

Abstract
Aptamer microarrays are a promising high-throughput method for ultrasensitive detection of multiple analytes, but although much is known about the optimal synthesis of oligonucleotide microarrays used in hybridization-based genomics applications, the bioaffinity interactions between aptamers and their targets is qualitatively different and requires significant changes to synthesis parameters. Focusing on streptavidin-binding DNA aptamers, we employed light-directed in situ synthesis of microarrays to analyze the effects of sequence fidelity, linker length, surface probe density, and substrate functionalization on detection sensitivity. Direct comparison with oligonucleotide hybridization experiments indicates that aptamer microarrays are significantly more sensitive to sequence fidelity and substrate functionalization and have different optimal linker length and surface probe density requirements. Whereas microarray hybridization probes generate maximum signal with multiple deletions, aptamer sequences with the same deletion rate result in a 3-fold binding signal reduction compared with the same sequences synthesized for maximized sequence fidelity. The highest hybridization signal was obtained with dT 5mer linkers, and the highest aptamer signal was obtained with dT 11mers, with shorter aptamer linkers significantly reducing the binding signal. The probe hybridization signal was found to be more sensitive to molecular crowding, whereas the aptamer probe signal does not appear to be constrained within the density of functional surface groups commonly used to synthesize microarrays.
Sensitive and accurate multiplexed protein measurements are fundamental for modern biomedical research and clinical practice. Immunoassays exploiting the diversity and specificity of antibody–antigen binding are the most commonly used and widely accepted methods for both single and multiplexed measurements.1 In recent years, however, aptamers—single-stranded nucleic acids generated by in vitro selection from combinatorial libraries to bind to specific target molecules (SELEX2−4)—are providing an alternative path to sensitive protein analysis. Perhaps one of the principal appeals of aptamer-based technology is that it leverages highly developed and versatile chemical synthesis of nucleic acids with in vitro selection to provide a purely chemical development pathway. The tool palette for aptamer synthesis includes not only natural DNA and RNA nucleoside monophosphates, but also non-natural building blocks with modifications at the 2′ position, such as 2′-O-methyl, 2′-fluoro-, and 2′-F-ANA;5−8 more profound sugar modifications, such as locked nucleic acid (LNA) and hexitol nucleic acid (HNA);9−11 or backbone modifications, such as phosphorodithioate linkages.12,13 The versatility of phosphoramidite chemistry also extends to the facile synthesis of complex microarrays, traditionally for genomics applications, but readily adaptable to arrays of aptamers, both for aptamer optimization14,15 and for aptamer-based multiplexed protein detection.16−18 The widespread use of oligonucleotide microarrays for high-throughput gene expression studies, as well as other hybridization-based genomics applications, provides the analytical, technological, and manufacturing infrastructure for the development of aptamer microarrays. Aptamer microarrays consist of aptamers immobilized on a solid substrate. Aptamer arrays are promising analytical tools because in vitro selection provides nucleic acid sequences with specificities and binding affinities comparable to those of monoclonal antibodies;19−21 however, they are more stable22 as well as easier to synthesize than antibody arrays because of the mature solid-phase oligonucleotide synthesis and spotting technology, and in situ synthesis of oligonucleotide microarrays using phosphoramidite chemistry.23−27
At the simplest level, oligonucleotide microarrays consist only of sequences of ∼25–60 nucleotides long immobilized on a substrate in a defined pattern. However, in the case of traditional hybridization-based microarray experiments, much effort has been devoted to understanding and optimizing technical parameters influencing aspects such as hybridization kinetics, efficiency, and signal intensity/noise ratio to maximize their analytical power. Clearly, for both aptamer arrays and hybridization-based arrays, sequence design is the most important consideration, but here, we consider the impact of synthesis parameters that also significantly affect microarray performance: microarray surface chemistry, oligonucleotide surface density, sequence fidelity, and surface-to-probe spacer length. We investigate the effect of these parameters on aptamer signal intensity and make direct comparisons with how the same parameters affect signal intensity in analogous hybridization experiments to gain insight into how to best adapt existing oligonucleotide microarray technology for aptamer-based bioaffinity applications.
Surface chemistry is used to modify the glass substrate to enable DNA attachment and, therefore, also determines the surface density of bound probes.28,29 Surface density strongly influences hybridization intensity and signal due to steric factors.30 In addition, the surface functionalization serves as a spacer, distancing the probes from the glass surface, which is known to increase hybridization efficiency.28,31,32 In addition, the surface functionalization changes surface electrostatics and hydrophobicity, which in turn influence hybridization and background intensity.33−35 Although the surface functionalization also serves as a spacer, additional distance between the surface and the array oligonucleotides can be introduced via specialized linker phosphoramidites or by oligonucleotide sequences, typically poly(dT).25,36−38 Finally, oligonucleotide sequence fidelity also plays a role in both hybridization and protein binding to aptamers. The relationship between the number and position of mismatches in the case of hybridization on microarrays is fairly predictable,39,40 but less so in the case of aptamers, in which the effect of mutations is highly variable.14
Experimental Section
Microarray Synthesis
DNA/aptamer microarrays were fabricated using maskless array synthesis (MAS) as described previously.27,41 Briefly, microarray substrates were used as received from the manufacturer, with the exception of the hydroxyl-functionalized substrates, which were Schott Nexterion Glass D slides functionalized with N-(3-triethoxysilylpropyl)-4-hydroxybutyramide (Gelest SIT8189.5). These slides were loaded into a metal staining rack and completely covered with a 500 mL solution consisting of 10 mg of the silane in 95:5 (v/v) ethanol/water and 1 mL of acetic acid. The slides were covered and gently agitated for 4 h and then rinsed twice for 20 min with gentle agitation in the same solution, but without the silane. The slides were then drained and cured overnight in a preheated vacuum oven (120 °C). The slides were stored in a desiccator cabinet until use.
Microarrays were synthesized directly on the slides using a maskless array synthesizer, which consists of an optical imaging system that uses a digital micromirror device to deliver patterned ultraviolet light near 365 nm to the synthesis surface. Microarray layout and oligonucleotide sequences are determined by selective removal of the 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) photocleavable 5′-hydroxyl protecting group on the oligonucleotides. Reagent delivery and light exposures are synchronized and controlled by a computer, which also stores and orders the display on the micromirror array. The chemistry is similar to that used in conventional solid-phase oligonucleotide synthesis. The primary modification is the use of NPPOC phosphoramidites. Upon absorption of a photon near 365 nm and in the presence of a weak organic base, 1% (m/v) imidazole in DMSO, the NPPOC group comes off, leaving a 5′-hydroxyl terminus that is able to react with an activated phosphoramidite during the next synthetic cycle. After synthesis, the microarrays were deprotected in 1:1 (v/v) ethylenediamine in ethanol for 2 h at room temperature, washed twice with water, and stored dry until use.
