

Abstract
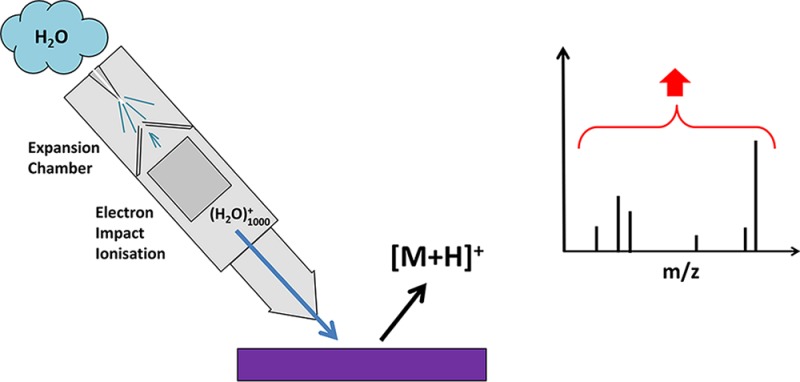
Low secondary ion yields from organic and biological molecules are the principal limitation on the future exploitation of time of flight-secondary ion mass spectrometry (TOF-SIMS) as a surface and materials analysis technique. On the basis of the hypothesis that increasing the density of water related fragments in the ion impact zone would enhance proton mediated reactions, a prototype water cluster ion beam has been developed using supersonic jet expansion methodologies that enable ion yields using a 10 keV (H2O)1000+ beam to be compared with those obtained using a 10 keV Ar1000+ beam. The ion yields from four standard compounds, arginine, haloperidol, DPPC, and angiotensin II, have been measured under static+ and high ion dose conditions. Ion yield enhancements relative to the argon beam on the order of 10 or more have been observed for all the compounds such that the molecular ion yield per a 1 μm pixel can be as high as 20, relative to 0.05 under an argon beam. The water beam has also been shown to partially lift the matrix effect in a 1:10 mixture of haloperidol and dipalmitoylphosphatidylcholine (DPPC) that suppresses the haloperidol signal. These results provide encouragement that further developments of the water cluster beam to higher energies and larger cluster sizes will provide the ion yield enhancements necessary for the future development of TOF-SIMS.
Time of flight-secondary ion mass spectrometry (TOF-SIMS) is now widely applied as a technique for the surface characterization of a wide range of technological and biological materials1.2 However, despite the introduction of metal cluster ion beams that have enabled higher yields with good spatial resolution although under static conditions (less than 1% of the surface can be used for analysis)3 and polyatomic primary beams such as SF5+, C60+, and Arn+ (n = 500 to ∼5000) that deliver analysis with much lower levels of bombardment induced damage that have moved TOF-SIMS analysis out of the static regime enabling depth profiling and 3D imaging,4,5 ion yields are still severely limited by the very low level of the ionization probability. The very best that can be hoped for bio-organic molecules under normal conditions is 10–4, with 10–5 and 10–6 being more normal.6 Consequently, under static conditions with an ion yield of 10–5 and instrument transmission of 10%, only ∼0.01 molecules (assuming a molecular size of 1 nm3) are available for detection from a 1 μm2 × 10 nm pixel of a pure compound. Even if analysis can go beyond static and can collect all the emitted ions from the pixel, only 1 would be available. Things are potentially better if a whole 1 μm voxel is consumed and everything can be collected; then, in principle, we would have 1000 ions for analysis. This capability requires an analytical process able to collect all or most of the ions generated from a pixel or voxel. However, it is not practical for the pulsed beam TOF-SIMS instruments to take advantage of this potential, and although an instrument that collects most of a continuous beam of ions generated from a sample has been developed in the form of the J105 Chemical Imager,7 analysis and imaging beyond 1 μm with good chemical identification is still limited in most situations. It is evident that there is enormous potential for increasing sensitivity and, thus, the range of application of SIMS if some method could be found to increase the ion yields by a factor of 10 to 100.
