

Abstract
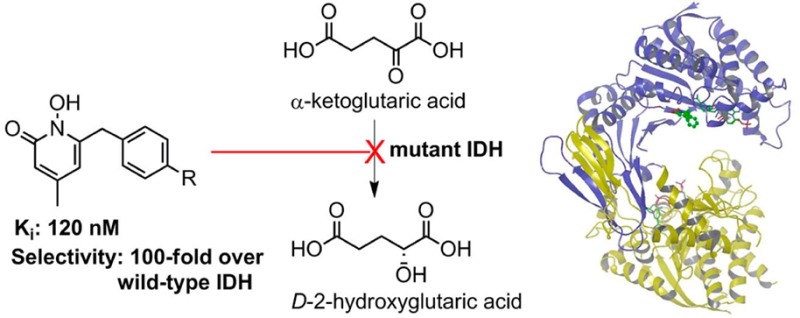
Mutations in isocitrate dehydrogenase (IDH), a key enzyme in the tricarboxylic acid cycle, have recently been found in ∼75% glioma and ∼20% acute myeloid leukemia. Different from the wild-type enzyme, mutant IDH1 catalyzes the reduction of α-ketoglutaric acid to d-2-hydroxyglutaric acid. Strong evidence has shown mutant IDH1 represents a novel target for this type of cancer. We found two 1-hydroxypyridin-2-one compounds that are potent inhibitors of R132H and R132C IDH1 mutants with Ki values as low as 120 nM. These compounds exhibit >60-fold selectivity against wild-type IDH1 and can inhibit the production of d-2-hydroxyglutaric acid in IDH1 mutated cells, representing novel chemical probes for cancer biology studies. We also report the first inhibitor-bound crystal structures of IDH1(R132H), showing these inhibitors have H-bond, electrostatic, and hydrophobic interactions with the mutant enzyme. Comparison with the substrate-bound IDH1 structures revealed the structural basis for the high enzyme selectivity of these compounds.
Keywords: Gene mutation, isocitrate dehydrogenase, enzyme inhibition, protein crystallography
Isocitrate dehydrogenases (IDH) are a family of Mg2+-dependent enzymes catalyzing the oxidative decarboxylation of isocitric acid (ICT) to α-ketoglutaric acid (α-KG),1 one of the key reactions in the tricarboxylic acid cycle providing aerobic organisms the majority of energy. Humans contain three IDH isozymes, i.e., IDH1 in cytoplasm and IDH2 and IDH3 in mitochondria. IDH1/2 use NADP+ as their cofactor, as schematically shown in Figure 1a.
Figure 1.

Enzyme reactions catalyzed by (A) wild-type IDH and (B) mutant IDH.
Mutations of IDH1/2 have recently been identified in several types of cancer.2−4 For example, IDH1 mutation is found in ∼75% low-grade gliomas (grade II and III) and secondary glioblastomas (grade IV glioma with a 5-year survival of <10%).5,6 About 20% acute myeloid leukemia (AML) also harbor IDH1/2 mutations.4 These mutations always occur in one DNA allele with the wild-type (WT) IDH1 gene in the other, suggesting the WT-IDH1 enzyme is also essential for the cancer cells. A special feature is that the identified IDH1 mutations are exclusively located in the Arg132 residue, with R132H being the predominant (∼93%).6 These genetic findings suggest IDH1 mutations play important roles in the initiation and/or progression of the cancer.
Biochemical investigation has revealed that all of the mutant IDH enzymes, including IDH1(R132H) and IDH1(R132C) (a frequent mutation in AML), are almost inactive in converting ICT to α-KG. Rather, these mutants were found to catalyze a new reaction, i.e., the reduction of α-KG to d-2-hydroxyglutaric acid (D2HG) using NADPH as the cofactor (Figure 1b).4,7,8 The direct physiological consequence of IDH mutation is a high cellular concentration of D2HG, the hallmark of this type of cancer. Further studies showed D2HG is an inhibitor of α-KG-dependent dioxygenases, including important epigenetic enzymes histone demethylases and 5-methylcytosine hydroxylases.3 The inhibition causes genome-wide hypermethylation of histone/DNA and blockage of cell differentiation, which could lead to tumorigenesis.9,10 Mutant IDH1 is therefore a novel target for intervention.11−13 It is of interest that, during the preparation of this letter, a series of diamide based compounds were disclosed to be the first inhibitors of mutant IDH1.14 Here, we report the discovery, activity, and synthesis of two novel, selective inhibitors of mutant IDH1. We also report the first X-ray crystal structures of inhibitor-bound IDH1(R132H), providing the exact inhibitor binding mode and the structural basis for the selectivity.
