

Abstract
Background
Multiple genes have been implicated by association studies in altering inflammatory bowel disease (IBD) predisposition. Paediatric patients often manifest more extensive disease and a particularly severe disease course. It is likely that genetic predisposition plays a more substantial role in this group.
Objective
To identify the spectrum of rare and novel variation in known IBD susceptibility genes using exome sequencing analysis in eight individual cases of childhood onset severe disease.
Design
DNA samples from the eight patients underwent targeted exome capture and sequencing. Data were processed through an analytical pipeline to align sequence reads, conduct quality checks, and identify and annotate variants where patient sequence differed from the reference sequence. For each patient, the entire complement of rare variation within strongly associated candidate genes was catalogued.
Results
Across the panel of 169 known IBD susceptibility genes, approximately 300 variants in 104 genes were found. Excluding splicing and HLA-class variants, 58 variants across 39 of these genes were classified as rare, with an alternative allele frequency of <5%, of which 17 were novel. Only two patients with early onset Crohn's disease exhibited rare deleterious variations within NOD2: the previously described R702W variant was the sole NOD2 variant in one patient, while the second patient also carried the L1007 frameshift insertion. Both patients harboured other potentially damaging mutations in the GSDMB, ERAP2 and SEC16A genes. The two patients severely affected with ulcerative colitis exhibited a distinct profile: both carried potentially detrimental variation in the BACH2 and IL10 genes not seen in other patients.
Conclusion
For each of the eight individuals studied, all non-synonymous, truncating and frameshift mutations across all known IBD genes were identified. A unique profile of rare and potentially damaging variants was evident for each patient with this complex disease.
Keywords: IBD-genetics; inflammatory bowel disease; crohn's disease; paediatric gastroenterology; ulcerative colitis; zollinger ellison syndrome,
Significance of this study.
What is already known on this subject?
Genome-wide association studies have implicated numerous candidate genes for inflammatory bowel disease (IBD), but evidence of causality for specific variants is largely absent. Furthermore, by design, genome-wide association studies are limited to the study of common variants and overlook the functionally detrimental variation imposed by rare/novel mutation.
Exome analysis is fully informative for the spectrum of variation within the protein coding sequence of genes. It has been used to successfully identify disease causing variants in Mendelian disorders, but its potential to identify the missing heritability in complex diseases such as paediatric IBD has not yet been realised.
What are the new findings?
This study examines genetic variants from the perspective of the patient rather than the gene—for each paediatric case a profile of deleterious variation is determined across a comprehensive panel of known IBD genes.
Paediatric IBD patients carry a wide spectrum of low frequency variants within candidate IBD genes.
In silico analyses indicate a substantial proportion of these mutations are potentially deleterious.
Consistent with complex inheritance, this small subset of patients with severe IBD exhibit a varied profile of mutation with limited sharing of specific variants across the set of eight exomes.
Significance of this study.
How might it impact on clinical practice in the foreseeable future?
Functional studies are required to confirm in silico assessment of variation impact on biology.
Even mutations confirmed to confer susceptibility must be considered among the full profile of disease predisposing variation present in any individual.
As the cost of next generation sequencing falls and the number of mutation profiles increases, there is clear potential for genetic characterisation of IBD phenotypic sub-types facilitating targeted therapeutic intervention/personalised medicine.
Introduction
Ulcerative colitis (UC) and Crohn's disease (CD) are the two main clinical phenotypes of inflammatory bowel disease (IBD), both resulting in chronic and relapsing inflammation. The incidence of IBD in the paediatric population of the UK is 5.2 per 100 000 children per year, with breakdown figures of 3.1 for CD, 1.4 for UC and 0.6 for IBD unclassified (IBDU).1 While the precise aetiology and pathogenesis is complex and incompletely understood, it is widely accepted that IBD occurs as the result of a dysregulated mucosal immune response to commensal gut flora in the genetically susceptible host.2 Familial aggregation of disease implies a strong genetic component,3 although environmental factors may play a greater role in ulcerative colitis.4
Over recent years, genome-wide association studies (GWAS) have been applied with huge success to identify common genes involved in both CD and UC. Genes with replicated evidence for strong association suggest that pathways involving disruption of the innate and adaptive immune system, compromised epithelial barrier function and impaired autophagy play a significant role in disease.2 However, despite the identification of over one hundred unique genes in IBD susceptibility, these common variants in combination account for less than a quarter of the genetic risk.5–7 The source of this missing heritability is the subject of much debate with various explanations: overestimates of original heritability statistics; underpowered GWAS studies (in terms of sample size and single nucleotide polymorphism (SNP) coverage) to detect common variants associated with decreasing effect sizes; poorly investigated epistatic and gene–environment interactions; and rare variation.8
Rare variants form the group of infrequent mutations that occur in <5% of the population. A large proportion of variants in this class occur at a much lower frequency (<0.1%), and many thousands are likely to be specific to ethnic groups, isolates, families or even individuals. Nevertheless, this class of variation harbours multiple penetrant disease mutations conferring medium to high risk. Rare variants escape detection by GWAS. BRCA1 and BRCA2 are examples of familial breast cancer genes that harbour many high risk variants but go undetected by GWAS. This is consequent to each of the disease causing mutations being shared by only a fraction of the patient group and so no common SNP can act as a proxy or ‘tag’ to flag the gene as causal. It is entirely plausible that a proportion of IBD and other complex disease heritability unaccounted for by common variation lies within higher risk rare variants. Furthermore, many of these mutations may lie within genes already implicated by association studies.