Microarray Linker-Length and Exposure-Gradient Experiments
The effect of spacer length and hybridization and aptamer binding were determined on microarrays synthesized with spots with stepwise increases in thymidine (dT) linker length (“linker gradients”) followed by either the St-2-1 streptavidin binding aptamer sequence42 or a sequence (QC25) of similar length and known to hybridize well (hybridization probes and aptamer sequences are given in Table 1). The effect of sequence fidelity on hybridization and aptamer binding were determined with microarrays synthesized with a light-exposure gradient. Spots synthesized with lower light exposure are insufficiently deprotected and therefore have deletion errors. These arrays were synthesized with a fixed-length dT 5mer linker and an exposure gradient between 0.2 and 18 J/cm2 at 365 nm. There is an exponential relationship between exposure and deprotection, with 6 J/cm2 corresponding to ∼99% NPPOC removal, and 12 J/cm2 corresponding to >99.9% removal (1% and <0.1% deletions per synthesis cycle, respectively).27
Table 1. Streptavidin Binding Aptamer Sequence St-2-1 and Mutant Sequences Derived from St-2-1, along with the Sequences Used in the Hybridization Experimentsa.
| name | sequence | length | affinity (%) |
|---|---|---|---|
| St-2-1 | ATT GAC CGC TGT GTG ACG CAA CAC TCA AT | 29 | 85 ± 3 |
| St-2-A | GCT ATT GAC CGC TGT GTG ACG CAA CAC TCA ATA GC | 35 | 86 ± 3 |
| St-2-T-1 | TTG ACC GCT GTG TGA CGC AAC ACT CAA | 27 | 73 ± 6 |
| St-2-T-2 | TGA CCG CTG TGT GAC GCA ACA CTC A | 25 | 25 ± 7 |
| St-2-T-3 | GAC CGC TGT GTG ACG CAA CAC TC | 23 | 23 ± 5 |
| St-2-R-1 | ATT GAC GCG TGT GAC GCA ACA CTC AAT | 27 | 60 ± 4 |
| St-2-R-2 | TAT TGA GTG TGA CGC AAC ACT CAA TA | 26 | 13 ± 7 |
| St-2-M-1 | ATT GAC CTC TGT GTG ACG CAA CAC TCA AT | 29 | 21 ± 5 |
| St-2-M-2 | ATT GAC CGC TGT GTG ACT CAA CAC TCA AT | 29 | 11 ± 8 |
| St-2-M-3 | ATT GAC CGC TGT GTA ACG CAA CAC TCA AT | 29 | 12 ± 3 |
| St-2-1_rev | TAA CTC ACA ACG CAG TGT GTC GCC AGT TA | 29 | |
| QC1 | CTG TTC TGC ATC CTG CCT TTA CAT T | 25 | |
| QC3 | GTT TGA GAC CAG TCT GAC CAA CAT G | 25 | |
| QC6 | TCT ACT ATC CCT AAG CCC ATT TCT C | 25 | |
| QC8 | GTT GTC ACA CAT ACA CTG CTC GAA A | 25 | |
| QC11 | CGG GCG GTC TCA ATC AAG CAT GGA TTA CGG TGT TTA CTC TGT CCT GCG GT | 50 | |
| QC13 | AGA GGA TGA CAA GGA CAC AAT CGT GCT CCC ATC TGT ATT CTT TAC GAA CT | 50 | |
| QC25 | GTC ATC ATC ATG AAC CAC CCT GGT C | 25 |
Binding affinity was determined by Bing et al. in a competition assay with FAM-labeled St-2-1. On our arrays, St-2-1_rev was used as a negative control sequence. The correlation between the affinity data and microarray binding data is given in Figure 6.
Exposure gradient and spacer gradient microarrays were hybridized in an adhesive chamber (SecureSeal SA200, Grace Biolabs) with a solution consisting of 0.3 pmol of 5′-Cy3-labeled probe, 40 μg of herring sperm DNA, and 200 μg of acetylated BSA in 400 μL of MES buffer (100 mM MES, 1 M NaCl, 20 mM EDTA, 0.01% Tween-20). After 2 h of rotation at 42 °C, the chamber was removed, and the microarrays were vigorously washed in a 50 mL centrifuge tube with 30 mL of nonstringent wash buffer (SSPE; 0.9 M NaCl, 0.06 M phosphate, 6 mM EDTA, 0.01% Tween-20) for 2 min and then, similarly, with stringent wash buffer (100 mM MES, 0.1 M NaCl, 0.01% Tween-20) for 1 min. Finally, the microarrays were dipped for a few seconds in the final wash buffer (0.1 × SSC) and then dried with a microarray centrifuge. Arrays were scanned with a Molecular Devices GenePix 4400A, and the intensity data were extracted with GenePix Pro.
Aptamer Array QC Hybridization
To determine the quality of the synthesized aptamer arrays, the arrays were hybridized with a mixture containing 15 different 25-mer and 50-mer Cy3-labeled QC oligomers (QC1-5, Sigma Genosis, UK; QC6-15, IDT, Belgium) varying in concentration from 0.01 to 100 pM. A solution of 5xSSC/0.1% SDS (SSC, AccuGene Lonza, Belgium; SDS, Sigma, USA) was used as the hybridization buffer. The hybridization mixture was preheated for 5 min at 95 °C, and a volume of 440 μL was used for hybridization in Agilent-one backing slides (G2534-60005, Agilent). Arrays were hybridized over 18 h in an Agilent oven at 45 °C. After hybridization, arrays were quickly rinsed with 6xSSPE/0.01% Tween-20 at room temperature (r.t.) (SSPE, AccuGene Lonza, Belgium; Tween-20, Sigma, Switzerland), washed at r.t. with 6xSSPE/0.01% Tween-20 for 1 min, washed at 45 °C with 0.6xSSPE/0.01% Tween-20 for 10 min, and washed at r.t. with 6xSSPE/0.01% Tween-20 for 10 min. Finally, arrays were dried by centrifugation and scanned at PMT10 using the Agilent High-Resolution C Scanner. Data were extracted using ImaGene 7.5 software.