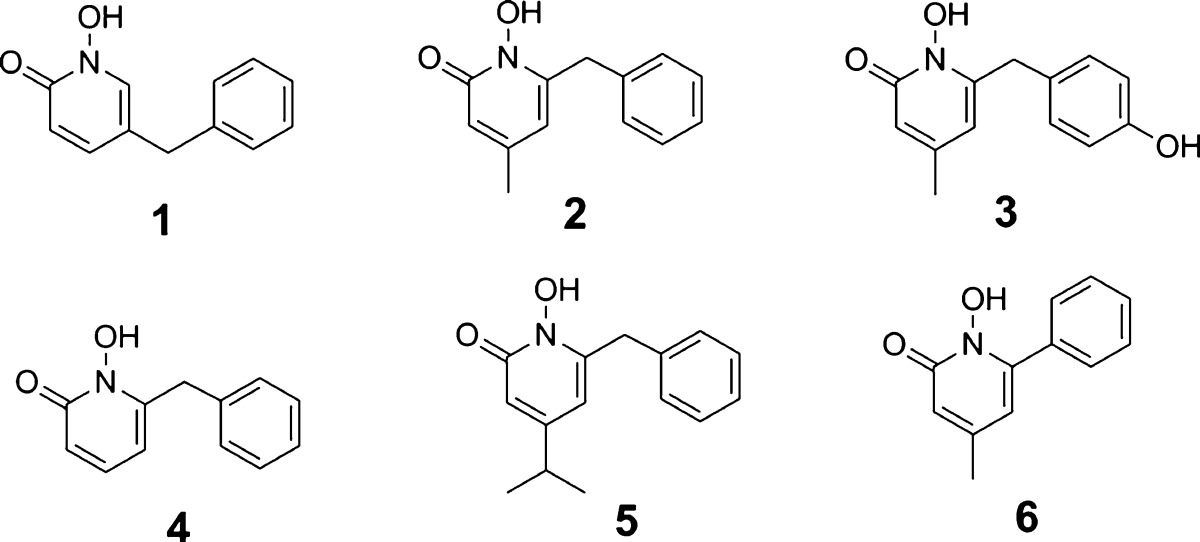
First, we expressed and obtained the recombinant proteins of WT IDH1, R132H, and R132C mutants (Supporting Information, Experimental Section), which were found to have comparable enzyme activity as those reported in previous studies,4,7,15 respectively. Our previous work on developing inhibitors of 1-deoxyxylulose-5-phosphate reductoisomerase,16,17 another Mg2+ and NADPH dependent reductase, provided a rich source of compounds that could target IDH1 mutants. Upon testing this focused library consisting of ∼130 compounds, 5-benzyl-1-hydroxypyridin-2-one (1) shown in Table 1 was identified as an inhibitor of IDH1(R132H) and IDH1(R132C) with Ki values of 8.2 and 6.6 μM, while it has no activity against WT IDH1 at 100 μM. Compound 1 is a drug-like molecule with a calculated log P value of 1.6 and is therefore of interest in the context of this study. Medicinal chemistry based on 1 has yielded 6-substituted 1-hydroxypyridin-2-one compounds 2 and 3 (Table 1) that are potent inhibitors of IDH1(R132H) with Ki values of 190 and 280 nM, respectively (Supporting Information Figure S1). It is remarkable that the 4-methyl group is of importance for the potency since compounds 4 and 5 with a 4-H and 4-isopropyl group, respectively, are ∼30× and 50× less active than 2. In addition, changing the 6-benzyl group in 2 to a phenyl group in compound 6 completely abrogates the inhibitory activity. These compounds also exhibited similar inhibitory activities against IDH1(R132C) mutant, with 2 and 3 being potent inhibitors of IDH1(R132C) with Ki values of 120 and 270 nM, respectively (Table 1).