Exome sequencing determines each letter of the genetic code at nearly all coding regions or exons in the genome (the ‘exome’), thereby generating the complete profile of coding variation. It has already proved its success in identifying causal mutations in an ever growing list of both recessive and dominant rare Mendelian disorders whereby sequencing of a small number of unrelated cases has been used to identify disease causing variants.9 One such case reported exome sequencing undertaken in a male child presenting at 15 months with intractable IBD; exome sequencing was used to successfully identify a causal mutation in the XIAP gene (X-linked inhibition of apoptosis gene) for which the child was hemizygous. After haematopoietic progenitor cell transplant treatment, as recommended for XIAP deficiency, the IBD resolved, suggesting that the Crohn's-like illness seen in this patient was driven by this single mutation.10
As next generation sequencing technology advances, it becomes increasingly affordable. Nevertheless, while costs remain in the region of several hundred pounds per sample, targeted analyses of those patient groups most likely to yield positive results is prudent. Prioritisation of cases with strong family history and/or patients representing the phenotypic ‘extreme’ of common traits is a useful strategy.11 One such example of an ‘extreme’ phenotype is paediatric disease in which onset is particularly early. Genetic susceptibility is thought to play a more important role in the aetiology of early-onset IBD than in late-onset IBD.12 This is supported by a higher rate of positive family history of IBD in patients with a younger age at diagnosis compared to the older age group, suggesting that an earlier presentation may be due to a higher burden of disease-causing mutations in the genomes of these affected children compared to those in whom disease manifests later in life.13 In addition, environmental confounding factors such as smoking are less likely to be exerting an influence on disease in paediatric cohorts. It has also been suggested that early-onset disease may in itself be a more aggressive phenotype; in CD, earlier age at diagnosis is associated with a greater need for surgery and increased small bowel disease.12–14
Two of the most comprehensive association studies investigating IBD have used adult cohorts, but a recent GWAS of 3246 early-onset IBD cases successfully identified five new loci associated with childhood susceptibility as well as replicating loci previously implicated in adult-onset disease.15 Early-onset disease genes have also been located using linkage analysis and candidate gene sequencing approaches undertaken in two unrelated consanguineous families.16 Despite distinct clinical and histopathological features of the CD and UC phenotypes, an estimated 30% of IBD-related loci are shared between both phenotypes.2 It is likely that further study of rare variation across implicated genes may uncover more commonality.
The application of exome sequencing to complex diseases is fraught with analytical difficulty; finding disease causing variants among the many innocent variants present in the genome has been likened to finding ‘needles in stacks of needles’.17 Targeting analyses to subsets of genes in patients with extreme phenotype is a practical approach to examining genetic influence in disease. In this study we apply next generation sequence technology to paediatric IBD (PIBD). The study is focused on a small cohort of eight paediatric patients with markedly early onset/severe disease. Patients are representative of the spectrum of IBD presentation, and limiting the study to this modest number makes data interpretable on a case-by-case basis. We focus on a comprehensive panel of known causal genes and for each patient describe their individual burden of rare and novel damaging variation.
Materials and methods
Recruitment of paediatric IBD cohort of patients
Children included in this study were selected from the ‘Genetics of Paediatric IBD’ cohort between October 2010 and October 2011. This cohort was recruited through tertiary referral paediatric IBD clinics at the University Hospital Southampton Foundation Trust. This hospital is the regional centre for paediatric gastroenterology, providing a tertiary paediatric gastroenterology and endoscopy service for the Wessex region, and draws on a patient population of 3.5 million. The service has a rolling database of over 300 paediatric IBD cases and approximately 50–70 patients are diagnosed each year. All children had a diagnosis of IBD and were aged between 5 and 18 years at time of recruitment, although their diagnosis may have been made at an earlier age. Diagnosis was established using the Porto criteria18; all children had compatible history, examination and laboratory investigation results, and infectious causes excluded. All were investigated with upper gastrointestinal endoscopy and ileo-colonoscopy. Written informed consent was obtained from the attending parent of all children, and the child where appropriate. In the initial recruitment interview, clinical data and venous blood samples (10 ml for DNA extraction and 8 ml for plasma extraction) were collected. Additional comprehensive clinical data were extracted from patient records. For each patient we gathered information on gender, dates of birth and initial diagnosis, disease extent currently and at diagnosis using the Paris classification,19 disease activity score at diagnosis (using the paediatric CD activity index (PCDAI) and the paediatric ulcerative colitis activity index (PUCAI)), height and weight currently and at first diagnosis, time to and date of first relapse, treatment history (use of steroids, immunomodulators, biological therapies, surgery), history of potential aetiological and modifying conditions such as smoking, gastrointestinal infection and other autoimmune disease, and family history.
Ethics statement
This study was approved by the Southampton and South West Hampshire Research Ethics Committee (REC) (09/H0504/125) and University Hospital Southampton Foundation Trust Research & Development (RHM CHI0497).
