

Abstract
We provide an overview of the methods used to label circulating cells for fluorescence detection by in vivo flow cytometry. These methods are useful for cell tracking in small animals without the need to draw blood samples, and are particularly useful for the detection of circulating cancer cells and quantification of circulating immune cells.
Keywords: In vivo flow cytometry, cell tracking, circulating tumor cells, circulating immune cells
1. Introduction
The longstanding goal of in vivo flow cytometry is to perform noninvasive, real-time detection and quantification of circulating cells in human subjects without the need to draw blood samples. Considerable challenges remain before this goal can be realized. In the meantime, in vivo flow cytometry has been established as a valuable research tool for monitoring circulating cells in preclinical animal studies [1–25]. In small animals that have limited blood volumes, frequent blood sampling can be difficult and may lead to adverse host stress response (see section 3 below). Several approaches have been developed using either fluorescence (1–17), Raman (18), photoacoustic (18–24), or photothermal (21,23,25) techniques to detect individual circulating cells in live animals (see Table 1). Here we focus on cell labeling methods for fluorescence-based in vivo flow cytometry that are widely used in animal studies. These methods include
Table 1.
Methods for labeling cells for in vivo flow cytometry. A listing of the labeling methods and the in vivo flow cytometers used, as well as the cell types studied in specific references cited in this review.
| LABELED ENTITY | METHOD | CELLS STUDIED | REFERENCES |
|---|---|---|---|
| Endogenous chromophores | |||
| (melanin) | PAFC/PTFC | mMel | Galanzha, EI et al., 2009 (23) |
| (hemoglobin) | PAFC/PTFC | RBCs | Zharov V, et al., 2006 (21) |
| In vivo fluorescent cell labeling | |||
| (ICG) | PAFC | Blood flow, Ret | Zharov V, et al., 2006 [21] |
| Fluorescent antibodies | |||
| (CD45) | IVFC | WBCs | Novak J, et al., 2004 [1] |
| (c-kit, sca-1) | IVFC | WBCs , HSCs | Boutrus S, et al., 2007 [7] |
| Fluorescent proteins | |||
| (Annexin V) | IVFC | Ann V+ Pr CA cells | Wei X, et al., 2005 [4] |
| Fluorescent vitamins | |||
| (Folate) | IVFC | huNP, OvCa, mLymph | He, W, et al., 2007 [15] |
| Ex Vivo membrane labeled tumor or blood cells | |||
| (DID) | IVFC | CD4+ T CELLS | Lee H, et al., 2006 [5] |
| IVFC | huALL | Sipkins DA, et al. 2005 [3] | |
| IVFC | MM | Alsayed Y, et al., 2007 [8] | |
| IVFC | WM | Leleu X, et al., 2007 [9] | |
| IVFC | RBCs | Novak J, et al., 2004 [1] | |
| IVFC | RBCs | Tkaczyk ER, et al., 2008 [17] | |
| RFC | Leukocytes | Alt C, et al., 2007 [6] | |
| Ex vivo cytoplasmic labeling of tumor cells | |||
| (calcein) | IVFC | MM | Azab A, et al., 2009 [10] |
| IVFC | WM | Roccaro AM, et al., 2010 [12] | |
| (Q dots) | IVFC | Br CA cells | Tkaczyk ER, et al., 2008 [17] |
| Transgenic fluorescent proteins in tumors or leukocytes | |||
| (GFP,DsRed) | IVFC | MM | Runnels JM, et al., 2011 [14] |
| IVFC | T cell populations in rejection | Fan Z, et al., 2010 [13] | |
| IVFC | huBr CA cells | Boutrus S, et al., 2007 [7] | |
| IVFC | mLuCa | Le TT, Huff TB, Chen, J-X, 2009 [16] | |
| Nanoparticles | |||
| (gold nanorods) | PAFC | free NP, bacteria | Zharov V, et al., 2007 [20] |
| PAFC/PTFC | free NP, huSqCa | Zharov VP, et al., 2006 [21] | |
| (magnetic nanoparticles) | PAFC | Br CA | Galanzha EI, et al., 2009 [22] |
| (carbon nanotubes) | RaFC | HeLa cells | Biris, et al., 2009 [18] |
| PAFC | free NP, bacteria | Zharov V, et al., 2007 [20] | |
| (folate conj-gold plated C nanotubes) | PAFC | Br CA | Galanzha EI, et al., 2009 [22] |
Abbreviations: Ann V, Annexin V; Br CA, Breast cancer; CA, cancer; DiD, DiIC18(5), GFP, green fluorescent protein; HeLa, cervicle cancer cells derived from patient Henrietta Lacks; HSCs, Hematopoietic stem cells; huALL, human acute lymphoblastic leukemia; huNP, human nasopharyngeal carcinoma; huSqCa, human squamous cell carcinoma; ICG, indocyanine green; IVFC, in vivo flow cytometer; mLCA, mouse lung carcinoma; mLL, mouse lymphocytic leukemia; mLuCa, mouse lung carcinoma; mLymph, mouse lymphoma; mMel. mouse melanoma; MM, multiple myeloma; NP, nanoparticles; Ov Ca, ovarian cancer; PAFC, photoacoustic flow cytometry; Pr CA, prostate cancer; PTFC, photothermal flow cytometry; Q dots, quantum dots; RaFC, Raman flow cytometry; RBCs, red blood cells; Ret, reticulocytes; RFC, retinal flow cytometer; WM, Waldenstrom’s macrglobulinemia; WBCs, white blood cells.