On-Array Streptavidin Binding Assay
To determine the optimal synthesis parameters for on-array aptamer binding experiments, arrays were used to monitor binding of streptavidin to aptamer sequences on the array after the quality check by QC hybridization. First, the arrays were prewetted with 5xSSC/0.01%-Tween-20 for 30 min at 45 °C. Next, the arrays were blocked with SuperblockT20 (Thermo Scientific) for 30 min at room temperature (r.t.). After blocking, the arrays were incubated with streptavidin (ProSpec, USA) using an incubation mixture containing 1xPBS/1 mM MgCl2/0.01% Tween-20/1% BSA/10 μg per mL streptavidin (PBS, Ambion, USA; MgCl2, Sigma, Germany; BSA, Sigma, USA). Incubation was performed at r.t. for 30 min. After streptavidin incubation, the arrays were rinsed three times with 1xPBS/1 mM MgCl2/0.05% Tween-20 and subsequently washed with the same buffer for 75 min at r.t., then the arrays were incubated for 30 min at r.t. with Cy5-labeled biotin, using a mixture containing 1xPBS/1 mM MgCl2/0.01% Tween-20/1 nM Biotin-dT5-Cy5 (IDT). After biotin incubation, the arrays were rinsed three times with 1xPBS/1 mM MgCl2/0.05% Tween-20, and subsequently washed with the same buffer for 30 min at r.t. After a final quick rinse with deionized water, the arrays were dried by centrifugation and scanned at PMT10 using the Agilent high-resolution C scanner. Data were extracted using ImaGene 7.5 software.
Results and Discussion
Influence of T-Spacer Length on Aptamer Binding vs DNA Hybridization
To test the influence of dT-spacers on the ability of the St-2-1 aptamer (Table 2) to bind to streptavidin, an array was synthesized on hydroxyl-functionalized substrates, with the aptamer sequence on spacers ranging in length from an oligo-dT 1mer to 25mers. The linkers were synthesized without capping steps to preserve equal probe density. To make a direct comparison with the influence of the spacers on DNA hybridization on the microarrays, the same array design was synthesized with the 25mer sequence (GTC ATC ATC ATG AAC CAC CCT GGT C). In both cases, the surface functionalization contributes ∼10 carbon–carbon bond lengths to the spacer, approximately equivalent to 2 dTs. Figure 1 summarizes the results of these experiments, which indicate that there is an optimum spacer length in both cases: dT5 for hybridization and dT11 for aptamer binding. In the case of hybridization on microarrays, it is known that a spacer improves hybridization by making the probe more accessible; on the other hand, excessively long spacers can hinder hybridization, presumably because ssDNA forms a random coil on the surface, allowing the probe to be covered, or “dissolved” in a mass of linker DNA.31
Table 2. St-2-1 Binding Data for Microarrays Synthesized on Different Substrates.
| dT10 spacer |
|||||
|---|---|---|---|---|---|
| surface | R2 a | ST-2-1 signal | ST-2-1/ST-2-1_rev | ST-2-1/backgrnd | rel SDb ST-2-1 |
| UltraGAPS | 0.90 | 154 | 8.5 | 9.2 | 33 |
| Schott E | 0.18 | 35 | 1.3 | 1.8 | 52 |
| Schott A+ | 0.83 | 56 | 2.3 | 2.7 | 97 |
| Hydroxyl | 0.72 | 48 | 3.0 | 3.4 | 55 |
R2 is the linearity with the binding affinity data from Bing et al.42
Rel SD of refers to the 14 replicates of ST-2-1 on the microarray.
Figure 1.
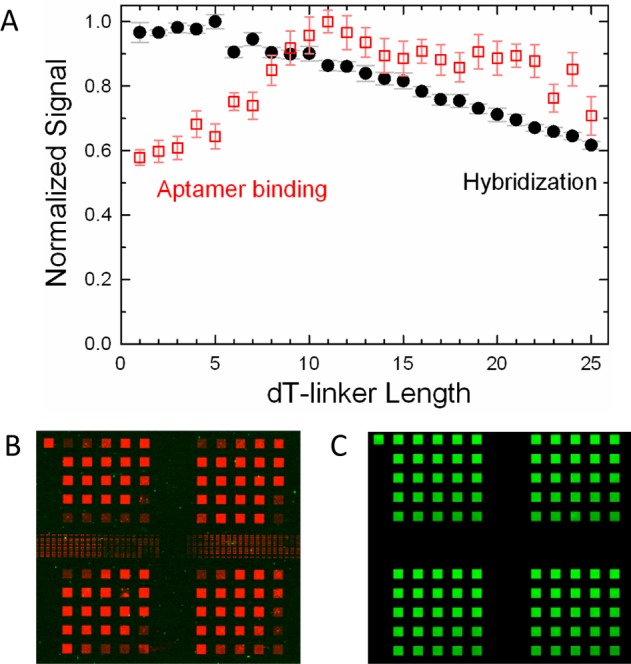
(A) Normalized fluorescent signals vs linker length from the St-2-1 aptamer binding assay (red empty squares), and from the equivalent 25mer hybridization experiment. (B) Microarray image of aptamer linker gradient showing four replicates and with the length increasing from left to right and from top to bottom in 25 steps from 1 to 25 dTs. (C) Equivalent image of hybridization experiment. Each of the square features on the microarray is synthesized using a 10 × 10 array of 16 μm DMD mirrors.
The effect of spacer length on hybridization can be clearly seen in experiments with radiolabeled DNA. Shchepinov et al.30 found a signal maximum with 10 couplings of a spacer phosphoramidite, equivalent to a total length of about 100 carbon–carbon bonds. Fluorescent labeling of the target sequence obscures the effect due to interactions between the fluorescent dye and the microarray substrate. Proximity to the surface greatly increases the fluorescence of cyanine dyes in hybridization experiments.37 We have also observed this effect with microarrays synthesized with oligonucleotides of increasing lengths terminated by a coupling with a Cy3 or Cy5 phosphoramidite (data not shown). The results in Figure 1 are the convolution of the two effects and indicate that the optimum spacer length for the aptamer is significantly longer than the optimum in the hybridization studies. That the St-2-1 aptamer binding signal decreases after 11 dTs is in contrast with the results for the IgE-binding aptamer, for which the signal is proportional to the oligo(dT) length up to at least dT 20mers.14 Lao et al.43 also found that the fluorescence signal from human α-thrombin HTQ and HTDQ aptamer binding was significantly higher for a dT12 vs a dT6 spacer, but did not explore other oligo(dT) lengths. In all of these cases, the labeling dyes used were cyanine dyes, which suggests that specific aptamer properties, rather than dye–substrate interactions, are the source of the differing spacer requirements, but we cannot exclude the possibility that differences between Cy3- and Cy5-labeling account for the difference. Nevertheless, it seems likely that the optimum spacer length for aptamer microarrays will need to be independently optimized for each aptamer and for each labeling dye.