A wide range of methods has been investigated over the years to try to increase secondary ion yields. The earliest was depositing analytes on silver foil that increased ion yields from some compounds via the formation of [M + Ag]+ ions.8 However, many analytes cannot be dispersed on a foil or film, and the approach is clearly unsuitable for imaging application. Other approaches have been to disperse silver or gold nanoparticles or MALDI type matrices on the samples.9−11 A degree of success in increasing [M + metal] or [M + H] ion yields has been obtained but rarely above a factor of 5 increase. The successes of these approaches have been quite sample specific, and once the additive is sputtered away, its benefits are lost. Since the advent of polyatomic beams, it has been observed that water in the sample, present either adventitiously or as a consequence of freezing the analyte in an ice can, under bombardment by C60+, gives rise to enhanced [M + H]+ yields.12−14 Enhancements in the range of 10 to 100 have been reported, but only very few molecules have been studied. These observations have been complemented by a study in which water was admitted to the analysis chamber just above the analyte15.16 An enhancement of 10× in the molecular ion of Irgafos 168 was observed with smaller increases for polymers. Using D2O, it has been suggested that these enhancements are associated with protonation activity in the presence of surface water, possibly as a consequence of the formation of H3O+ in the bombardment region. Such a suggestion accords with the phenomenon underlying proton transfer reaction mass spectrometry (PTRMS) and selected ion flow tube MS (SIFTMS), usually used to detect organics in the atmosphere.17,18 Because the proton affinity of most organic molecules is higher than that of water, 691 kJ/mol, H3O+ is highly reactive to many organic analytes such that the kinetics are determined by the collision rates and significant protonated ion yields are obtained. On the basis of this thinking, we are in the process of developing a water cluster primary ion beam by ionization of a neutral cluster formed by supersonic jet expansion (as distinct from an electrospray type method19,20) using a similar methodology as for argon cluster beams.21−23 The hypothesis being that, in addition to benefits of enhanced low damage sputtering by a large cluster beam, sputtering with such a beam will generate a high density of water molecules and fragments in the emission zone that will enhance the formation of [M + H] ions and possibly [M – H] ions. If the density of proton donating species can be made high enough, ion yields might saturate to a relatively uniform level and this may have the benefit of ameliorating the other bugbear of SIMS and other desorption mass spectrometries, the matrix effect.
Comparing TOF-SIMS Spectra from 10 keV Ar1000+ and (H2O)1000+ Results
This Letter reports on initial data from our prototype water beam system based on a previous argon beam design23 that has been mounted on the J105 TOF-SIMS instrument.7 This very preliminary data shows that, relative to yields obtained from 10 keV Ar1000+, [M + H]+ ion yields from arginine, [Asn1 Val5] angiotensin II, dipalmitoylphosphatidylcholine (DPPC), and the drug haloperidol using singly charged 10 keV (H2O)1000+ are all enhanced by ∼10× or more. Further experimental details can be found in the online Supporting Information.
In Figure 1, the positive ion SIMS spectra from the lipid DPPC obtained using Ar1000+ and (H2O)1000+ primary beams under 1 × 1011 ions cm–2 dose conditions are compared. These analysis conditions are somewhat above static, resulting in the removal of 5% to 10% of the surface volume sampled (termed static+). Broadly, the spectra obtained are very similar. There are two obvious differences: first, the ion yields under water bombardment compared to argon are very significantly increased, m/z 184 by a factor 16 and [M + H]+m/z 735 by a factor 17. Second, the ratio of the [M + H]+ to the [M + Na]+ peaks has reversed, from 0.7 under argon to 2.0 under water. It is clear that water bombardment has a profound effect on the ion formation process and, while all ion yields are enhanced, protonation is favored. Figure 2 summarizes the data obtained from all the samples under the same dose conditions. In all cases, the enhancement relative to argon is around 10 or more. DPPC and haloperidol show the highest enhancements of close to 20; indeed, in the case of haloperidol, the yield of the [M + H]+, m/z 376 ion was so high the detector system was saturated. Hence, we have plotted the lower [M – OH]+ species at m/z 358.