Direct labeling of specific cell populations by injecting fluorescently conjugated antibodies or antibody fragments into the circulation.
Ex vivo isolation and purification of specific cell populations from donor animals, labeling the cells with fluorescent cell tracers, followed by adoptive transfer into recipient animals. This method can also be used to label cultured cell lines or primary human cells before they are introduced into animals.
Use of animals expressing fluorescent proteins (FP) in specific cell populations, or transducing ex vivo cultured cells to express FP before injection into host animals.
2. Direct labeling of circulating cells with fluorescent IgG antibodies or Fab antibody fragments
If the cell population of interest expresses a specific cell surface marker, and an antibody recognizing the cell surface marker is available, then the antibody can be conjugated to a fluorophore and injected into the circulation for direct cell labeling in vivo. For example, anti-CD45 antibody can be used to label all circulating leukocytes [1], anti-CD3 antibody can be used to label the T cell population, and anti-CD4 antibody can be used to label just the CD4+ T cell subset. A large selection of fluorescently conjugated antibodies is available from commercial suppliers. There are also commercial conjugation kits that can be used to attach a fluorophore such as FITC, Cy5, or any of the AlexaFluor dyes, to an antibody of one’s choice.
The amount of antibodies to inject depends on the abundance of the target cells one expects to find in the circulation but is typically on the order of 10 μg per mouse. Assuming the mouse blood volume is 2 mL, the concentration of fluorescent antibodies in the blood immediately after injection is about 33 nM, which is readily detectable as background fluorescence signal. The real signal from labeled cells needs to exceed the background in order to be detected. The signal increases with time as the antibody binds to the target cells, while the background decreases with time as the free (unbound) antibody is cleared from the circulation. The optimum time for cell detection after antibody injection will depend on the kinetics of antibody binding as well as on the clearance time of the unbound antibody, and can take from minutes to hours depending on the antibody. The clearance kinetics of injected antibodies from the plasma has been examined in pre-clinical and clinical studies using radionuclide-conjugated antibodies directed against circulating cell types. Typically the clearance is biphasic. For example, anti-mouse Thy1.1 (T cell marker) initially cleared with a t½ of less than 3 hrs followed by t½ of 34 hrs (26), and anti-human CD45 initially cleared with t½ equal to 1.4 hours, but at later times t½ was 26.7 hours (27). The terminal clearance rate for anti-CD45 was not unlike the clearance rate for non-specific control antibody (t½ = 27.3 hours). In addition, in the latter study, peak antibody plasma concentrations for anti-CD45 were 3.75 times lower than those observed for non-specific antibody when measured as percentages of injected dose. Taken together, these data point to initial antibody clearance rates as largely determined by antibody binding to accessible targets in vivo while later clearance rates reflect saturation of target cells and tissues.
As in all antibody-labeling studies, it is important to perform proper control experiments using isotype IgG in order to take into account potential nonspecific labeling. This is important since injected antibody can be taken up by endocytic cells or by cells with Fc receptors. In an in vivo flow cytometer, cells with such non-specific uptake capacities will be falsely counted as labeled cells. The best way to eliminate such false positive count is to use a double labeling scheme where the specific and the isotype control IgG are labeled with two distinct fluorophores and co-injected into the circulation. The specifically labeled cells (single positive) can then be distinguished from the nonspecifically labeled cells (double positive because both specific and control IgGs will be taken up) using a two-channel in vivo flow cytometer [7].
Another important consideration is that antibody binding can lead to cell activation or cell death through antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), both requiring binding of the Fc region of the antibody to host Fc receptors [28, 29]. We have investigated the effect of antibody binding to circulating cells by in vivo flow cytometry. We noted that after injection of Cy5-conjugated anti-CD4 antibody, the number of circulating cells in the Cy5 channel showed an initial increase during the first 3 hours, followed by a steady decrease in the ensuing 24 hours (Figure 1A, solid line). Similar kinetics were observed after injection of anti-CD45 antibody [1]. The initial increase in cell count is attributed to a combination of a) binding of the antibody to the target cells and b) clearance of the unbound antibody, allowing the labeled cells to be detected as the background signal decreases, while the slower decline in cell number is attributed to the depletion of target cells after antibody labeling.
Figure 1.