Role of Sequence Fidelity and Surface Density in Aptamer Binding vs Hybridization
The tolerance of aptamer microarrays to sequence error is important for maximizing the binding signal. Sequence fidelity is particularly relevant in the case of in situ microarray synthesis, for which postsynthesis oligonucleotide purification is not possible. Phosphoramidite chemistry has been optimized to yield average stepwise coupling efficiencies well above 99% for DNA monomers and ∼99% for RNA monomers, for both solid-phase synthesis and in situ synthesis on microarrays,27,44 but other sources of error, most prominently depurination, reduce the overall yield further.45 Spotted microarrays may use HPLC- or gel electrophoresis-purified oligonucleotides to reach a sequence purity greater than ∼85%, although the value for this level of purity has not been established for aptamer applications or for traditional microarray applications. In addition to lacking the option of oligonucleotide purification, in situ microarray synthesis results in a significantly higher error rate as a result of the additional complexity associated with the simultaneous synthesis of large numbers of sequences.25,27 Here, we take advantage of the high degree of control afforded by maskless array synthesis (MAS) to assess the sequence fidelity requirement of hybridization vs protein binding to aptamers.
When a single, short sequence is synthesized on a microarray substrate with MAS, the minimal complexity results in very high fidelity oligonucleotides. This is a result of the elimination of stray light effects present when multiple sequences are synthesized simultaneously as well as by the intrinsically low rate of depurination and other side reactions in the MAS chemistry, which does not use acidic conditions and requires minimal exposure to oxidants.27 Synthesis errors can then be introduced in a controlled manner by reducing the UV light exposure, which results in decreased NPPOC cleavage and, hence, the introduction of deletion errors.
Figure 2 (black squares) shows the results from a hybridization experiment on a microarray synthesized with a light exposure gradient between 0.2 and 18 J/cm2 at 365 nm. The fraction of NPPOC groups cleaved with a given light exposure is a first-order exponential with the form 1-e–t/τ, where t is the light exposure and τ is a rate constant of ∼1.3 J/cm2.27 The fraction of sequences with no deletion errors is therefore ∼(1 – e–t/τ)n, where n is the sequence length. Microarray features (“spots”) that receive the lowest exposures have a very low surface density of hybridizable oligonucleotides (“hybridizable” is defined here as capable of forming a hybrid under typical stringent hybridization conditions), but the hybridization signal increases rapidly to reach a maximum around 6 J/cm2. At this exposure, ∼5% of the NPPOC groups remain attached (12 J/cm2 is required for >99.9% photodeprotection). This is a clear indication that microarrays used in hybridization experiments have a very high error tolerance. For the 25mer sequence used in these experiments, the 6 J/cm2 optimum exposure results in ∼75% of sequences with at least one deletion error. Surface density of oligonucleotides plays a role in this effect, as it is known that hybridization efficiency decreases quickly with oligonucleotide surface density, presumably due to steric crowding.31,46
Figure 2.
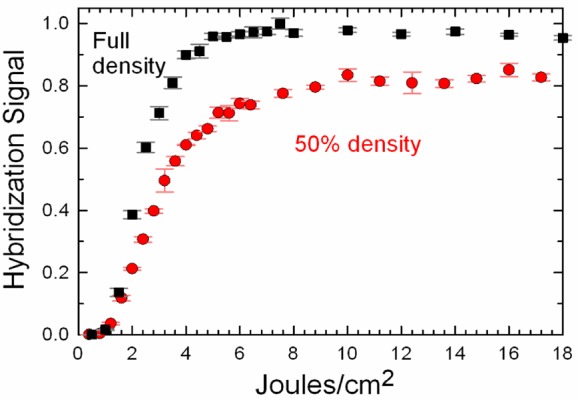
Normalized hybridization fluorescent signals for a single 25mer sequence synthesized with a photodeprotection light exposure gradient between 0.2 and 18 J/cm2. The black squares are from a microarray synthesized for maximum surface density of oligonucleotides. The red circles are from an equivalent experiment, but with the oligonucleotide surface density reduced by 50% by a partial light exposure followed by capping. The microarray layout is the same as for the data shown in Figure 1, but with 25 exposure steps replacing the 25 linker steps.
We hypothesized that microarray hybridization efficiency remains constant above a certain oligonucleotide fidelity threshold because then the critical parameter is the density of hybridizable oligonucleotides, rather than just the density of the oligonucleotides. This is because the single-to-double-stranded transition doubles both the surface density of oligonucleotides and the density of charged groups, hindering further hybridization. In addition, the much higher persistence length of dsDNA (∼50 nm47) vs ssDNA (∼2 nm48) may restrict diffusion near the surface and thereby inhibit further hybridization.
To test this hypothesis, we reduced the surface density by 50% after the linker synthesis using a light exposure corresponding to a 50% NPPOC cleavage (2 J/cm2) and capped the resulting 5′-OH groups with a dimethoxytrityl-dT phosphoramidite (DMT-dT) coupling.49 The same 25mer sequence as above was then synthesized with an exposure gradient, and the results are plotted alongside those of the full density experiment in Figure 2 (red circles). These data indicate that the hybridization signal reaches ∼80% of the intensity of the full density results. In addition, the light exposure resulting in maximum signal shifts higher, to at least 10 J/cm2 or perhaps more, as the slope remains slightly positive up to 18 J/cm2. These results are compatible with our hypothesis. Specifically, the signal is much higher than 50%, indicating that hybridization on the full density array, and at exposures greater than 6 J/cm2, is constrained by molecular crowding. In addition, the optimum light exposure of the 50% density data is much higher, indicating that with reduced molecular crowding, the increase in sequence fidelity resulting from greater light exposures leads to increased hybridization. The optimum light exposure also depends on sequence length, with light exposure gradients of 60mers reaching a maximum hybridization signal at ∼3 J/cm2 (Supporting Information Figure S1), which is also in agreement with the crowding hypothesis, since longer sequences result in a greater mass of DNA on the surface.
The St-2-1 aptamer binds to the 60 kDa protein streptavidin, a much bulkier macromolecule than the 8 kDa complementary 25mer sequence in the hybridization experiments. In addition, the St-2-1 aptamer forms a double-stranded structure with two loops, which may further increase molecular crowding at the microarray surface. As a result, we hypothesized that St-2-1 aptamer microarrays would have a lower optimal oligonucleotide density. We also hypothesized that the aptamer array would be less error-tolerant than the comparable hybridization array. The sensitivity of aptamers to sequence fidelity was explored by Katilius et al.,14 who used in situ synthesized microarrays to determine the effect of mutations on the affinity of the IgE-binding aptamer. They found that the majority of single mutations result in near complete loss of binding. Similar experiments exploring the effects of defects on hybridization affinity, also on microarrays, indicated that hybridization is significantly less error-sensitive.40
To test the role of sequence fidelity and surface density in aptamer microarrays, we synthesized the St-2-1 aptamer with the same light exposure gradient as in the hybridization experiment above. Figure 3 (black squares) shows that the optimum exposure, 11 J/cm2, is almost twice that of the hybridization experiment, confirming that the aptamer is more sensitive to errors. Another feature of these data is that after the optimum exposure, the aptamer binding signal drops significantly, which may be an indication of molecular crowding. To test whether the aptamer binding signal is constrained by crowding, the same array was resynthesized with the surface density reduced by 50% after the linker synthesis using the same partial exposure and capping procedure as above. The signal intensity of the 50% density array is shown in Figure 3 (red circles). The data show that the binding signal is reduced by approximately 50%, indicating that, unlike the hybridization results, binding is not significantly constrained by molecular crowding at the normal microarray oligonucleotide surface densities. The curves in Figure 3 exhibit a clear drop at very high light exposures. Since the maxima and the extent of subsequent decline is similar for both surface densities, the effect cannot be attributed to crowding.