Selection of samples
Eight patient samples from our PIBD cohort as previously described were selected for exome sequencing for this study. These eight patients were selected based on age of diagnosis, disease severity or positive family history in a first degree relative. Selection criteria and patient phenotypic characteristics are summarised in table 1.
Table 1.
Summary of patient phenotypes and characteristics (specific selection criteria are in bold)
| Sample ID | Age at diagnosis (years) | Sex | Disease | Phenotype description and selection criteria | Ethnicity | Family history |
| Proband 1 | 11 | Male | CD | Severe disease requiring surgery/ Stricturing ileo-colonic disease requiring right hemicolectomy within 6 months of diagnosis. | White British | – |
| Proband 2 | 7 | Female | CD | Early age of onset/non-stricturing, non-penetrating mild to moderate pancolitis, disease resistant to treatment. | White British | – |
| Proband 3 | 6 | Male | CD | Early age of onset/non-stricturing, non-penetrating granulomatous colitis and duodenitis. Mother diagnosed CD aged 21 years. | White British | + |
| Proband 4 | 6 | Female | CD | Early age of onset/non-penetrating pancolitis with possible ileo-caecal stricture. | White British | – |
| Proband 5 | 13 | Male | CD | Non-stricturing, non-penetrating, colitis. Family history including maternal CD and maternal grandparental UC. | White British | + |
| Proband 6 | 9 | Male | UC | Severe left sided colitis, also with oral pemphigus. | White British | – |
| Proband 7 | 2 | Male | UC | Early age of onset/mild to moderate pancolitis. | White British | – |
| Proband 8 | 3.5 | Male | IBDU | Early age of onset/left sided colitis. | Iraqi | – |
CD, Crohn's disease; IBDU, inflammatory bowel disease unclassified; UC, ulcerative colitis.
DNA and plasma extraction
Genomic DNA was extracted from EDTA anticoagulated peripheral venous blood samples using the salting out method. Plasma was isolated from lithium–heparin anticoagulated peripheral venous blood samples using standard methods.
Exome sequencing
Targeted exome capture was performed using the SureSelect Human All Exon 50Mb kit (Agilent). The Illumina HiSeq system was used to generate sequence data. These steps were conducted at the Wellcome Trust Centre for Human Genetics at Oxford University. The resultant paired end sequencing data were aligned against the human genome reference sequence 18 (hg18) using the Novoalign software (2.06.09MT, Novocraft Technologies, Selangor, Malaysia). Duplicate reads, resulting from PCR clonality or optical duplicates, and reads mapping to multiple locations were excluded from downstream analysis. Depth and breadth of sequence coverage was calculated with custom scripts and the BedTools package.20 Single nucleotide substitutions and small insertion deletions were identified and quality filtered within the SamTools software package21 and in-house software tools. Variants were annotated with respect to genes and transcripts with the Annovar tool.22 Summary statistics for exome sequencing, mapping and coverage are shown in supplementary table 1 (available online only). Data from the 1000 Genomes Project (1KG) phase I (2010 November release) were utilised using LiftOver (University of California Santa Cruz Genome Browser, http://genome.ucsc.edu/cgi-bin/hgLiftOver) for the conversion of 2010 November coordinates to hg18. Variants were characterised as novel if they were previously unreported in the dbSNP129, dbSNP132, 1KG data and our 22 in-house reference exomes (supplementary table 2). Southampton reference exomes for evaluating the burden of mutation comprised independent DNA samples from unrelated individuals who were exome sequenced on the same platform at the same time as part of other local projects. Each reference exome was from a patient with a distinct clinical diagnosis but no history of gastrointestinal or autoimmune disease. The clinical phenotypes of the 22 reference exomes included 10 with leukaemia, 5 with lymphoma, 4 with Beckwith–Wiedemann syndrome and 3 with macrocephaly malformation syndrome.
The National Heart Lung and Blood Institute Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu/EVS/) (Feb 2012) was used as a reference dataset for rare variant allele frequency in a European American population (table 2). This project contains exome data from approximately 3500 European American individuals taken from 12 disease cohorts with a range of heart, lung or blood disorders.
Table 2.