Figure 1.

Positive ion spectrum of DPPC: (a) Acquired with 10 keV Ar1000+ with ion dose of 1 × 1011 ions cm–2; (b) acquired with 10 keV (H2O)1000+ with ion dose of 1 × 1011 ions cm–2.
Figure 2.

(a) Comparison of signal intensity of 10 keV (H2O)1000+ and 10 keV Ar1000+ for pure compounds. (b) Ratio of secondary ion intensities of 10 keV(H2O)1000+ relative to 10 keV Ar1000+ for pure compounds Arg(inine), Halo(peridol), dipalymitoylphosphatidylcholine (DPPC), and [Asn1 Val5] Angio(tensin II).
The yield enhancement of [M + H]+ from angiotensin II is almost exactly 10. The enhancement of the fragment ions follows quite closely the enhancement of the molecular ion. For example, m/z 254, which is the b2-NH3 fragment, is also enhanced by a factor 10. This seems to be true for most of the molecules we have studied. The fragment to molecular ion ratio is very similar under both beams. This behavior would be expected if the water beam predominantly enhances the yield of the [M + H]+ ion and most fragment formation results from fragmentation of this ion. Very early24,25 and recent MSMS studies from this laboratory would support this expectation. It is not true for all fragment ions, e.g., the m/z 95 from haloperidol where the ratio changes from ∼0.4 under argon to ∼0.2 under water. This may cast light on a changing mechanism of fragmentation with the involvement of water fragments in the near surface region.
It is interesting that the water cluster analysis of a second DPPC sample that had a higher yield of [M + Na]+ under the argon beam showed only a 10× enhancement of the [M + H]+ ion, and the [M + Na]+/[M + H]+ ratio which was about 1.0 under argon did not change. The interference of alkali metal ions in the formation of protonated secondary ions has been studied in this laboratory, and this work shows that salt can inhibit the formation of these [M + H]+ ions26,27 As in these earlier studies, the present results might suggest that the extent of salt inhibition is concentration related.
Although at this stage we have not carried out detailed depth profiling or sputter yield measurement, all of the samples were subjected to extended sputtering under water cluster bombardment up to a dose of about 1.7 × 1012 ions cm–2. For most of the samples, the signal profiles up to this dose were quite stable. Assuming a molecular volume of about 1 nm3 and a sputter yield of ∼50 to 100 molecules per primary impact,28 the 1.7 × 1012 ions cm–2 dose is well beyond the static limit and almost all the molecules in a 1 μm2 × 10 nm surface volume will be removed. In light of our interest in subμm analysis, Table 1 compares the number of molecular secondary ions detected under our instrumental conditions from a 1 μm2 surface using the argon and water beams under the static+ dose of 1 × 1011 ions cm–2 and when the whole surface layer is sputtered by 1.7 × 1012 ions cm–2 and most of the resulting secondary ions are collected.
Table 1. Comparison of the Number of Detectable “Molecular” Secondary Ions from a 1 μm2 Area under Static+ and Whole Area Analysis Conditions Using 10 keV Ar1000+ and 10 keV (H2O)1000+ Primary Beams.
| static+ conditions (1 × 1011 ions cm–2) |
whole 1 μm2 area consumed |
|||
|---|---|---|---|---|
| compound | argon | water | argon | water |
| arginine m/z 175 | 0.17 | 1.4 | 2.6 | 21.0 |
| haloperidol m/z 358 | 0.06 | 1.3 | 0.9 | 13.4 |
| DPPC m/z 184 | 0.033 | 0.55 | 0.51 | 6.4 |
| DPPC m/z 735 | 0.001 | 0.02 | 0.02 | 0.24 |
| angiotensin II m/z 1032 | 0.09 | 0.84 | 1.09 | 6.5 |
It is clear that the yields/1 μm2 under our static+ conditions are around 5 to 10 times the expectations for strict static conditions. Given ionization probabilities in the order of 10–5, they are in line with the higher ion dose. The present water beam improves the situation by a factor of 10, but the ability to accumulate all or most of the ions from a given area for analysis starts to lift the ion yield into a useful region. If the sputter yield is 50 to 100 per impact, the ionization probability under the water beam is ≥10–4.