Figure 3.
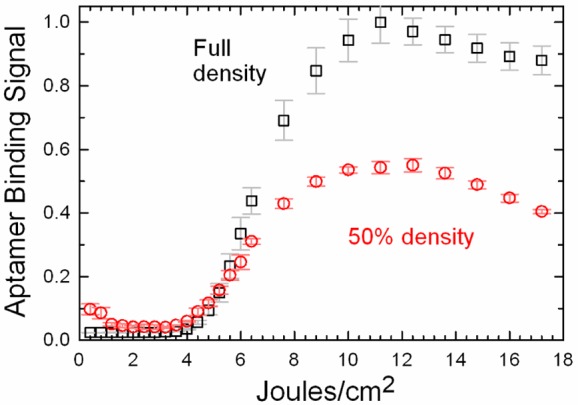
Normalized streptavidin binding signal for a St-2-1 aptamer sequence synthesized with a photodeprotection light exposure gradient between 0.2 and 18 J/cm2. The black squares represent data from a microarray synthesized with a maximum surface density of oligonucleotides. The red circles are from an equivalent experiment, but with a microarray with an oligonucleotide surface density reduced by 50%.
By 12 J/cm2, the photodeprotection is essentially complete (∼99.9%), and additional exposure would not significantly improve the sequence fidelity. Since aptamers appear to be much more sensitive to sequence error than hybridization probes, we speculate that the drop in signal is a consequence of errors introduced by the very high light exposure itself. It is well-known that high doses of UV-A are capable of inducing both single-strand DNA breaks and pyrimidine dimers.50 This result indicates that methods to increase the aptamer surface density, for example, by using substrates functionalized at a higher density (see below) or by the use of branching phosphoramidites, could be effective in increasing aptamer binding signal.
Influence of the Substrate on Aptamer Binding and DNA Hybridization
Surface chemistry is necessary to modify the glass surface to enable the initial phosphoramidite coupling reaction. This surface functionalization determines the surfaced density of bound oligonucleotides. Functionalization chemistry may also affect hybridization by increasing the distance to the glass surface and by changing the surface electrostatics and hydrophobicity, properties which are also known to influence hybridization and nonspecific target binding and, hence, background intensity.33−35 To evaluate the effect of surface chemistry on both aptamer binding and hybridization, we synthesized microarrays containing a variety of hybridization probes and aptamers on four different types of functionalized glass substrates: Corning UltraGAPS (Gamma Amino Propyl Silane), Schott Nexterion Slide A+ (GAPS; 1.0 ± 0.3 × 1012 molecules/cm2), Schott Nexterion ring-opened Slide E (epoxysilane, 5.6 ± 0.3 × 1012 molecules/cm2). Surface densities were provided by Schott, but not Corning. The fourth substrate was functionalized in-house as described in the Experimental Section and have a surface hydroxyl density of about 2.7 × 1012 molecules/cm2.49
Figure 4 shows sections of fluorescent images from the same microarray design synthesized on the four substrate types. The microarrays include both aptamers and hybridization probes but were synthesized with light exposures of 11 J/cm2 to maximize aptamer binding signals. To maximize their comparability, all four microarrays were synthesized consecutively from the same batch of reagents and monomers, and the hybridization assays, followed by the aptamer binding assay, were performed on the four arrays in parallel. Differences in signal and background intensity are apparent for both the aptamer binding signal (Red/Cy5) and hybridization signals (Green/Cy3). The charts in Figure 5 compare the aptamer binding and hybridization signals between microarrays synthesized on the different surfaces and with a dT10 spacer. There is a substantial substrate-dependent difference in aptamer signal, with the UltraGAPS giving a much higher signal than the other substrates. Table 2 gives additional data on the ratios of signal-to-control, signal-to-background, and relative standard deviations of the aptamer signal on the four substrates, and the UltraGAPS substrate leads to substantially better results in all these categories. Since both the UltraGAPS and the A+ substrates are γ-aminopropyl silane functionalized, the higher signal from the UltraGAPS slide may indicate that this substrate is manufactured with a higher density of functional groups than the A+ substrates. Among the three substrates with known surface densities of functional groups, there is no correlation between surface density and aptamer binding signal. However, we cannot exclude that differences among the substrates, other than surface density, are responsible.
Figure 4.

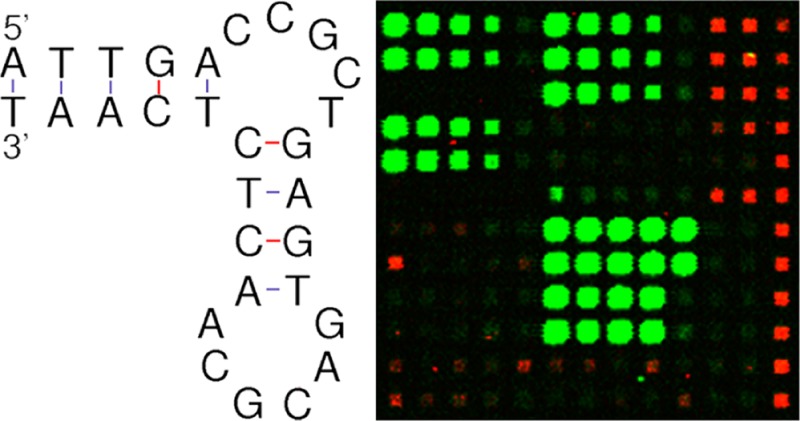
Fluorescent images of sections of microarrays synthesized on different substrates. (A) Corning UltraGAPS, (B) Schott Epoxy ring-opened, (C) Schott A+, (D) Schott Glass D hydroxyl-functionalized with N-(3-triethoxysilylpropyl)-4-hydroxybutryramide. Green features are hybridization signals from Cy3-labeled sequences. Red features are from Cy5-labeled biotin binding to the streptavidin–aptamer pairs. A scheme identifying the sequences corresponding to the spots can be found in the Supporting Information. The arrays were synthesized with light exposures of 11 J/cm2 and with 32 × 32 μm features (4 DMD mirrors) separated by gaps of 48 μm. All images were acquired with the same scanner settings.