Characterisation of non-synonymous, stopgain and indel variants with an alternative allele frequency of <0.05 or not reported in 1000 genomes across 39 known IBD genes
|
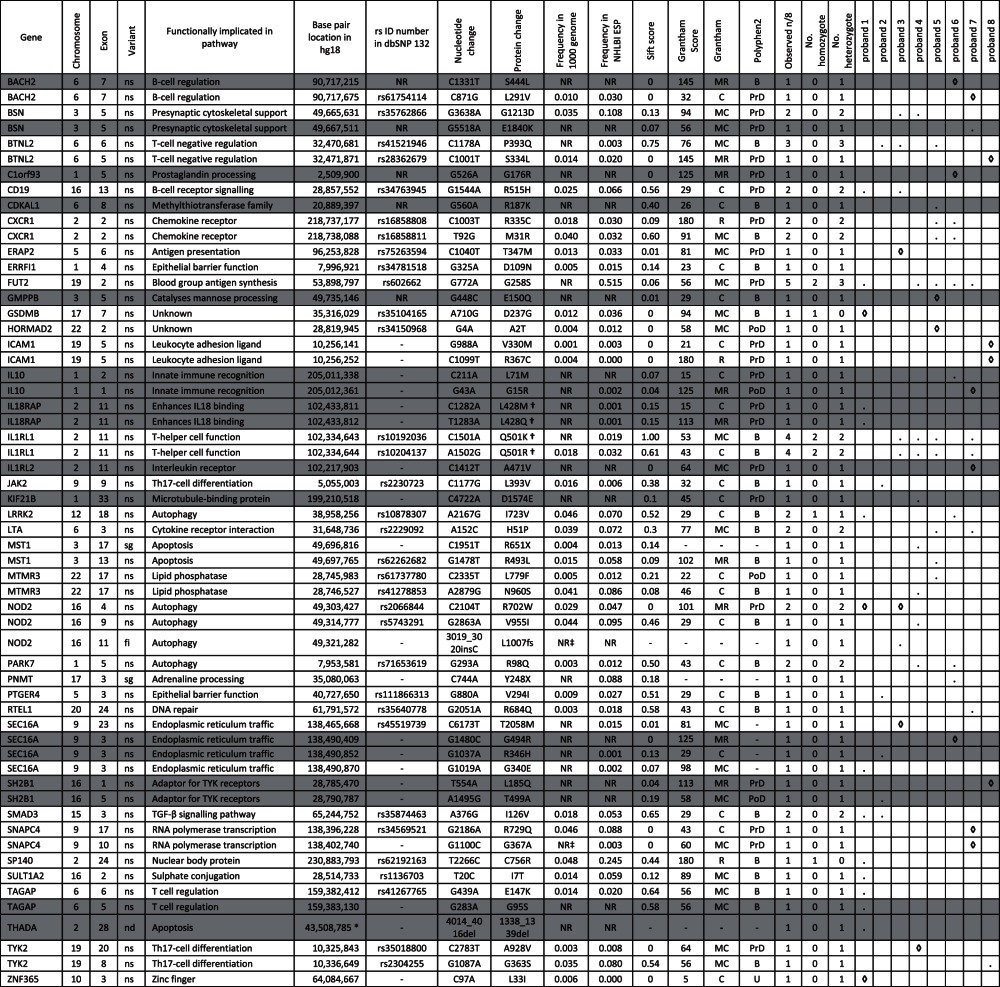
Novel variants are shown in grey.
Where a specific variant is present in a proband, this is indicated by a dot (.).
Where a specific variant is present in a proband and has a SIFT score of <0.05 this is indicated by a ◊.
*Indicates the first bp location of a 3-bp deletion.
†Indicates a dinucleotide variant (that for IL18RAP results in a codon change from CTG>AAG, resulting in p.L428K amino acid change).
‡NR indicates variants that despite not being reported in dbSNP132 or 1000 genomes, are reported in dbSNP129 or seen in our in-house reference exomes and are therefore not characterised as novel.
B, benign; C, conservative; fi, frameshift insertion; MC, moderately conservative; MR, moderately radical; nd, non-frameshift deletion; NR, not reported; ns, non-synonymous; PoD, possibly damaging; PrD, probably damaging; R, radical; sg, stopgain; U, unknown.
Selection of a panel of known IBD genes
We constructed a panel of high priority genes previously shown to be strongly associated with IBD. Our aim was to include all genes with convincing evidence for disease causality in previous studies. Selection was based on the findings of two genome-wide meta-analyses of IBD,5 6 one genome-wide association study of early-onset IBD,15 and one linkage study in consanguineous families with early-onset IBD.16 Gene names were cross referenced with the Human Genome Nomenclature Committee to ensure that the most up-to-date versions of gene names were applied (http://www.genenames.org/). Our consolidated panel represented 169 genes (supplementary table 3).
Evaluation of spectrum of mutation and predicted functional impact
Exome data from our eight patients were cross-referenced against our gene panel described above. Synonymous variants were excluded from analysis due to their decreased likelihood of functional effect on protein. SIFT (‘sorting intolerant from tolerant’) scores23 were annotated using Annovar, or where scores were missing, were derived indirectly using the database of non-synonymous functional prediction.24 A small number of additional missing scores were obtained from the SIFT server at http://sift.jcvi.org. SIFT is a sequence homology-based tool that predicts whether an amino acid substitution is likely to affect protein function. Variants with SIFT scores of <0.05 are considered ‘deleterious’, and SIFT therefore allows prioritisation of amino acid changes by ranking according to score.
We examined in silico predictions from the Polyphen2 (Polymorphism Phenotyping v2) server at http://genetics.bwh.harvard.edu/pph2/bgi.shtml.25 Polyphen2 uses a probability model to generate thresholds and classify polymorphisms as benign, possibly damaging or probably damaging, based on 11 predictive features relating to sequence, phylogenetic and structural information which characterise the substitution. Additional functional predictions of the result of each amino acid change were derived from Grantham scores,26 which predict the effect of amino acid substitutions according to chemical properties including polarity and molecular volume. The Grantham distance, d, between two amino acids is classified as conservative (0<d≤50), moderately conservative (50<d≤100), moderately radical (100<d≤150) or radical (d>150).27 Radical changes predicted by these scores are linked to clinical phenotypes.28
Burden of mutation
Using only novel variants or variants with an alternative allele frequency of <0.05 in the 1000 genomes data, a χ2 contingency test was performed to test for an excess of rare potentially deleterious variants (non-synonymous and frameshift indels) compared to neutral synonymous variants, within the panel of known IBD genes in our eight cases compared to 22 reference exome samples from non-IBD patients.