Table 1. Structures and Biological Activities of 1–6.
| IDH enzyme Ki (μM) |
cytotoxicity (μM) | |||
|---|---|---|---|---|
| R132H | R132C | WT | WI-38 | |
| 1 | 8.2 | 6.6 | >50 | >50 |
| 2 | 0.19 | 0.12 | 12.3 | >50 |
| 3 | 0.28 | 0.27 | 16.8 | >50 |
| 4 | 5.9 | 10.5 | >50 | >50 |
| 5 | 9.5 | 14.7 | >50 | >50 |
| 6 | >50 | >50 | >50 | >50 |
We tested the activity of compounds 2 and 3 against WT-IDH1 to determine the enzyme selectivity since compounds that also strongly block WT-IDH1 are undesirable, given the important physiological role of this enzyme. As shown in Table 1, compounds 2 and 3 exhibited very weak activity against WT-IDH1 with Ki values of 12.3 and 16.8 μM, showing a high selectivity of >60-fold, despite only a single amino acid difference between the two enzymes. Moreover, as also can be seen in Table 1, neither 2 nor 3 exhibits high cytotoxicity against human WI-38 fibroblast cells with EC50 values of >50 μM. HT1080 fibrosarcoma cells, which harbor an IDH1(R132C) mutation,14 were treated with the most potent inhibitor 2 for 48 h. Compound 2 was found to be able to inhibit the production of D2HG with an EC50 value of 2.4 μM, using a reported HPLC-MS method.7 This compound is therefore a useful probe to investigate the cancer biology of IDH1 mutation.
Next, we investigated how the novel inhibitors 2 and 3 bind to the mutant IDH1 and what is the structural basis for the selectivity using X-ray crystallography. Previous crystallographic studies of R132H mutant as well as WT IDH1 proteins in complex with isocitric acid (ICT) have revealed the critical role of Arg132 for the WT enzyme.15 Without the bound ICT, mutant IDH1(R132H) exhibits an open conformation with an entrance channel of ∼20 Å wide. Upon ICT binding, the protein still adopts the open conformation and, as shown in Supporting Information Figure S2a, ICT is located in the ligand binding site I with H-bond and electrostatic interactions with Arg100, Ser94, and Thr77, as well as NADP. The mutated His132 is not involved in ICT binding as it is ∼9.5 Å away. In contrast, WT-IDH1 behaves differently. While it adopts an open conformation without the bound ICT, upon ligand binding, the protein undergoes a considerable conformational change transitioning to a closed state with an entrance channel of ∼13 Å wide. ICT is located in the ligand binding site II, which is also the catalytic site ∼6.5 Å away from the site I (Supporting Information Figure S2b). Of interest is that Arg132 is now in the active site, forming two H-bonds/electrostatic interactions with the two carboxyl groups of ICT (Supporting Information Figure S2c). The role of Arg132 is therefore to help pull ICT to the catalytically active site II, which occurs concomitantly with the protein transitioning to the closed state. This provides an explanation as to why IDH1(R132H) loses 95% of its ability to convert ICT to α-KG. It is likely due to the shorter side chain of His as well as the chemical nature of its imidazole group (as compared to the guanidine in Arg). However, the IDH1(R132H):α-KG structure shows a different binding mode.7 As shown in Supporting Information Figures S2d,e, it mimics the overall structure of the WT-IDH1:ICT complex. The mutant enzyme exhibits a closed conformation with the bound α-KG located in the ligand binding site II, which is the catalytic site of the mutant enzyme. His132 has no contacts with α-KG (5.5 Å away).
We determined the X-ray structures of IDH1(R132H) in complex with NADPH and inhibitors 2 or 3 to a resolution of 3.3 Å similar to that in the previously reported structures.15 Statistics for diffraction data and structure refinement are shown in Supporting Information Table S1 and the overall structures of the two complexes illustrated in Figures 2a and S3a, Supporting Information. The protein crystallizes as a homodimer with both subunits exhibiting an open conformation with a ∼20 Å wide protein entrance channel. An NADPH molecule was found in each of the subunits with a similar binding characteristics as that in the IDH1(R132H):ICT. However, only one inhibitor can be clearly located in the protein based on electron density and omit maps (Supporting Information Figures S4). Figures 2b and S3b, Supporting Information, show the close-up views of the binding sites of 2 and 3, respectively. Compounds 2 and 3 bind to IDH1(R132H) in a very similar manner consistent with their chemical structures. The 4-methyl-1-hydroxypyridin-2-one ring is comfortably located in a pocket surrounded by Arg100, Ser94, Thr77, Asn96, Arg109, and NADPH. The two O atoms of 2 are ∼2.7 Å from the two N atoms of the guanidine group of Arg100, indicating strong H-bonds and electrostatic interactions. The planar −CONH2 of Asn96 is located right underneath the pyridine ring of 2, with the distance of ∼4.1 Å. The 4-methyl group of 2/3 fits into a predominantly hydrophobic cavity, 3.7–4.1 Å from −CH3 of Thr77 and −CH2– and −O– of Ser94. Loss of these interactions could account for the reduced activities of compounds 4 and 5 with a 4-H and 4-i-Pr, which are either too small or too big to be favorable at this position. The 6-benzyl group of both inhibitors is involved in hydrophobic interactions with the pyridine ring of NADPH. Although these interactions could also contribute to the high affinity binding of the inhibitors, lack of direct binding of the 6-benzyl group to the protein (4.5 Å away from Arg109) renders the electron density of the moiety less strong than that of the 1-hydroxypyridin-2-one core. In addition, the 6-phenyl group of compound 6 would have a severe steric repulsion with NADPH, if its 1-hydroxypyridin-2-one ring maintains the above-mentioned favorable interactions with IDH1(R132H). This should account for the loss of activity for compound 6.