Kinetics of cell depletion detected by IVFC is influenced by the fluorescent probe used. A, B. Cy5 conjugated antibody against CD4 was injected intravenously into BALB/c mice, and fluorescent cells were counted using the IVFC to quantify the number of circulating fluorescent T cells as a function of time. A. The circulation time of the Cy5-CD4 labeled T cells was significantly enhanced by pre-treatment of the mice with Fc block (a mixture of antibodies directed against mouse CD16 and CD32) in a dose-dependent fashion. B. Likewise, enzymatic removal of the Fc portion of the anti-CD4 antibody enhanced the longevity of the fluorescent T cells in the circulation by tens of hours when compared to the results obtained using whole antibody (dashed square vs diamond solid lines). However, syngeneic T cells labeled with the lipophilic membrane dye DiD persisted in the circulation for days (dashed circle line). C. Depletion of T cell numbers over time is due to loss of fluorescent Fab fragment binding rather than removal of the cells from the circulation as is demonstrated by the reduction in fluorescent signal intensity over time. D. Cells labeled with DiD did not lose signal intensity. For A: n = 3 mice; B: n = 5 mice; C,D: Plots from two representative mice injected with either Fab fragment directed against CD4 or DiD labeled donor T cells. Each data point was measured three times. CD4 antibody and Fab fragment injections were done at 1mg/kg.
The involvement of Fc receptors in the observed depletion of the antibody-labeled cells was confirmed using Fc Block, which is a mixture of anti-CD16 & anti-CD32 antibodies used to block the Fc receptors in the host cells. When mice were pre-treated with injections of Fc Block prior to injection of the fluorescent antibodies, the circulation time of the labeled cells increased significantly in a dose-dependent manner (Figure 1A, dashed lines). Antibody-mediated cell depletion was almost completely prevented when mice were pre-treated with an Fc Block dose of 100 μg per mouse.
We next investigated whether modification of the antibody can retain its cell labeling specificity while minimizing the adverse effects of antibody binding. We used a commercially available reagent to cleave and remove the Fc portion of the IgG through enzymatic digestion. The enzyme papain works by cleaving the Fc region on the amino-terminal side of the disulfide bonds to produce two identical Fab molecules. The purified Fab molecules were then conjugated to the fluorescent dye Cy5 and injected into BALB/c mice in order to label circulating lymphocytes. As shown in Figure 1B, cells labeled with anti-CD4 Fab fragments have significantly longer circulation time compared to cells labeled with intact anti-CD4 antibodies (note the time axis is in days rather than hours).
Did the removal of the Fc portion of the antibody simply delay cell clearance from the circulation? In principle, the reduction in the detected cell number can mean either (a) cells are being depleted from the circulation, or (b) cells remain in the circulation but the fluorescent label is lost from the cell surface. To investigate the latter possibility as a possible explanation for the decrease in cell count over time, we examined the mean fluorescence intensity of the signal peaks detected by the in vivo flow cytometer as a function of time. As shown in Figure 1C, in the case of Fab fragments, the reduction in cell count can indeed be explained by the decrease in the mean fluorescent intensity of the detected peaks over time, resulting in fewer peaks being identified as positive cells. The fact the Fab fragment is more easily lost from the cell surface compared to intact antibodies is not surprising since the Fab fragment has only one binding site as opposed to the bivalent intact antibodies.
In summary, circulating cells can be readily detected by in vivo injection of fluorescent antibodies that target specific cell surface markers. However, labeled cells will be captured by host cells through the interaction of the Fc receptors with the antibody, leading to rapid clearance (within 24 hours) of the labeled cells from the circulation. To monitor cells without altering their normal physiology, it is preferable to use antibody fragments (Fab) from which the Fc sequences have been removed. The single chain Fab also avoids cross-linking the target molecules and therefore avoids cell activation. The monovalent Fab does have a lower affinity compared to the intact (bivalent) antibodies so the probe is more likely to be dissociated from the target cells over time.
In addition to antibodies, other injectable probes can be used to target specific cell surface epitopes. For example, when cells undergo apoptosis, the phosphatidylserine that is normally on the inner leaflet of the plasma membrane becomes exposed on the outer cell surface, and can be recognized by the apoptotic cell probe Annexin V. Injection of Annexin V conjugated to Alexa Fluor 647 has been used to detect circulating apoptotic cells [4]. In an elegant study, He et al [15] used folate-fluorophore conjugates to label circulating cancer cells that overexpress the folate receptor (FR) and detected the labeled cells by two-photon in vivo flow cytometry. The unbound folate conjugates have shorter circulation time compared to anti-FR antibodies, thus reducing the background fluorescence signal. The use of folate conjugates also avoids underestimation of circulating cancer cells [15] caused by antibody-mediated cell depletion as described above.