Results
Exome sequencing
On average, each PIBD exome had 78% of mappable bases of the Gencode defined exome represented by coverage of at least 20 reads (supplementary table 1). For each patient approximately 23 000 variants were found. After exclusion of synonymous variants, approaching 13 000 variants were found per patient, of which approximately 300 were novel (supplementary table 2).
Characterisation of mutations in genes known to be associated with IBD
Across all eight exomes, we found 332 variants (excluding synonymous) among 104 of our panel of 169 genes (supplementary table 4). Of these, approximately 40% (122) were found in HLA class genes. Seventeen were novel variants not previously reported in public databases or our own in-house database of non-IBD patient reference exomes.
Table 2 describes the set of variants remaining after removal of splicing, common (where the alternative allele frequency in 1000 genomes is reported as >0.05) and HLA variants. Fifty-eight variants within 39 genes remain, of which 17 are novel.
The χ2 analysis to test for an excess of deleterious rare variants in known and candidate IBD genes in IBD cases listed in table 2 compared to 22 reference exomes did not reach statistical significance (supplementary table 5).
Crohn's disease patient profiles
Only two patients with early onset CD exhibit rare potentially deleterious variations within NOD2.
Proband 1 was diagnosed with CD aged 11 years and required a right hemicolectomy for extensive ileo-caecal stricturing. He is a heterozygote carrier of the NOD2 R702W variant that is associated with a twofold increase in odds ratio of CD.29 In addition he harbours potentially damaging mutations in GSDMB and ZNF365 and a dinucleotide variant of undetermined functionality on one chromosomal copy of the IL18RAP gene. The presence of ileal disease and a stenotic phenotype in this patient is also consistent with his NOD2 variant profile.29
Proband 2 carries a novel variant in each of the SEC16A and SH2B1 genes. This patient also has a rare variant in JAK2; however, SIFT scoring suggests none of these mutations are likely to be particularly deleterious.
Proband 3 is the second patient with NOD2 variation and carries both the R702W variant and the L1007 frameshift insertion. Carriage of two or more high risk alleles in NOD2 confers a 17-fold increased risk of IBD.29 Exome analysis cannot determine if both variants have been co-inherited on the same chromosome. Proband 3 additionally possesses potentially deleterious variants in ERAP2 and SEC16A.
Proband 4 presented with severe disease aged 6 years. She carries the NOD2 V955I variant, but this is predicted to be innocuous as is her private variant in KIF21B. She is a heterozygote for a number of previously seen variants with borderline (∼0.05) SIFT scores (FUT2, MTMR3). The most distinct rare (frequency of 0.003) and potentially deleterious variant observed in this patient is the A928V variant in the TYK2 gene.
Proband 5 possesses one variant in the GMPBB gene and another in HORMAD2, both estimated by SIFT to be harmful. The former is ascertained as novel to this individual, whereas the latter occurs in <0.5% of chromosomes studied in the thousand genomes project, but in just over 1% of the 3500 exomes tested in Exome Variant Server.
UC and IBDU patient profiles
Proband 6 has a histological diagnosis of UC and carries novel deleterious mutations in the BACH2, C1orf93 and SEC16A genes. A fourth novel variant in the IL10 gene also has a low SIFT score.
Proband 7 is a boy, diagnosed aged 2, and similar to our other UC patient, exhibits a potentially functionally detrimental mutation in BACH2 and a second very rare and possibly damaging mutation in IL10. The IL1RL2 and SNAPC4 genes are also apparently compromised in this individual.
Proband 8 was diagnosed at a young age with IBDU, and possesses two possibly harmful variants in ICAM1, one in BTNL2 and a novel deleterious variant in SH2B1.
Predicted functional impact
Figure 1 illustrates relationships between SIFT, Grantham and Polyphen2 scores for all non-synonymous variants in table 2. There is particularly close agreement between SIFT and Polyphen2 scores as noted previously.30 Agreement with Grantham scores is less clear, but there is striking concordance between the vast majority of variants with a SIFT score >0.2 (benign) being independently designated benign by Polyphen2 and conservative by Grantham. Notably, two variants are classified as radical by Grantham and probably damaging by SIFT and/or Polyphen2—CXCR1 (R335C) and ICAM1 (R367C)—with the latter being classified as radical/damaging by all three criteria.
Figure 1.

In silico functional predictions.
