Cruz JGP, Delwing-Dal Magro D. Mate tea prevents oxidative stress in the blood and hippocampus of rats with acute or chronic ethanol administration. Oxidative Medicine and Cellular Longevity. 2012 doi: 10.1155/2012/314758.314758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo S, Yan JQ, Yang TB, Yang XQ, Bezard E, Zhao BL. Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson’s disease through inhibition of ROS-NO pathway. Biological Psychiatry. 2007;62(12):1353–1362. doi: 10.1016/j.biopsych.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Shi W, Huang LY, Wang RZ, et al. Time course of oxyhemoglobin induces apoptosis in mice brain cells in vivo . Acta Neurochirurgica. 2008;(104):23–26. [Google Scholar]
- 21.Zhu X, Chen C, Ye D, et al. Diammonium glycyrrhizinate upregulates PGC-1α and protects against Aβ1-42-induced neurotoxicity. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0035823.e35823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh BK, Tripathi M, Chaudhari BP, Pandey PK, Kakkar P. Natural terpenes prevent mitochondrial dysfunction, oxidative stress and release of apoptotic proteins during nimesulide-hepatotoxicity in rats. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034200.e34200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen K, Zhang Q, Wang J, et al. Taurine protects transformed rat retinal ganglion cells from hypoxia-induced apoptosis by preventing mitochondrial dysfunction. Brain Research. 2009;1279:131–138. doi: 10.1016/j.brainres.2009.04.054. [DOI] [PubMed] [Google Scholar]
- 24.Xinmin D, Yunyou D, Chaosheng P, et al. Dexamethasone treatment attenuates early seawater instillation-induced acute lung injury in rabbits. Pharmacological Research. 2006;53(4):372–379. doi: 10.1016/j.phrs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Kintner DB, Begum G, et al. Endoplasmic reticulum Ca2+ signaling and mitochondrial Cyt c release in astrocytes following oxygen and glucose deprivation. Journal of Neurochemistry. 2010;114(5):1436–1446. doi: 10.1111/j.1471-4159.2010.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Kong Q. Evaluation of centrosome abnormalities and p53 inactivation in chemical induced hepatocellular carcinogenesis. Neoplasma. 2009;56(2):169–176. doi: 10.4149/neo_2009_02_169. [DOI] [PubMed] [Google Scholar]
- 27.Garcia JH, Wagner S, Liu KF, Hu XJ, Mohr JP. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats: statistical validation. Stroke. 1995;26(4):627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Gleichmann M, Cheng A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron. 2008;60(5):748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos RX, Correia SC, Wang X, et al. Alzheimer’s disease: diverse aspects of mitochondrial malfunctioning. International Journal of Clinical and Experimental Pathology. 2010;3(6):570–581. [PMC free article] [PubMed] [Google Scholar]
- 30.Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. Journal of Neurotrauma. 2010;27(7):1311–1320. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Human molecular genetics. 2009;18(2):R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–153. doi: 10.1016/j.neuroscience.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 33.Hail N, Lotan R. Cancer chemoprevention and mitochondria: targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Molecular Nutrition and Food Research. 2009;53(1):49–67. doi: 10.1002/mnfr.200700527. [DOI] [PubMed] [Google Scholar]
- 34.Gomez-Pinilla F, Nguyen TT. Natural mood foods: the actions of polyphenols against psychiatric and cognitive disorders. Nutritional Neuroscience. 2012;15(3):127–133. doi: 10.1179/1476830511Y.0000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy PH. Mitochondrial dysfunction in aging and Alzheimer’s disease: strategies to protect neurons. Antioxidants and Redox Signaling. 2007;9(10):1647–1658. doi: 10.1089/ars.2007.1754. [DOI] [PubMed] [Google Scholar]
- 36.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 37.Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. British Journal of Pharmacology. 2012;167(4):699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen D, Gao F, Li B, et al. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. Journal of Biological Chemistry. 2010;285(49):38214–38223. doi: 10.1074/jbc.M110.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu C, Wang X, Xu F, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death and Differentiation. 2005;12(2):162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- 40.Matz PG, Fujimura M, Lewen A, Morita-Fujimura Y, Chan PH. Increased cytochrome c-mediated DNA fragmentation and cell death in manganese-superoxide dismutase-deficient mice after exposure to subarachnoid hemolysate. Stroke. 2001;32(2):506–515. doi: 10.1161/01.str.32.2.506. [DOI] [PubMed] [Google Scholar]
- 41.Hong JT, Ryu SR, Kim HJ, et al. Protective effect of green tea extract on ischemia/reperfusion-induced brain injury in Mongolian gerbils. Brain Research. 2001;888(1):11–18. doi: 10.1016/s0006-8993(00)02935-8. [DOI] [PubMed] [Google Scholar]
- 42.Yang CS, Lambert JD, Sang S. Antioxidative and anti-carcinogenic activities of tea polyphenols. Archives of Toxicology. 2009;83(1):11–21. doi: 10.1007/s00204-008-0372-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bastianetto S, Krantic S, Chabot JG, Quirion R. Possible involvement of programmed cell death pathways in the neuro-protective action of polyphenols. Current Alzheimer Research. 2011;8(5):445–451. doi: 10.2174/156720511796391854. [DOI] [PubMed] [Google Scholar]