3. Adoptive transfer of ex vivo purified and labeled cells
Many experiments use cultured cells, or cells harvested from a donor animal or even patient cell samples, that are then injected into recipient animals to study their in vivo function. For example tumor cell lines or tumor cells obtained from cancer patients are commonly used to study their metastatic properties after injection into the circulation, and immune cells are frequently isolated from donor mice are adoptively transferred into recipient mice to determine their immunologic function. In these cases, a cell tracker dye such as DiIC18(5) (known as DiD, a lipophilic dye that intercalates into the plasma membrane and fluoresces in the far red) can be used to label the cells before injection, and in vivo flow cytometry can be used to monitor the circulating cell number over time [3, 8–12]. For example, subsets of immune cells can be isolated from donor animals, purified, labeled ex vivo with a fluorescent dye such as DiD, and reintroduced into recipient animals in a procedure called adoptive transfer. Using this method, CD4+ T lymphocytes were isolated from extracted lymph nodes and spleens of BALB/c mice via negative selection using immunomagnetic beads (i.e. the beads were used to pull down all cells that are not CD4+ T cells). The purified CD4+ T cells were then labeled ex vivo with DiD and injected into BALB/c recipients. Unlike antibody labeling, DiD labeled cells were not depleted and remain in the circulation for more than a week (Figure 1B, circles). The slight decrease followed by a slight increase in circulating cell count during the first 7 days after adoptive transfer likely reflects the initial cell “homing” to the lymph nodes followed by their recirculation kinetics. Unlike Fab antibody fragments, the DiD label stayed on the cell membrane with no apparent loss of signal intensity over time (Figure 1D).
A similar technique was used to assess the effect of repeated collection of blood samples from mice. T cells were isolated and purified from the lymph nodes and spleen as described above. Cells were labeled with 10 μM of DiD for 30 min at 37°C, followed by intravenous injection into cognate BALB/c mice at a dose of 5 × 106 cells per mouse. After overnight equilibrium, the in vivo flow cytometry was carried out to detect the number of donor T cells in the peripheral blood. One group of mice was then bled by tail snip and ~100 μl of blood was collected. The circulating, fluorescently labeled donor T cells were enumerated again by in vivo flow cytometry 4 hours later. The procedure was repeated at 8 and 24 hours and at each time 50 μl of blood was collected, followed by in vivo flow cytometric analysis 4 hours later. The results (Figure 2) suggest that repeated blood sampling invokes a stress response that reduces the number of circulating lymphocytes. It is well documented that the absolute number of peripheral blood T cells decreases rapidly and significantly (40~70% lower than baseline) during traumatic stress in both humans and animals [30–32]. Studies with 51Cr-labeled lymphocytes suggested that a decrease in egress of lymphocytes, rather than increased homing or cell death was the mechanism for the lymphopenia induced by traumatic stress [31]. Our results confirm that for frequent monitoring of circulating cells, in vivo flow cytometry is superior to conventional methods that require repeated collection of blood samples.
Figure 2.
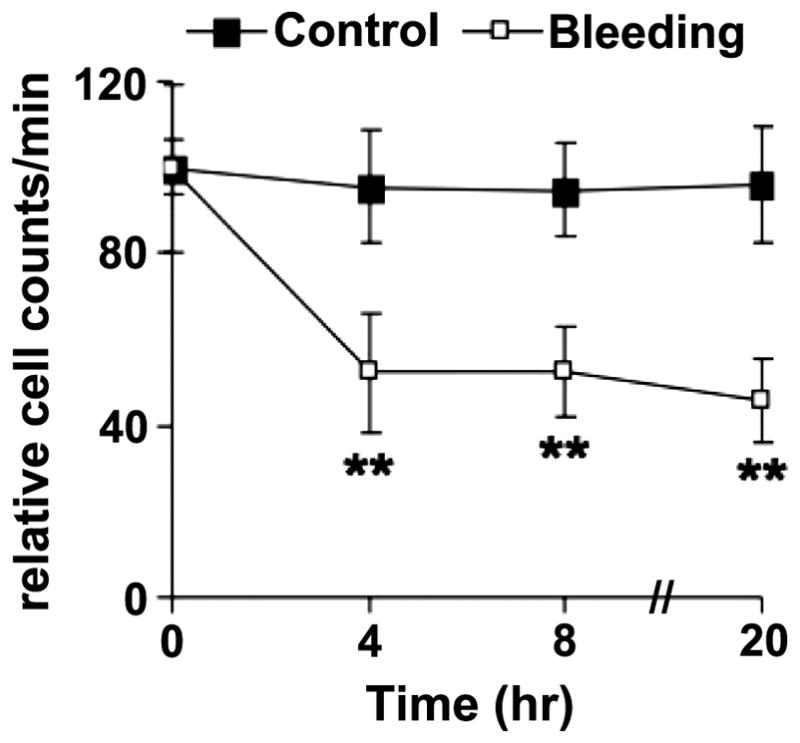
Depletion of circulating lymphocyte numbers after repeated bleeding is quantified by IVFC. T cells were isolated and purified from the lymph nodes and spleen, labeled with 10 μM of DiD, and injected intravenously into syngeneic BALB/c mice at a dose of 5 × 106 cells per mouse. After overnight equilibrium, in vivo flow cytometry was carried out to detect the number of donor T cells in the peripheral blood of the recipient mice. The experimental mice (n= 6) were then bled by tail snip and ~100 μl of blood was collected. Four hours later, the circulating fluorescently labeled donor T cells were enumerated again by in vivo flow cytometry. The procedure was repeated at 8 and 24 hours except that at these time points 50 μl of blood were collected, followed by in vivo flow cytometric analysis 4 hours later. Control mice (n=6) were not bled. The results are shown as the relative cell counts of circulating cells per minute. **P<0.01.