Figure 5.
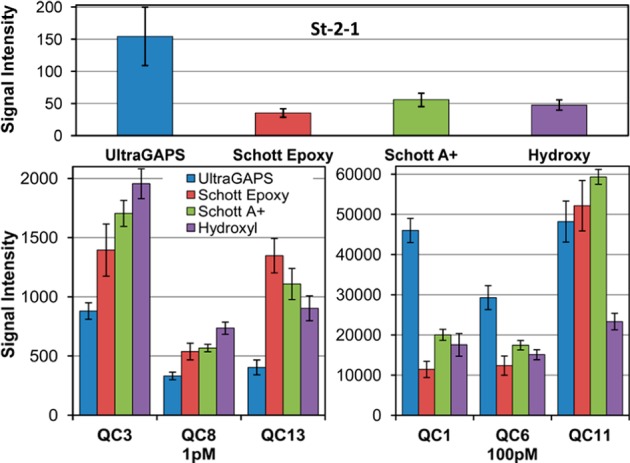
Aptamer binding and hybridization signal comparison between microarrays synthesized with four different surface chemistries (left to right): Corning UltraGAPS/amino-modified; Schott E/epoxy-modified, ring-opened; Schott A+/amino-modified; and in-house hydroxyl-functionalized. Top: St-2-1 aptamer–streptavidin binding signal. Bottom: hybridization signal for three probes hybridized with 1 and 100 pM complementary sequences. Error bars are the standard deviation among replicates.
Hybridization data from the same microarrays are also shown in Figure 5. Each microarray included the probes described in Table 1: QC1, and QC11, the complementary sequences to which were included in the hybridization solution at a concentration of 100 pM. The complementary sequence probes QC3, QC8, and QC13 were present in the hybridization solution at a concentration of 1 pM. Whereas the UltraGAPS substrate performed substantially better than the other substrates in the aptamer binding assay, none of the substrates was clearly superior in the hybridization data. There is a clear indication in the data that the relationship between hybridization signal and substrate is heterogeneous and sequence-specific, suggesting that the surface chemistry significantly influences both hybridization and aptamer binding.
On-Array Streptavidin Binding Assay under Optimized Conditions
To test the suitability of the optimized synthesis conditions (11mer dT linker, 11J/cm2 light deprotection) for on-array aptamer screening, a panel of streptavidin-binding aptamers were chosen as a model. In 2010, Bing et al. described the streptavidin-binding aptamer St-2-1 and a series of St-2-1-derived mutated sequences and their binding affinity for streptavidin measured in a competition assay with FAM-labeled St-2-1.42 The percentage FAM-labeled St-2-1 replaced by the tested aptamer was determined as a measure of binding affinity for the aptamer (Table 1). In Figure 6, the on-array signals for the St-2-1 variants of Table 1 are plotted against the affinity data described in Bing et al., resulting in a correlation coefficient of 0.92.
Figure 6.
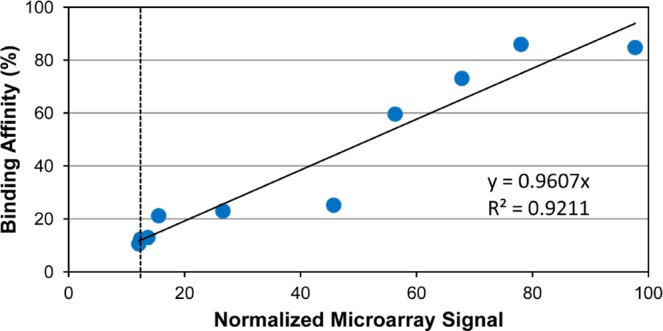
Correlation between data of an on-array aptamer binding assay (14 replicate spots on array) and an off-array competition experiment published by Bing et al. Blue dots are the St-2-1 and the mutated variants of St-2-1 shown in Table 1. Dotted line indicates the signal of the negative control St-2-1_rev.
Figure 7 shows the effect of single mutations in the 29-mer St-2-1 aptamer sequence. For every position, all possible variants were tested, resulting in 87 mutants. The single mutations appear to be less critical in the terminal stem sequence when the mutation is at a distance of four or more bases from the bulge (Figure 7, blue bars). Single mutations in the central stem between both bulges are highly critical (Figure 7, red bars). This is in agreement with the findings of Bing et al., who found that the hairpin bulge structure is critical for streptavidin binding. Most single mutations in the bulges are critical. In their paper, Bing et al. describe that G8, G15, and G18 are important to maintain good binding, and we observe the same in our data. Replacing the G at these positions by one of the other bases lowers the binding affinity to the level of the negative control. Mutant T10C appears to be the only mutant with the same binding affinity as St-2-1.
Figure 7.