The second major impetus for these studies was that the use of water primary beams might modify matrix effects in the case of [M ± H] ions. We have shown that the use of the hydrogen containing a polyatomic ion beam, coronene C24H12+, can help to lift the matrix effect.29 Previous studies have shown that DPPC exerts a very significant suppression effect on the formation of the [M + H]+ ion of haloperidol30.31 As a preliminary study, a 1:10 mixture of haloperidol in DPPC was prepared by dissolving appropriate amounts of the two compounds in a methanol chloroform mixture and spin-casting the resulting solution onto a silicon wafer. Figure S.1, Supporting Information, summarizes the outcome of a comparative analysis of this mixture with the argon and water beams. The expected mixture ratio 0.09YHal/(0.09YHal + 0.091YDPPC) has been calculated using the observed m/z 358 ion yield from pure haloperidol, YHal, and the m/z 735 ion yield from pure DPPC, YDPPC, under the argon and water beams. This is compared with the observed YHalmix/(YHalmix + YDPPCmix) from the mixture. Similar data for DPPC is also presented in Figure S.1, Supporting Information. It can be seen that under argon bombardment there is significant suppression of the haloperidol ion yield ratio by almost 5× from the expected 0.86 down to 0.19 and a corresponding enhancement of DPPC by more than 6× from 0.13 to 0.81. Under water bombardment however, the mixture ratio for haloperidol is close to 50% of that expected. In contrast, the DPPC ratio is still some 5 times higher than expected. After a dose of 1.7 × 1012 ions cm–2, the relative enhancement for haloperidol is maintained if not somewhat increased, so surface segregation effects are minimal. It is clear that, while there is a real benefit under water cluster bombardment, there is still some way to go to lift the matrix effect entirely. This suggests that DPPC is still benefiting significantly compared to haloperidol from the extra protonation capability and that saturation of protonation species in the emission region has not been reached, so there is still more benefit to be had if cluster sizes can be increased.
Discussion and Conclusions
The data we report makes it clear that the water cluster beam provides an approximately 10 times or more increase in ion yield for a range of molecules over the yields observed with 10 keV argon clusters of the same nuclearity (n = 1000). Under these conditions, there is also a benefit in a reduced matrix suppression effect. These observations are consistent with the experimental observations reported from earlier studies on the benefits of ice matrices and water dosing of samples that resulted in increased [M + H]+ yields. Furthermore, MD simulations from Garrison’s group have suggested that hydrogen species generated by a primary ion impact in the emission zone may well give rise to protonated molecules that are in a sense preformed to be sputtered out by subsequent primary particle impacts.32 The data presented in this paper is understandable in the light of this idea such that a water cluster impact would result in an increased density of hydrogen, protons, oxygen atoms and ions, and hydroxyl ions. The resulting ion–molecule reactions in the emission zone would be those expected to result in increased secondary ion formation and subsequent release. It would clearly be extremely interesting to be able to use a D2O cluster beam to clarify the mechanisms of protonation and other reactions in the emission zone. Similarly, the use of 18O enriched water could possible provide further insights, although at some expense!