Discussion
In this study we have applied exome sequencing, which allows the screening of the complete spectrum of variation within protein coding genes. There is abundant evidence that such regions are likely to be highly enriched for disease causing variation.31 We have focused on the identification of rare and novel variation within genes known to contain causal variants or identified as candidate genes for IBD. Excluding HLA variants and considering only rare non-synonymous, stop-gain mutations and indels, we uncovered 58 variants across 39 genes, of which 17 were not previously reported. Of these, 35% (20 variants) have SIFT scores under 0.05, 12 of these are also classified as probably damaging by Polyphen2 and five of these (BTNL2: S334L; C1orf93: G176R; ICAM1: R367C; NOD2: R702W and SH2B1: L185Q) are also classified as moderately radical or radical by Grantham score. One variant, CXCR1 (R335C), has a borderline SIFT score of 0.09 and is classified as probably damaging by Polyphen2 and radical by Grantham score. These variants may compromise protein function and contribute to the PIBD phenotype in these patients.
Our study included five patients with childhood onset CD. The variant profiles show that four of these patients carry potentially deleterious mutations in one or more IBD candidate genes. One child had a 17-fold increased risk of IBD on the basis of his NOD2 profile alone. Others in this group bear variants with likely impact on antigen presentation (ERAP2), endoplasmic reticulum trafficking (SEC16A) and T-helper cell differentiation. A variant in the IL18RAP gene was recently reported by Rivas et al 7 to carry a threefold OR for CD, and variants in the same gene have also been implicated in coeliac disease.32 We identify a rare, non-synonymous, two-base pair mutation in this gene in one of our severely affected early onset CD cases. Our study examined only two patients with a clear diagnosis of UC and intriguingly we observe unique, potentially deleterious variation in both the B-cell regulatory gene BACH2 and IL10 genes in both patients. Interestingly, defective IL10 functioning is already recognised in UC pathogenesis,33 34 whereas although other components of B-cell signalling (IL7R and IRF5) have shown previous association with UC,6 variation in BACH2 has shown previous association with CD only. Our patient with undetermined IBD is the only patient with rare ICAM1 variants. This gene, in which our IBDU patient carries two functionally damaging variants, plays a role in cell-mediated inflammation and has been identified as a therapeutic target in IBD.35
Assessing our results obtained for each individual in our cohort with IBD, we can see clearly that it is possible to generate an individualised variant profile for each patient. Individualised profiles are already being usefully applied to refine disease diagnosis. For example, Franke et al 36 reported recently on a whole genome sequencing undertaken on a 47-year-old patient diagnosed with CD in her 20s. Her case was particularly severe, as she had failed standard treatments including anti-TNF, had undergone multiple bowel resections, and required intermittent parenteral nutrition. Sequencing in this patient revealed multiple ‘hits’ in the autophagy pathways. This prompted in-depth mycobacterial diagnostics and ultimately resulted in a diagnosis of chronic active Mycobacterium avium infection.
Although suggestive and interesting mutation profiles have emerged from our small panel, it is clear that our picture is far from complete. Proband 3 displays rare variation across many genes, but not one of these appears to have potential functional consequence. Furthermore, in 65 genes previously linked to IBD, we identified no variants in our eight probands. It is possible that these genes do not contribute to disease in this small group, consistent with a high degree of genetic heterogeneity in this complex disease. It is also possible that limitations of sequencing technology or the analytical pipeline could have resulted in failure to call true variants. By focusing our analysis on exomes, we rely on the fact that many of the non-coding SNP variants previously implicated by GWAS simply flag coding variants in the genomic vicinity. Protein-coding genes harbour about 85% of the mutations with large effects for disease-related traits,37 but it is entirely possible that restriction of the exome capture to coding regions might have overlooked non-coding variants with significant impact on protein expression. By tabulating rare and novel variants, we are focusing attention on those variants hypothesised to have larger effect sizes on the assumption that such variants confer significant genetic contribution to childhood severe and/familial disease.38 However, for any complex disease, multiple common susceptibility variants, each contributing very modest effect sizes, should not be ignored.