Figure 2.
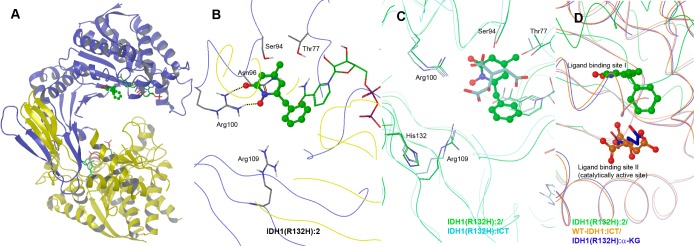
(A) Overall structure of IDH1(R132H):2 (ball and stick model). (B) Close-up view of the active site of IDH1(R132H):2. (C) The active sites of the aligned structures of IDH1(R132H):2 (with carbon and protein chain shown in green) and IDH1(R132H):ICT (in cyan), showing 2 is located in the ligand binding site I. (D) The aligned structures of IDH1(R132H):2 (in green), WT-IDH1:ICT (in orange), and IDH1(R132H):α-KG (in blue).
We next examined the binding site of 2/3 with respect to those of ICT and α-KG in mutant and WT IDH1. Thus, the structure of IDH1(R132H):2 complex was aligned with that of IDH1(R132H):ICT, showing these two structures are very similar with a root-mean-square deviation (rmsd) of 0.8 Å for the matching Cα atoms. The flexible loop 133–141 for both structures, including the catalytically important Tyr139 for the conversion of αKG to D2HG, is largely unordered. As shown in Figure 2c, compound 2 occupies the ICT binding site I in mutant IDH1. Although there are no major conformational changes for the key residues Arg100, Ser94, Thr77, and Asn96 (omitted in Figure 2c for clarity), 2 (Ki: 0.19 μM) binds to IDH1(R132H) with a much higher affinity than ICT (Kd >200 μM),15 likely due to increased hydrophobic and electrostatic interactions. Structural alignment of IDH1(R132H):2 with WT-IDH1:ICT as well as IDH1(R132H):α-KG further confirmed that 2 is not located in the catalytically active binding site II (Figure 2d). These studies suggest that the strong binding of 2/3 stabilizes IDH1(R132H) in the catalytically inactive, open conformation state. It also blocks α-KG from binding to the site II as well as protein conformational change to its closed state.
Docking using the program Glide18 was performed to investigate the observed high selectivity. First, compound 2 was docked into the IDH1(R132H):2 structure, and the 10 lowest-energy docking structures shown in Supporting Information Figure S5a are tightly clustered and mimic the crystal structure of 2, especially with respect to the conformation of the 4-methyl-1-hydroxypyridin-2-one ring. Next, docking suggested 2 could not bind favorably to the binding site II in WT IDH1. As shown in Supporting Information Figure S5b, 4 out of the 10 lowest-energy docking structures are not located in site II. In the remaining 6 docking structures, the phenyl ring, rather than the more favorable 1-hydroxypyridin-2-one ring, is located in the arginine-rich pocket. Moreover, there have been no WT-IDH1 structures with a ligand bound to the site I, suggesting Arg132 destabilizes this binding site. These studies provide a rationale for why compounds 2 and 3 exhibit weak activity against WT IDH1.