Other beam systems have been investigated in the past with the aim of enhancing protonated organic molecule ion yields. Massive clusters of glycerol were successful in desorbing and detecting large peptides, much larger than can be detected in routine SIMS today;33 however, the development did not take off possibly because of the technical difficulties of handling the glycerol beam. Desorption electrospray ionization (DESI) uses a spray of charged droplets to desorb and ionize under atmospheric conditions.34 Proton exchange reactions are important in this process, but yields relative to SIMS are difficult to determine. Giant water cluster beams generated by electrospray methods35,36 have been successful in generating SIMS type spectra and would be expected to deliver similar benefits to the cluster beam discussed here. As we have argued, the authors suggest that the mechanism of ion formation involves proton transfer that is greatly enhanced in the impact region. There are challenges in handling this beam, and it has been difficult to determine the degree of signal enhancement relative to other ion beams from the data reported.
What do the results reported here allow us to anticipate with future developments of water cluster beams? We are looking for ion yields that enable useful analysis of multiple compounds within a submicrometer pixel area. The present studies have been exploratory to assess how far it is worth pursuing the objective of a workable routine water cluster beam. Table 1 shows that useful enhancements have been made such that, if SIMS instrumentation capable of accumulating the whole yield from a pixel is used, the water beam would provide ∼10 molecules per pixel. On the basis of comparisons between 10 and 20 keV Ar1000 data we have obtained in this laboratory, we would expect at least a factor of 5 increase if the water beam energy is increased to 20 keV. We would hope to obtain another factor of 10× improvement by increasing the cluster size to 10 000. This would provide a very useful benefit for analysis by taking whole pixel yields into the 100s of ions, but only future experiments will prove this. Similarly, we would hope that these enhanced conditions would enable the matrix effect benefits to be increased too.
Acknowledgments
This research was funded by the UK Engineering and Physical Sciences Research Council, EPSRC under grant EP/K01353X/1.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Vickerman J. C.; Briggs D.. TOF-SIMS - Materials Analysis by Mass Spectrometry; IM Publications and SurfaceSpectra Ltd: Manchester, UK: 2013. [Google Scholar]
- Fletcher J. S.; Vickerman J. C. Anal. Chem. 2013, 85, 610–639. [DOI] [PubMed] [Google Scholar]
- Briggs D.; Wootton A. B. Surf. Interface Anal. 1982, 4, 109–115. [Google Scholar]
- Mahoney C. M. Mass Spectrom. Rev. 2010, 29, 247–293. [DOI] [PubMed] [Google Scholar]
- Wucher A. Appl. Surf. Sci. 2006, 252, 6482–6489. [Google Scholar]
- Cheng J.; Kozole J.; Hengstebeck R.; Winograd N. J. Am. Soc. Mass. Spectrom. 2007, 18, 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. S.; Rabbani S.; Henderson A.; Blenkinsopp P.; Thompson S. P.; Lockyer N. P.; Vickerman J. C. Anal. Chem. 2008, 80, 9058–9064. [DOI] [PubMed] [Google Scholar]
- Heile A.; Lipinsky D.; Wehbe N.; Delcorte A.; Bertrand P.; Felten A.; Houssiau L.; Pireaux J. J.; De Mondt R.; Van Royen P.; Van Vaeck L.; Arlinghaus H. F. Surf. Interface Anal. 2008, 40, 538–542. [Google Scholar]