SIFT, Polyphen2 and Grantham scores provide an indication of potential causality but they must be interpreted with caution, particularly for complex traits. Kumar et al 39 describe in silico prediction such as SIFT as effective for monogenic disease, but consider such tools to be less effective for lower penetrance variants associated with complex diseases. Furthermore, one study compiled in silico prediction scores and found pairwise agreement between all methods to be in the range 60–70%, implying fairly substantial disagreement.24 These and other studies underpin the difficulty in ascribing functional evidence and translational importance of genetic variants, and the particular difficulty in heterogeneous complex disease. However, it is notable that published evidence demonstrates a clear functional impact for two of the six variants listed above as having an overall deleterious score by two or more of the in silico measures. The CXCR1 gene R335C variant has been previously implicated in chronic obstructive pulmonary disease and asthma.40 The two CXCR1 mutations listed in table 2 (R335C and M31R) are in tight linkage disequilibrium and both are known to alter the structure and charge of the protein at the respective positions. The N-terminus of CXCR1 protein has been identified as potentially important for receptor–ligand binding, leading to the suggestion that the M31R variant may affect this interaction. This led to the hypothesis that both polymorphisms could impact receptor function through alterations in structure.41 The upper right quadrant of figure 1 indicates those variants where all three in silico prediction tools are concordant in ascribing detrimental effects of the variant. Mutations such as the rare R376C ICAM1 variant may modify the function of the encoded glycoprotein expressed on immune and endothelial cells and should be prioritised for functional assessment. Another non-synonymous variant highlighted by the in silico scores is the NOD2 R702W variant which, together with the NOD2 L1007fs variant, has been found to impair the activation of the NF-κB pathway in response to muramyl dipeptide (MDP), a bacterial wall component, with the L1007fs mutant unable to respond.42 NOD2 is localised to the cell membrane but the L1007fs polymorphism disrupts this association and thus the protein has cytoplasmic distribution. Forcing the L1007fs mutant protein to associate with the plasma membrane does not lead to activation of the NF-κB pathway in response to MDP; thus it is not the localisation of the NOD2 mutant, but rather an inability to respond to MDP, that affects induction of the NF-κB pathway. The L1007fs mutation has been shown to produce a truncated protein with impaired function.43 The NOD2 R702W variant occurred in four of the 22 non-IBD reference exomes, representing a higher than expected frequency. Although the reference exomes were composed of germline DNA from patients with diverse diagnoses (various lymphomas, leukaemias and congenital growth disorders), all four of these IBD negative controls had a diagnosis of chronic lymphocytic leukaemia. Interestingly, a population based cohort study of 47 679 Swedish patients with CD or UC, reported a 20% increased risk of haematopoietic cancers in these patients.44 However, the role of NOD2 polymorphisms has been further investigated in a variety of cancers, with most finding no association.45 Recently, however, Sivakumaran et al 46 found abundant evidence for pleiotropy in complex disease, defined as one gene having an effect on multiple phenotypes. The authors identified many genes harbouring variants associated with CD and other immune-mediated phenotypes. These associations include a CD association with chronic lymphocytic leukaemia, through the SP140 gene (within which a rare variant is listed in table 2). Other gene/disease associations linked with CD include BACH2 with type 1 diabetes and coeliac disease, IL18RAP in coeliac disease, IL1RL1 with eosinophil count and coeliac disease, MST1 with UC and primary sclerosing cholangitis, ZNF365 with breast cancer, and NOD2 with leprosy, among many others.47 All of these genes contain rare variants listed in table 2 within the eight patients we have exome sequenced.
The abundance of potentially damaging variants arising from next generation sequencing renders interpretation of the potential impact of disease challenging. However, focusing on early onset and other forms of ‘severe’ phenotype, including familial cases, coupled with our ability to filter variants identified with increasingly large and reliable databases of apparently neutral variants, offers the prospect of identifying important rare variants involved in complex traits such as IBD. This is the first study whereby a cohort of patients have been exome sequenced with the specific aim of generating a unique and personalised profile of rare variants across known disease genes for each patient. The rare variant profiles presented here provide a relatively small number of potential causal variants and include many mutations classed as deleterious by in silico prediction, a number of potential compound heterozygotes and a number of variants for which there is established functional evidence of roles in disease. These data, assessed from the perspective of individual patients, provide one of the first glimpses of personal mutation profiles and establish a foundation to elucidate the disease significance of these variants in future next-generation sequencing analyses of PIBD patients.
Supplementary Material
Acknowledgments
The authors would like to thank Nikki J Graham from the DNA laboratory in Human Genetics & Genomic Medicine, University of Southampton; and David Buck and Lorna Gregory from the Wellcome Trust Centre for Human Genetics, Oxford University.
Footnotes
Contributors: KC was responsible for analysis, and with AEW, interpretation of data, drafting of the manuscript, critical revision of article and final approval. RMB, NA and CW were responsible for acquisition of data, critical revision and final approval of article. AC, JG, RU-G, WT, JWH and MS were responsible for interpretation of data, critical revisions and final approval. SE was responsible for conception, design, acquisition of data, analysis and interpretation of data, drafting, revision and approval of the final manuscript.
Funding: This project was supported by: NIHR Biomedical Research Unit (Nutrition, Diet & Lifestyle), University Hospital Southampton NHS Foundation Trust with specific thanks to Liz Blake, Senior Paediatric Research Sister, and Rachel Haggarty, Senior Children's Research Nurse; University Hospital Southampton Foundation Trust R&D; and the Crohn's in Childhood Research Association (CICRA).
Competing interests: None.
Patient consent: Obtained.