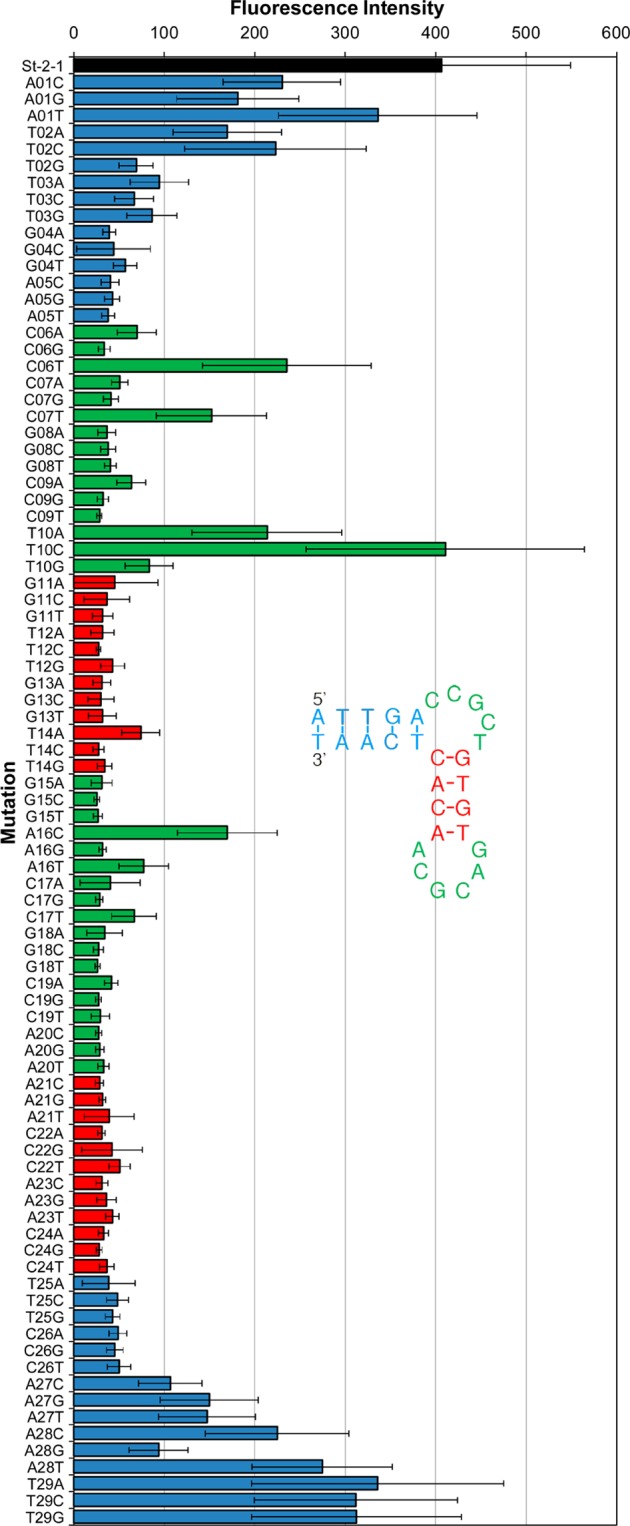
Effect of mutations in the 29-mer St-2-1 aptamer sequence on the binding affinity. Binding was tested using an on-array streptavidin binding assay. Black bar, St-2-1; blue bars, mutation in the terminal stem; green bars, mutation in the bulges; red bars, mutation in the sequences between the bulges. Error bars are based on 12 replicates on the array. Signal negative control St-2-1_rev =28.4.
Figure 8 shows the results of mutants that had one base pair replacement in the double stranded parts of St-2-1. All 27 possible base pair replacements were present on the array (12 replicates). From the results, it can be seen that base pair T14A21 is most critical, since every other combination tested at this position resulted in a significant loss of signal in the streptavidin binding assay. Furthermore, there are a few other replacements that lower the binding affinity significantly, at positions A5T25 (replacement by C5G25 or T5A25) and T12A23 (replacement by C12G23). The insets in Figure 8 show the secondary structure of the variants A12T23, G12C23, and C12G23. In accordance with the data from the on-array streptavidin-binding assay, mutant C12G23 is structurally distinct from St-2-1 and has a much reduced binding affinity, whereas mutants A12T23 and G12C23 are structurally similar to St-2-1 and retain a similar binding affinity (DNA structures from http://mfold.rna.albany.edu/?q=mfold/dna-folding-form).51 Together, these data show that aptamer arrays are a powerful tool for aptamer screening.
Figure 8.
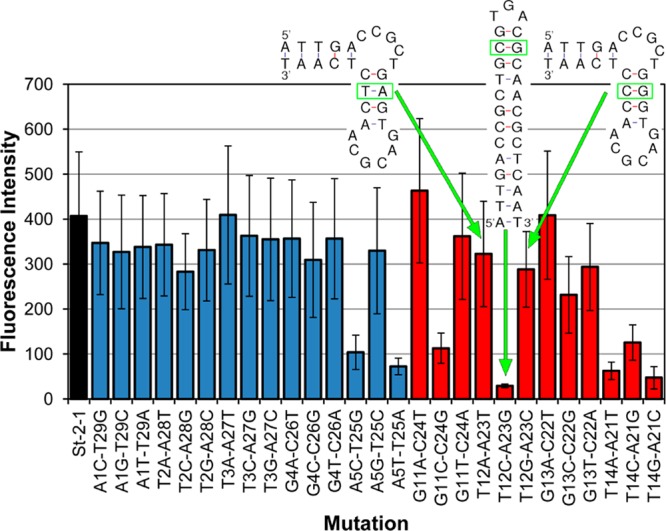
Effect of base pair replacements in the double-stranded parts of St-2-1. Binding was tested using an on-array streptavidin binding assay. Black bar, St-2-1; blue bars, mutation in the terminal stem; red bars, mutation in the sequence between the bulges. Error bars are based on 12 replicates on the array. Secondary structure of the variants A12T23, G12C23, and C12G23. The structural variant C12G23 correlates with reduced streptavidin binding on-array.
Conclusions
Using the versatility of in situ synthesis of oligonucleotide microarrays, we have explored the impact of synthesis parameters on aptamer microarray performance and made direct comparisons with similar hybridization-based arrays. The results indicate that, relative to traditional hybridization assays, aptamer microarray detection can be significantly improved by increasing the spacer length and by maximizing oligonucleotide sequence fidelity. Aptamer microarrays also appear to be less sensitive than hybridization microarrays to molecular crowding, proving a pathway for further improvement. The functionalization chemistry of the glass substrate also significantly affects the aptamer binding signal, either by modifying the oligonucleotide surface density, or via electrostatic or hydrophobic interactions with the aptamers or target protein. Aptamers are likely to prove more heterogeneous than hybridization probes in regard to optimum microarray parameters because of their varied 3-D structures and modes of interactions with their targets, and therefore, additional synthesis optimizations with a variety of aptamers will be necessary to understand the full extent of variability.