- Delcorte A. Appl. Surf. Sci. 2006, 252, 6582–6587. [Google Scholar]
- Prabhakaran A.; Yunus S.; Wehbe N.; Bertrand P.; Delcorte A. Surf. Interface Anal. 2011, 43, 74–77. [Google Scholar]
- Wu K. J.; Odom R. W. Anal. Chem. 1996, 68, 873–882. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Winograd N. Anal. Chem. 2005, 77, 3651–3659. [DOI] [PubMed] [Google Scholar]
- Conlan X. A.; Lockyer N. P.; Vickerman J. C. Rapid Commun. Mass Spectrom. 2006, 20, 1327–1334. [DOI] [PubMed] [Google Scholar]
- Piwowar A. M.; Fletcher J. S.; Kordys J.; Lockyer N. P.; Winograd N.; Vickerman J. C. Anal. Chem. 2010, 82, 8291–8299. [DOI] [PubMed] [Google Scholar]
- Mouhib T.; Delcorte A.; Poleunis C.; Bertrand P. J. Am. Soc. Mass. Spectrom. 2010, 21, 2005–2010. [DOI] [PubMed] [Google Scholar]
- Mouhib T.; Delcorte A.; Poleunis C.; Bertrand P. Surf. Interface Anal. 2013, 45, 46–49. [Google Scholar]
- Smith D.; Spanel P. Mass Spectrom. Rev. 2005, 24, 661–700. [DOI] [PubMed] [Google Scholar]
- Blake R. S.; Monks P. S.; Ellis A. M. Chem. Rev. 2009, 109, 861–896. [DOI] [PubMed] [Google Scholar]
- Beuhler R. J.; Friedman L. J. Chem. Phys. 1982, 77, 2549–2557. [Google Scholar]
- Beuhler R.; Friedman L. Chem. Rev. 1986, 86, 521–537. [Google Scholar]
- Ninomiya S.; Ichiki K.; Seki T.; Aoki T.; Matsuo J. Nucl. Instrum. Methods Phys. Res, Sect. B 2009, 267, 2601–2604. [Google Scholar]
- Takaoka G. H.; Kawashita M. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2008, 38, 111–117. [Google Scholar]
- Rabbani S.; Barber A. M.; Fletcher J. S.; Lockyer N. P.; Vickerman J. C. Anal. Chem. 2011, 83, 3793–3800. [DOI] [PubMed] [Google Scholar]
- a Leggett G. J.; Vickerman J. C.; Briggs D. Surf. Interface Anal. 1990, 16, 3–8. [Google Scholar]; b Leggett G. J.; Vickerman J. C. Int. J. Mass Spectrom. Ion Processes 1992, 122, 281–319. [Google Scholar]
- Leggett G. J.; Briggs D.; Vickerman J. C. Surf. Interface Anal. 1991, 17, 737–744. [Google Scholar]
- Jones E. A.; Lockyer N. P.; Vickerman J. C. Anal. Chem. 2008, 80, 2125–2132. [DOI] [PubMed] [Google Scholar]
- Piwowar A. M.; Lockyer N. P.; Vickerman J. C. Anal. Chem. 2009, 81, 1040–1048. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Wucher A.; Winograd N. J. Phys. Chem. B 2006, 110, 8329–8336. [DOI] [PubMed] [Google Scholar]
- Biddulph G. X.; Piwowar A. M.; Fletcher J. S.; Lockyer N. P.; Vickerman J. C. Anal. Chem. 2007, 79, 7259–7266. [DOI] [PubMed] [Google Scholar]
- Jones E. A.; Lockyer N. P.; Vickerman J. C. Int. J. Mass Spectrom. 2007, 260, 146–157. [Google Scholar]
- Jones E. A.; Lockyer N. P.; Vickerman J. C. Appl. Surf. Sci. 2006, 252, 6727–6730. [Google Scholar]
- Brenes D. A.; Postawa Z.; Wucher A.; Blenkinsopp P.; Garrison B. J.; Winograd N. Surf. Interface Anal. 2013, 45, 50–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornett D. S.; Lee T. D.; Mahoney J. F. Rapid Commun. Mass Spectrom. 1994, 8, 996–1000. [DOI] [PubMed] [Google Scholar]
- Ifa D. R.; Wu C.; Ouyang Z.; Cooks R. G. Analyst 2010, 135, 669–681. [DOI] [PubMed] [Google Scholar]
- Asakawa D.; Fujimaki S.; Hashimoto Y.; Mori K.; Hiraoka K. Rapid Commun. Mass Spectrom. 2007, 21, 1579–1586. [DOI] [PubMed] [Google Scholar]
- Asakawa D.; Hiraoka K. Rapid Commun. Mass Spectrom. 2011, 25, 655–660. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.