Adoptively transferred tumor cells may also be monitored short-term using cell tracker dyes and in vivo flow cytometry. Leukemia and multiple myeloma are both hematologic malignancies that can spread to multiple locations in the bone marrow via the circulation. We have used in vivo flow cytometry to study how leukemic cells and multiple myeloma cells adhere to the bone marrow vasculature and exit the circulation. We have shown that circulating leukemic cells recognize the bone marrow vascular endothelium expressing a chemokine (chemical attractant) called stromal-derived factor-1 (SDF-1) [3]. The interaction of SDF-1 with the chemokine receptor CXCR4 on the leukemic cell surface causes the leukemic cells to arrest on the vascular endothelium and eventually transmigrate into the bone marrow stroma. By monitoring the kinetics of cell depletion from the circulation, we can evaluate the effectiveness of therapeutic agents aiming to block the SDF-1/CXCR4 interaction [3]. The same interaction has also been shown to facilitate multiple myeloma cell binding to the bone marrow vascular endothelium [8–12]. Figure 3A shows that, in addition to the SDF-1/CXCR4 interaction, the integrin VLA-4 is also important for multiple myeloma cell binding to the bone marrow vasculature, as pre-treatment of the myeloma cells with anti-VLA-4 antibody significantly increased their circulation time.
Figure 3.
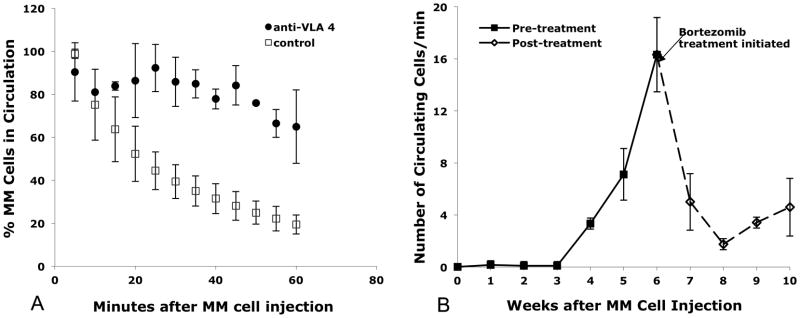
Enumeration of circulating multiple myeloma cells immediately after injection (A) and over the course of the disease and following treatment (B) by IVFC. A. Treatment with anti-VLA-4 antibody significantly prolonged the circulation time of multiple myeloma cells after injection. DiD-labeled MM.1S cells injected into the tail veins of BALB/c mice were dramatically depleted from the circulation within an hour of injection unless homing was abrogated by treatment with an agent that interferes with binding through the CXCR4/SDF-1 pathway, such as anti-VLA 4 antibody (control, n = 4 mice; VLA 4 antibody treated, n = 2 mice). For every data point, at least three 60-second measurements were taken per mouse over a five-minute period, counted using Matlab, averaged and plotted above. Bars indicate standard errors. B. The number of circulating GFP+ multiple myeloma cells increased during tumor growth, but decreased in response to therapy. Weekly in-vivo flow cytometry sessions were repeatedly performed on three SCID/Bg mice that had been injected with MM.1S-GFP+ cells. For each weekly measurement, ten one-minute traces were taken, the number of GFP + cells were counted, averaged and plotted against time. Beginning in the sixth week post cell injection, bortezomib was administered twice-weekly at the clinically relevant concentration of 1 mg/kg. Solid line indicates cell counts accumulated before the mice were treated. Dashed line indicates cell counts after twice-weekly bortezomib treatment. Figure 3A,B reproduced from [14] with permission.
4. Cell labeling using fluorescence proteins
Advances in molecular biology have led to the development of reporter gene constructs that encode fluorescent proteins (FP) in many types of cells or whole animals. To study cancer growth and response to therapy, for example, cancer cell lines can be transduced to express the green fluorescent protein (GFP), before they are injected or implanted in recipient animals [14]. One major advantage of using FP to label cells is that as long as the FP is stably expressed, the cells will continue to be detectable and are therefore suitable for long-term monitoring of tumor growth, whereas cell labeled with a fluorescent dye such as DiD will become dimmer every time the cells undergo division, and can become undetectable after several divisions. Figure 3B shows long-term monitoring of tumor growth, as reflected by the increased number of circulating tumor cells during the first 6 weeks after injection of GFP-expressing multiple myeloma cells. At that time point the mice received treatment with bortezomib (velcade), and the tumor burden dropped off precipitously [14]. These results demonstrated the power of in vivo flow cytometry to follow tumor growth and response to therapy continuously in individual animals over several months.
Mouse models in which fluorescent proteins are expressed in specific cell populations are particularly useful for in vivo imaging applications. For example CX3CR1-GFP mice are reporter mice whose monocytes/macrophages selectively express the GFP. In LysM::GFP mice, GFP is expressed in both macrophages and granulocytes. In MHC Class II GFP mice, the antigen-presenting cells including dendritic cells and B cells express the GFP. In all these mice, GFP cells can be detected in the circulation and in vivo flow cytometry can be used to monitor the GFP cells without the need for further labeling.