Finally, we describe the synthesis of compounds 1–6 (Scheme 1). 6- or 5-Bromo-2-methoxypyridine was coupled with benzylmagnesium chloride using a palladium-catalyzed coupling reaction. Using our previous method,16 the benzyl products were oxidized to a pyridine oxide, followed by removal of the methyl group to give compounds 1 or 4. An acyl chloride was reacted with ethyl 3-methyl-2-butenate in the presence of AlCl3 to afford compound 7 as a mixture of stereoisomers, which was cyclized to become a single compound pyron-2-one 8. Since the reaction of 8 with hydroxylamine occurred in a very poor yield (0–20%), 8 was converted to more reactive pyron-2-thione 9,19 which was reacted readily with hydroxylamine to give compounds 2, 3, or 6 in 67–73% yield. 2,6-Dihydroxy-isonicotinic acid was treated with POBr3 followed by methanolysis to give dibromo-ester 10, whose 4-methoxycarbonyl group was converted to an isopropenyl in 11 by treatment with MeMgBr followed by an elimination reaction. One of the bromo groups of 11 was substituted by a −OMe, and the other was reacted with n-BuLi and benzaldehyde to give compound 12. It was hydrogenated and dehydroxylated using Et3SiH20 to produce 4-isopropyl-2-methoxy-pyridine 13, which was then oxidized and deprotected to afford compound 5.
Scheme 1. Synthesis for Compounds 1–6.

Reagents and conditions: (i) BnMgCl, Pd(dppf)Cl2; (ii) 3-chloroperoxybenzoic acid; (iii) AcCl, reflux, then MeOH; (iv) RCOCl, AlCl3; (v) H2SO4/AcOH; (vi) P4S10; (vii) NH2OH, pyridine; (viii) POBr3, 130 °C, then MeOH; (ix) MeMgBr; (x) methanesulfonyl chloride, NEt3; (xi) NaOMe; (xii) n-BuLi, tetramethylethylene diamine, THF, −78 °C, then benzaldehyde; (xiii) H2, Pd(OH)2/C; (xiv) Et3SiH, trifluoroacetic acid, 50 °C.
In summary, recent biological studies have shown mutant IDH1 is a drug target for certain cancer. Using rational compound screening and medicinal chemistry, two 1-hydroxypyridin-2-one compounds 2 and 3 were found to be potent inhibitors of IDH1(R132H) and IDH1(R132C), the frequent mutations in cancer, with Ki values as low as 120 nM. These compounds exhibit >60-fold selectivity over WT-IDH1 and do not have cytotoxicity against human cells, representing novel chemical probes for cancer biology studies of mutant IDH1. In addition, we report the first crystal structures of IDH1(R132H):inhibitor complexes, showing 2 and 3 are located in the ligand binding site I, with several strong H-bond, electrostatic and hydrophobic interactions with the protein. Inability of these inhibitors to bind to the catalytically active site II could account for their high selectivity. Moreover, this work suggests targeting the binding site I in mutant IDH1 is a viable strategy for future rational design of potent and selective inhibitors of mutant IDH1, and our structures could serve as a useful platform for this purpose.
Acknowledgments
We thank the staff of the X-ray Crystallography Facility at Baylor College of Medicine for assistance in data collection.
Glossary
Abbreviations
- AML
acute myeloid leukemia
- IDH
isocitrate dehydrogenase
- ICT
isocitric acid
- α-KG
α-ketoglutaric acid
- WT-IDH1
wild-type IDH1 enzyme
- IDH1(R132H)
R132H mutant IDH1 enzyme
- D2HG
d-2-hydroxyglutaric acid
Supporting Information Available
Supplementary Figures S1–5, Supplementary Table S1, and Experimental Section. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors of the IDH1(R132H):2 and 3 complexes have been deposited in Protein Data Bank as entries 4I3L and 4I3K, respectively.
Author Contributions
§ These authors contributed equally to this work.