Acknowledgments
M.M.S. gratefully acknowledges financial support from the University of Vienna, the Faculty of Chemistry of the University of Vienna, and the Austrian Science Fund (FWF P23797).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kingsmore S. F. Nat. Rev. Drug Discovery 2006, 5, 310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D. L.; Joyce G. F. Nature 1990, 344, 467–468. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Rhie A.; Kirby L.; Sayer N.; Wellesley R.; Disterer P.; Sylvester I.; Gill A.; Hope J.; James W.; Tahiri-Alaoui A. J. Biol. Chem. 2003, 278, 39697–39705. [DOI] [PubMed] [Google Scholar]
- Green L. S.; Jellinek D.; Bell C.; Beebe L. A.; Feistner B. D.; Gill S. C.; Jucker F. M.; Janjic N. Chem. Biol. 1995, 2, 683–695. [DOI] [PubMed] [Google Scholar]
- Dowler T.; Bergeron D.; Tedeschi A. L.; Paquet L.; Ferrari N.; Damha M. J. Nucleic Acids Res. 2006, 34, 1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleavey G. F.; Watts J. K.; Alain T.; Robert F.; Kalota A.; Aishwarya V.; Pelletier J.; Gewirtz A. M.; Sonenberg N.; Damha M. J. Nucleic Acids Res. 2010, 38, 4547–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshkin A. A.; Singh S. K.; Nielsen P.; Rajwanshi V. K.; Kumar R.; Meldgaard M.; Olsen C. E.; Wengel J. Tetrahedron 1998, 54, 3607–3630. [Google Scholar]
- Obika S.; Nanbu D.; Hari Y.; Morio K.-i.; In Y.; Ishida T.; Imanishi T. Tetrahedron Lett. 1997, 38, 8735–8738. [Google Scholar]
- Hendrix C.; Rosemeyer H.; Verheggen I.; Van Aerschot A.; Seela F.; Herdewijn P. Chem.—Eur. J. 1997, 3, 110–120. [Google Scholar]
- King D. J.; Bassett S. E.; Li X.; Fennewald S. A.; Herzog N. K.; Luxon B. A.; Shope R.; Gorenstein D. G. Biochemistry 2002, 41, 9696–9706. [DOI] [PubMed] [Google Scholar]
- Yang X.; Bassett S. E.; Li X.; Luxon B. A.; Herzog N. K.; Shope R. E.; Aronson J.; Prow T. W.; Leary J. F.; Kirby R.; Ellington A. D.; Gorenstein D. G. Nucleic Acids Res. 2002, 30, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katilius E.; Flores C.; Woodbury N. W. Nucleic Acids Res. 2007, 35, 7626–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N. O.; Tok J. B.-H.; Tarasow T. M. PLoS ONE 2008, 3, e2720 EP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett J. R.; Cho E. J.; Ellington A. D. Methods 2005, 37, 4–15. [DOI] [PubMed] [Google Scholar]
- Collett J. R.; Cho E. J.; Lee J. F.; Levy M.; Hood A. J.; Wan C.; Ellington A. D. Anal. Biochem. 2005, 338, 113–123. [DOI] [PubMed] [Google Scholar]
- Cho E. J.; Collett J. R.; Szafranska A. E.; Ellington A. D. Anal. Chim. Acta 2006, 564, 82–90. [DOI] [PubMed] [Google Scholar]
- Brody E. N.; Gold L. Rev. Mol. Biotechnol. 2000, 74, 5–13. [DOI] [PubMed] [Google Scholar]
- Hesselberth J.; Robertson M. P.; Jhaveri S.; Ellington A. D. Rev. Mol. Biotechnol. 2000, 74, 15–25. [DOI] [PubMed] [Google Scholar]
- Conrad R.; Keranen L. M.; Ellington A. D.; Newton A. C. J. Biol. Chem. 1994, 269, 32051–32054. [PubMed] [Google Scholar]
- Wilson D. S.; Nock S. Curr. Opin. Chem. Biol. 2001, 6, 81–85. [DOI] [PubMed] [Google Scholar]
- Fodor S. P.; Read J. L.; Pirrung M. C.; Stryer L.; Lu A. T.; Solas D. Science 1991, 251, 767–773. [DOI] [PubMed] [Google Scholar]
- Singh-Gasson S.; Green R. D.; Yue Y.; Nelson C.; Blattner F.; Sussman M. R.; Cerrina F. Nat. Biotechnol. 1999, 17, 974–978. [DOI] [PubMed] [Google Scholar]
- LeProust E. M.; Peck B. J.; Spirin K.; McCuen H. B.; Moore B.; Namsaraev E.; Caruthers M. H. Nucleic Acids Res. 2010, 38, 2522–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey J. G.; Mitra D.; Somoza M. M.; Cerrina F.; Damha M. J. J. Am. Chem. Soc. 2009, 131, 8496–8502. [DOI] [PubMed] [Google Scholar]
- Agbavwe C.; Kim C.; Hong D. G.; Heinrich K.; Wang T.; Somoza M. M. J. Nanobiotechnol. 2011, 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaucage S. L. Curr. Med. Chem. 2001, 8, 1213–1244. [DOI] [PubMed] [Google Scholar]
- Halliwell C. M.; Cass A. E. G. Anal. Chem. 2001, 73, 2476–2483. [DOI] [PubMed] [Google Scholar]
- Shchepinov M. S.; Case-Green S. C.; Southern E. M. Nucleic Acids Res. 1997, 25, 1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern E.; Mir K.; Shchepinov M. Nat. Genet. 1999, 21, 5–9. [DOI] [PubMed] [Google Scholar]
- Maskos U.; Southern E. M. Nucleic Acids Res. 1992, 20, 1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J.-G.; Kokpinar O.; Friehs K.; Stahl F.; Scheper T. Anal. Chem. 2008, 80, 7372–7378. [DOI] [PubMed] [Google Scholar]
- Taylor S.; Smith S.; Windle B.; Guiseppi-Elie A. Nucleic Acids Res. 2003, 31, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainrub A.; Pettitt B. M. J. Am. Chem. Soc. 2003, 125, 7798–7799. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lee H. J.; Corn R. M. Anal. Chem. 2007, 79, 1082–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Hurek T.; Reinhold-Hurek B. Nucleic Acids Res. 2005, 33, e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G.; Lübbecke M.; Walter J.-G.; Stahl F.; Scheper T. Chem. Eng. Technol. 2011, 34, 2022–2028. [Google Scholar]
- Naiser T.; Kayser J.; Mai T.; Michel W.; Ott A. Phys. Rev. Lett. 2009, 102, 218301. [DOI] [PubMed] [Google Scholar]
- Naiser T.; Ehler O.; Kayser J.; Mai T.; Michel W.; Ott A. BMC Biotechnology 2008, 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbavwe C.; Somoza M. M. PLoS ONE 2011, 6, e22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing T.; Yang X.; Mei H.; Cao Z.; Shangguan D. Bioorg. Med. Chem. 2010, 18, 1798–1805. [DOI] [PubMed] [Google Scholar]
- Lao Y. H.; Peck K.; Chen L. C. Anal. Chem. 2009, 81, 1747–1754. [DOI] [PubMed] [Google Scholar]
- Wang T.; Oehrlein S.; Somoza M. M.; Perez J. R. S.; Kershner R.; Cerrina F. Lab Chip 2011, 11, 1629–1637. [DOI] [PubMed] [Google Scholar]
- Septak M. Nucleic Acids Res. 1996, 24, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. W.; Heaton R. J.; Georgiadis R. M. Nucleic Acids Res. 2001, 29, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. B.; Cui Y.; Bustamante C. Science 1996, 795–798. [DOI] [PubMed] [Google Scholar]
- Murphy M. C.; Rasnik I.; Cheng W.; Lohman T. M.; Ha T. Biophys. J. 2004, 86, 2530–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Phillips M. F.; Cerrina F.; Smith L. M. Langmuir 2009, 25, 6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieman T. A.; Nicholson W. L. Appl. Environ. Microbiol. 2000, 66, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Nucleic Acids Res. 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.