Alternatively, donor cells can be isolated from reporter mice that express FP and adoptively transferred into the host animals. This approach has been used to study the response of T cells to transplantation from a mismatched donor (allogeneic transplantation). In a mouse model of pancreatic islet transplantation (an experimental procedure to treat type I diabetes by transplanting functional pancreatic islets containing β-cells that secrete insulin in response to elevated glucose level in the blood), host T cells will attack the foreign tissue (allograft) leading to graft rejection unless they are kept in check by immunosuppressive therapy or by tolerance induction. Tolerance is a state in which the host immune system is “instructed” to accept tissue from a specific donor, while retaining the capacity to defend against other foreign pathogens (unlike immunosuppression). In this model, tolerance was induced by giving mice rapamycin plus anti-CD154 antibody treatment. Islet allograft survived in the treated mice for the duration of the experiment (>45 days) while in untreated mice the graft was rejected by 15 days after transplantation [13]. The rejection has been shown to be mediated by CD4 T cells. However, CD4 T cells contain both the effector T (Teff) cells that attack and destroy foreign grafts, as well as regulatory T (Treg) cells that protect the grafts. It was hypothesized that rapamycin plus anti-CD154 antibody treatment induced tolerance by inducing massive conversion of T cells to the regulatory phenotype. A reporter mouse was created in order to visualize the converted T cells. In these mice the Teff cells express the red FP (dsRed), the pre-existing (natural) Treg cells express the green FP (GFP), and the newly converted (induced) Treg cells express both the GFP and the dsRed and will be detected as double-positive (yellow) cells. In vivo flow cytometry was used in combination with in vivo confocal microscopy to observe the kinetics of these T cell populations in the circulation and in the graft of these tri-color-coded reporter mice. Figure 4A shows an example of all three T cell subsets detected in a single 3-second trace. Double positive (induced Treg cells) were observed in the circulation as early as 4 days after transplantation.
Figure 4.

Simultaneous detection of circulating Teff cells (DsRed), natural Treg cells (GFP), and induced Treg cells (DsRed+GFP+ double positive) in the tricolor-coded reporter mice used to demonstrate rejection/tolerance of pancreatic islet graft. A. Representative in vivo flow cytometry trace showing the identification of single positive endogenous natural Treg (green box), Teff (red boxes) and double-positive induced Treg (yellow box) cells. The second peak in the DsRed channel occurred about 45 ms before the second peak in the GFP channel. As this time difference was greater than the uncertainty of the measurements, these two peaks were distinguished as separate cells and not a double-positive iTreg cell. B,C. Pancreatic islet grafts from DBA/2 mice were placed under the renal capsules of allogeneic C57BL/6 mice expressing GFP Treg and DsRed Teff cells. The numbers of nTreg (GFP), Teff (DsRed) and iTreg (DsRed+GFP double positive) were followed daily in the peripheral blood using in vivo flow cytometry until graft rejection occurred in the control animals. Note the ten-fold difference in cell counts between the mouse treated with CD154-specific mAb plus rapamycin (tolerance-induced) (B) and the untreated immunocompetent mouse (C). Each curve was derived by serial analysis of the same blood vessel of the same animal over time. Reproduced from [13] with permission.
By performing in vivo flow cytometry measurements everyday during the first week and again on day 14 after transplantation, we obtained the kinetics of the three circulating T cell sub-populations for both the rapamycin plus anti-CD154 antibody-treated mice (Figure 4B) and the control untreated mice (Figure 4C). These results are striking in two ways. First, in both treated and untreated mice, the number of red (Teff) cells greatly exceeds the number of green (natural Treg) and yellow (induced Treg) cells. Second, in the untreated animal (Figure 4C), the number of Teff cells continued to increase until the graft was rejected (at day 14) while in the treated animal (Figure 4B) the absolute number Teff cells was much lower (note the ten-fold difference in the vertical scales of the two plots) and was declining between day 7 and day 14. Tolerance induction, at least in this model, was accompanied by reducing the number of graft-attacking Teff cells, rather than increasing the number of graft-producing Treg cells [13]. This study is a prime example of using in vivo flow cytometry to help biologists answer mechanistic questions that were previously difficult to address because the kinetics of T cell expansion was not known (therefore it was not known what would have been the optimum time point to draw blood for ex vivo analysis). The results also suggest that early assessment of graft survival or failure may be possible by monitoring the Teff and Treg populations in the peripheral circulation a few days after transplantation.