This work was supported by a grant (R01NS080963) from National Institute of Neurological Disorders and Stroke (NINDS/NIH) to Y.S. and a grant (Q1279) from Robert Welch Foundation to B.V.V.P.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Xu X.; Zhao J.; Xu Z.; Peng B.; Huang Q.; Arnold E.; Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [DOI] [PubMed] [Google Scholar]
- Parsons D. W.; Jones S.; Zhang X.; Lin J. C.; Leary R. J.; Angenendt P.; Mankoo P.; Carter H.; Siu I. M.; Gallia G. L.; Olivi A.; McLendon R.; Rasheed B. A.; Keir S.; Nikolskaya T.; Nikolsky Y.; Busam D. A.; Tekleab H.; Diaz L. A. Jr.; Hartigan J.; Smith D. R.; Strausberg R. L.; Marie S. K.; Shinjo S. M.; Yan H.; Riggins G. J.; Bigner D. D.; Karchin R.; Papadopoulos N.; Parmigiani G.; Vogelstein B.; Velculescu V. E.; Kinzler K. W. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Yang H.; Liu Y.; Yang Y.; Wang P.; Kim S. H.; Ito S.; Yang C.; Xiao M. T.; Liu L. X.; Jiang W. Q.; Liu J.; Zhang J. Y.; Wang B.; Frye S.; Zhang Y.; Xu Y. H.; Lei Q. Y.; Guan K. L.; Zhao S. M.; Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S.; Cairns R. A.; Minden M. D.; Driggers E. M.; Bittinger M. A.; Jang H. G.; Sasaki M.; Jin S.; Schenkein D. P.; Su S. M.; Dang L.; Fantin V. R.; Mak T. W. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.; Parsons D. W.; Jin G.; McLendon R.; Rasheed B. A.; Yuan W.; Kos I.; Batinic-Haberle I.; Jones S.; Riggins G. J.; Friedman H.; Friedman A.; Reardon D.; Herndon J.; Kinzler K. W.; Velculescu V. E.; Vogelstein B.; Bigner D. D. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C.; Meyer J.; Balss J.; Capper D.; Mueller W.; Christians A.; Felsberg J.; Wolter M.; Mawrin C.; Wick W.; Weller M.; Herold-Mende C.; Unterberg A.; Jeuken J. W.; Wesseling P.; Reifenberger G.; von Deimling A. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [DOI] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; van der Heiden M. G.; Su S. M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P. S.; Patel J.; Wise D. R.; Abdel-Wahab O.; Bennett B. D.; Coller H. A.; Cross J. R.; Fantin V. R.; Hedvat C. V.; Perl A. E.; Rabinowitz J. D.; Carroll M.; Su S. M.; Sharp K. A.; Levine R. L.; Thompson C. B. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Ward P. S.; Kapoor G. S.; Rohle D.; Turcan S.; Abdel-Wahab O.; Edwards C. R.; Khanin R.; Figueroa M. E.; Melnick A.; Wellen K. E.; O’Rourke D. M.; Berger S. L.; Chan T. A.; Levine R. L.; Mellinghoff I. K.; Thompson C. B. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S.; Rohle D.; Goenka A.; Walsh L. A.; Fang F.; Yilmaz E.; Campos C.; Fabius A. W.; Lu C.; Ward P. S.; Thompson C. B.; Kaufman A.; Guryanova O.; Levine R.; Heguy A.; Viale A.; Morris L. G.; Huse J. T.; Mellinghoff I. K.; Chan T. A. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner J. R.; Chinnaiyan A. M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [DOI] [PubMed] [Google Scholar]
- Dang L.; Jin S.; Su S. M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [DOI] [PubMed] [Google Scholar]
- Garber K. Oncometabolite? IDH1 discoveries raise possibility of new metabolism targets in brain cancers and leukemia. J. Natl. Cancer Inst. 2010, 102, 926–928. [DOI] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan Q.; Shi D.; Biller S. A.; Su S. M. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Zhong C.; Peng Y.; Lai Z.; Ding J. Molecular mechanisms of ″off–on switch″ of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 2010, 20, 1188. [DOI] [PubMed] [Google Scholar]
- Deng L.; Sundriyal S.; Rubio V.; Shi Z.; Song Y. Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-d-xylulose-5-phosphate reductoisomerase. J. Med. Chem. 2009, 52, 6539. [DOI] [PubMed] [Google Scholar]
- Deng L.; Endo K.; Kato M.; Cheng G.; Yajima S.; Song Y. Structures of 1-deoxy-d-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes. ACS Med. Chem. Lett. 2011, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glide, version 5.5; Schrödinger, LLC: New York, 2010. [Google Scholar]
- Deodhar K. D.; Kekare M. B.; Pednekar S. R. A facile synthesis of 2-pyridones by nucleophilic reaction on pyran2-thiones. Synthesis 1985, 328–331. [Google Scholar]
- Batt D. G.; Maynard G. D.; Petraitis J. J.; Shaw J. E.; Galbraith W.; Harris R. R. 2-Substituted-1-naphthols as potent 5-lipoxygenase inhibitors with topical antiinflammatory activity. J. Med. Chem. 1990, 33, 360–370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.