Ethics approval: This study was approved by the Southampton & South West Hampshire Research Ethics Committee (REC) (09/H0504/125).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Sawczenko A, Sandhu BK, Logan RF, et al. Prospective survey of childhood inflammatory bowel disease in the British Isles. Lancet 2001;357:1093–4 [DOI] [PubMed] [Google Scholar]
- 2. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011;474:307–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bengtson MB, Solberg C, Aamodt G, et al. Familial aggregation in Crohn's disease and ulcerative colitis in a Norwegian population-based cohort followed for ten years. J Crohns Colitis 2009;3:92–9 [DOI] [PubMed] [Google Scholar]
- 4. Spehlmann ME, Begun AZ, Burghardt J, et al. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflamm Bowel Dis 2008;14:968–76 [DOI] [PubMed] [Google Scholar]
- 5. Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010;42:1118–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 2011;43:246–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 2011;43:1066–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bodmer W, Tomlinson I. Rare genetic variants and the risk of cancer. Curr Opin Genet Dev 2010;20:262–7 [DOI] [PubMed] [Google Scholar]
- 9. Gilissen C, Hoischen A, Brunner HG, et al. Unlocking Mendelian disease using exome sequencing. Genome Biol 2011;12:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 2011;13:255–62 [DOI] [PubMed] [Google Scholar]
- 11. Day-Williams AG, Zeggini E. The effect of next-generation sequencing technology on complex trait research. Eur J Clin Invest 2011;41:561–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Ridder L, Weersma RK, Dijkstra G, et al. Genetic susceptibility has a more important role in pediatric-onset Crohn's disease than in adult-onset Crohn's disease. Inflamm Bowel Dis 2007;13:1083–92 [DOI] [PubMed] [Google Scholar]
- 13. Biank V, Broeckel U, Kugathasan S. Pediatric inflammatory bowel disease: clinical and molecular genetics. Inflamm Bowel Dis 2007;13:1430–8 [DOI] [PubMed] [Google Scholar]
- 14. Lacher M, Kappler R, Berkholz S, et al. Association of a CXCL9 polymorphism with pediatric Crohn's disease. Biochem Biophys Res Commun 2007;363:701–7 [DOI] [PubMed] [Google Scholar]
- 15. Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet 2009;41:1335–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 2009;361:2033–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 2011;12:628–40 [DOI] [PubMed] [Google Scholar]
- 18. IBD Working Group of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Inflammatory bowel disease in children and adolescents: recommendations for diagnosis–the Porto criteria. J Pediatr Gastroenterol Nutr 2005;41:1–7 [DOI] [PubMed] [Google Scholar]
- 19. Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis 2011;17:1314–21 [DOI] [PubMed] [Google Scholar]
- 20. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Handsaker B, Wysoker A, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 2011;32:894–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974;185:862–4 [DOI] [PubMed] [Google Scholar]
- 27. Li WH, Wu CI, Luo CC. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol 1984;21:58–71 [DOI] [PubMed] [Google Scholar]
- 28. Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 2003;(33 Suppl):228–37 [DOI] [PubMed] [Google Scholar]
- 29. Economou M, Trikalinos TA, Loizou KT, et al. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol 2004;99:2393–404 [DOI] [PubMed] [Google Scholar]
- 30. Rudd MF, Williams RD, Webb EL, et al. The predicted impact of coding single nucleotide polymorphisms database. Cancer Epidemiol Biomark Prev 2005;14:2598–604 [DOI] [PubMed] [Google Scholar]
- 31. Lehne B, Lewis CM, Schlitt T. Exome localization of complex disease association signals. BMC Genomics 2011;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 2010;42:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet 2008;40:1319–23 [DOI] [PubMed] [Google Scholar]
- 34. Festen EA, Stokkers PC, van Diemen CC, et al. Genetic analysis in a Dutch study sample identifies more ulcerative colitis susceptibility loci and shows their additive role in disease risk. Am J Gastroenterol 2010;105:395–402 [DOI] [PubMed] [Google Scholar]
- 35. Philpott JR, Miner PB., Jr Antisense inhibition of ICAM-1 expression as therapy provides insight into basic inflammatory pathways through early experiences in IBD. Expert Opin Biol Ther 2008;8:1627–32 [DOI] [PubMed] [Google Scholar]
- 36. Franke A, Kuehbacher T, Nikolaus S, et al. The complete individual genome of a Female Crohn's disease patient—What can you Learn? Gastroenterol 2011;140(5 Suppl 1):S-90 [Google Scholar]
- 37. Majewski J, Schwartzentruber J, Lalonde E, et al. What can exome sequencing do for you? J Med Genet 2011;48:580–9 [DOI] [PubMed] [Google Scholar]
- 38. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008;40:695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar S, Dudley JT, Filipski A, et al. Phylomedicine: an evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet 2011;27:377–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stemmler S, Arinir U, Klein W, et al. Association of interleukin-8 receptor alpha polymorphisms with chronic obstructive pulmonary disease and asthma. Genes Immun 2005;6:225–30 [DOI] [PubMed] [Google Scholar]
- 41. Vasilescu A, Terashima Y, Enomoto M, et al. A haplotype of the human CXCR1 gene protective against rapid disease progression in HIV-1+ patients. Proc Natl Acad Sci U S A 2007;104:3354–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lecine P, Esmiol S, Metais JY, et al. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem 2007;282:15197–207 [DOI] [PubMed] [Google Scholar]
- 43. Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001;411:603–6 [DOI] [PubMed] [Google Scholar]
- 44. Askling J, Brandt L, Lapidus A, et al. Risk of haematopoietic cancer in patients with inflammatory bowel disease. Gut 2005;54:617–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yazdanyar S, Nordestgaard BG. NOD2/CARD15 genotype, cardiovascular disease and cancer in 43,600 individuals from the general population. J Intern Med 2010;268:162–70 [DOI] [PubMed] [Google Scholar]
- 46. Sivakumaran S, Agakov F, Theodoratou E, et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet 2011;89:607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lees CW, Barrett JC, Parkes M, et al. New IBD genetics: common pathways with other diseases. Gut 2011;60:1739–53 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.