5. Future prospects
From the examples described in the previous sections, it is clear that in vivo flow cytometry fills a biological need as a research tool for cell tracking in live animals. At this stage of the development, the major limitation of the technique is its inability to detect very rare circulating cells. In its most common implementation, a small artery in the mouse ear is used for measurement. The vasculature of the ear is readily accessible, and the same blood vessel can be identified from day to day for longitudinal monitoring. However, the total blood volume flowing through a single 30 μm diameter artery in a 1 min scan is <1 μL, or less than 10−3 of the total blood volume in a mouse. That means on average we need about 1000 fluorescent cells in the entire circulation in order to detect 1 cell/min with the in vivo flow cytometer. This number is somewhat improved with the development of the retinal flow cytometer that can detect circulating cells in all the major retinal blood vessels (5 artery/vein pairs) around the optic disc of the eye [6]. Compared to the original in vivo flow cytometer, the number of detected cells using the retinal flow cytometer was approximately 5 times higher [6], but the lower detection limit (~200 fluorescent cells per mL of blood) is still two orders of magnitude higher than needed to detect very rare circulating cells, such as mobilized blood stem cells.
For eventual human applications, either (i) endogenous signals will need to be identified that are specific markers for the cell population of interest [16,21,23], or (ii) molecular probes will need to be developed that label specific human cell populations and are approved for human use. If it is an injectable probe, there needs to be justification for probe injection rather than blood draw. A reasonable justification would be if a single injection of the probe results in prolonged labeling of the circulating cells, enabling longitudinal monitoring without altering the function of the labeled cells. In addition, light intensity will need to be carefully monitored and kept below the safe exposure limit for human use. In this regard, more sensitive detection scheme need to be explored, including photoacoustic or photothermal detection schemes (18–25) as possible alternatives.
Acknowledgments
The authors wish to thank Juwell Wu for generous help with figure preparation for this manuscript.
Footnotes
This research was funded by NIH P01 CA111519, R01 CA125690, R21 HL098750, and R01 HL0095722.
References
- 1.Novak J, Georgakoudi I, Wei X, Prossin A, Lin CP. An in vivo flow cytometer for real-time detection and quantification of circulating cells. Optics Lett. 2004;29:77–79. doi: 10.1364/ol.29.000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgakoudi I, Solban N, Novak J, Rice WL, Hasan T, Lin CP. In vivo flow cytometry: A new method for the quantification of circulating tumor cells. Cancer Res. 2004;64:5044–47. doi: 10.1158/0008-5472.CAN-04-1058. [DOI] [PubMed] [Google Scholar]
- 3.Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumor engraftment. Nature. 2005;435:969–73. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei X, Sipkins DA, Pitsillides CM, Novak J, Georgakoudi I, Lin CP. Real-time detection of circulating apoptotic cells by in vivo flow cytometry. Mol Imaging. 2005;4:415–6. doi: 10.2310/7290.2005.05148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee H, Alt C, Pitsillides C, Puoris’haag M, Lin CP. In vivo imaging flow cytometer. Opt Exp. 2006;14:7789–800. doi: 10.1364/oe.14.007789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alt C, Veilleux I, Lee H, Pitsillides CM, Côté D, Lin CP. Retinal flow cytometer. Opt Lett. 2007;32:3450–3452. doi: 10.1364/ol.32.003450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boutrus S, Greiner C, Hwu D, Chan M, Kuperwasser C, Lin CP, Georgakoudi I. Portable two-color in vivo flow cytometer for real-time detection of fluorescently-labeled circulating cells. J Biomed Opt. 2007;12:020507. doi: 10.1117/1.2722733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM, Jia X, Lu G, Timm M, Kumar A, Hedin KE, Cote D, Veilleux I, Hedin KE, Roodman GD, Witzig TE, Kung AL, Hideshima T, Anderson KC, Lin CP, Ghobrial IM. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12) -dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leleu X, Jia X, Runnels J, Ngo HT, Moreau A-S, Farag M, Spencer JA, Pitsillides CM, Hatjiharissi E, Roccaro A, O’Sullivan G, McMillin DW, Moreno D, Kiziltepe T, Carrasco R, Treon SP, Hideshima T, Anderson KC, Lin CP, Ghobrial IM. The Akt pathway regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2007;110:4417–4426. doi: 10.1182/blood-2007-05-092098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azab AK, Runnels JM, Pitsillides C, Moreau A-S, Azab F, Leleu XP, Jia X, Wright R, Ospina B, Carlson AL, Alt C, Burwick N, Roccaro AM, Ngo HT, Farag M, Melhem MR, Sacco A, Munshi NC, Hideshima T, Rollins BJ, Anderson KC, Kung AL, Lin CP, Ghobrial I. The CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, Roccaro AM, Ngo HT, Melhem MR, Sacco A, Jia X, Anderson KC, Lin CP, Rollins BJ, Ghobrial IM. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009;114:619–629. doi: 10.1182/blood-2009-01-199281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roccaro AM, Sacco A, Husu EM, Pitsillides C, Vesole S, Azab AK, Azab F, Melhem M, Ngo HT, Quang P, Maiso P, Runnels J, Liang M-C, Wong K-K, Lin C, Ghobrial IM. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood. 2010;115:559–569. doi: 10.1182/blood-2009-07-235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan Z, Spencer JA, Lu Y, Pitsillides CM, Singh G, Kim P, Yun SH, Toxavidis V, Strom TB, Lin CP, Koulmanda M. In vivo tracking of “color-coded” effector, natural and induced regulatory T cells in the allograft response. Nat Med. 2010;16:718–722. doi: 10.1038/nm.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Runnels JM, Carlson AL, Pitsillides C, Thompson B, Wu J, Spencer JA, Kohler JMJ, Azab A-K, Moreau A-S, Rodig SJ, Kung A, Anderson KC, Ghobrial IM, Lin CP. Characterization of multiple myeloma engraftment, growth and response to therapy using complementary in vivo optical technologies. J Biomed Optics. 2011;16:011006. doi: 10.1117/1.3520571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He W, Wang H, Hartmann LC, Cheng JX, Low PS. In vivo quantification of rare circulating tumor cells by multiphoton intravital flow cytometry. Proc Natl Acad Sci U S A. 2007;104:11760–11765. doi: 10.1073/pnas.0703875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le TT, Huff TB, Cheng J-X. Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer. 2009;9:42. doi: 10.1186/1471-2407-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tkaczyk ER, Zhong CF, Ye JY, Myc A, Thomas T, Cao Z, Duran-Struuck R, Luker KE, Luker GD, Norris TB, Baker JR. In vivo monitoring of multiple circulating cell populations using two-photon flow cytometry. Opt Commun. 2008;281:888–894. doi: 10.1016/j.optcom.2007.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biris AS, Galanzha EI, Li ZR, Mahmood M, Xu Y, Zharov VP. In vivo Raman flow cytometry for real-time detection of carbon nanotube kinetics in lymph, blood, and tissues. J Biomed Opt. 2009;14:021006. doi: 10.1117/1.3119145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudryashov SI, Allen SD, Galanzha EI, Galitovskaya E, Zharov VP. Photoacoustics of individual live cells and particles. Proc SPIE. 2006;6086:143–154. [Google Scholar]
- 20.Zharov V, Galanzha E, Shashkov E, Kim J-W, Khlebtsov NG, Tuchin VV. Photoacoustic flow cytometry: principle and application for real-time detection of circulating single nanoparticles, pathogens, and contrast dyes in vivo. J Biomed Opt. 2007;12:051503. doi: 10.1117/1.2793746. [DOI] [PubMed] [Google Scholar]
- 21.Zharov VP, Galanzha EI, Shashkov EV, Khlebtsov NG, Tuchin VV. In vivo photoacoustic flow cytometry for monitoring of circulating single cancer cells and contrast agents. Opt Lett. 2006;31:3623–5. doi: 10.1364/ol.31.003623. [DOI] [PubMed] [Google Scholar]
- 22.Galanzha EI, Shashkov EV, Kelly T, Kim JW, Yang LL, Zharov VP. In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells. Nat Nanotechnol. 2009;4:855–860. doi: 10.1038/nnano.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galanzha EI, Shashkov EV, Spring PM, Suen JY, Zharov VP. In vivo, noninvasive, label-free detection and eradication of circulating metastatic melanoma cells using two-color photoacoustic flow cytometry with a diode laser. Cancer Res. 2009;69:7926–7934. doi: 10.1158/0008-5472.CAN-08-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu M, Wang LV. Photoacoustic imaging in biomedicine. Rev Sci Instrum. 2006;77:041101. [Google Scholar]
- 25.Zharov VP, Galanzha EI, Tuchin VV. In vivo photothermal flow cytometry: Imaging and detection of cells in blood and lymph flow. J Cell Biochem. 2006;97:916–932. doi: 10.1002/jcb.20766. [DOI] [PubMed] [Google Scholar]
- 26.Badger CC, Krohn KA, Shulman H, Flournoy N, Berstein ID. Experimental Radioimmunotherapy of Murine Lymphoma with 131I-labeled Anti-T-Cell Antibodies. Canc Res. 1986;46:6223–6228. [PubMed] [Google Scholar]
- 27.Matthews DC, Appelbaum FR, Eary JF, Fisher DR, Durack LD, Hui TE, Martin PJ, Mitchell D, Press OW, Storb R, Bernstein ID. Phase I Study of 131I-Anti-CD45 Antibody Plus Cyclophosphamide and Total Body Irradiation for Advanced Acute Leukemia and Myelodysplastic Syndrome. Blood. 1999;97:1237–1247. [PubMed] [Google Scholar]
- 28.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toft P, Svendsen P, Tonnesen E, Rasmussen JW, Christensen NJ. Redistribution of lymphocytes after major surgical stress. Acta Anaesthesiol Scand. 1993;37:245–249. doi: 10.1111/j.1399-6576.1993.tb03708.x. [DOI] [PubMed] [Google Scholar]
- 31.Fosse E, Opdahl H, Aakvaag A, Svennevig JL, Sunde S. White blood cell populations in patients undergoing major vascular surgery. Scand J Thorac Cardiovasc Surg. 1985;19:247–252. doi: 10.3109/14017438509102726. [DOI] [PubMed] [Google Scholar]
- 32.Bolton PM, Kirov SM, Donald KJ. The effects of major and minor trauma on lymphocyte kinetics in mice. Aust J Exp Biol Med Sci. 1979;57:479–492. doi: 10.1038/icb.1979.49. [DOI] [PubMed] [Google Scholar]