

Abstract
Glucagon-like peptide 1 (GLP-1) agonists improve myocardial function and insulin sensitivity in the setting of chronic heart failure. Endogenously produced GLP-1 peptide (7-36) is rapidly cleaved by dipeptidyl peptidase 4 (DPP4) to the 9-36 peptide, which lacks anti-hyperglycemic activity. To elucidate the effect of increased endogenous GLP-1 during heart failure progression, the DPP4 inhibitor saxagliptin or vehicle was administered by daily oral gavage to female TG9 mice, a transgenic model of dilated cardiomyopathy, starting at day of life 42, just prior to the development of detectable contractile dysfunction. Saxagliptin treatment inhibited DPP4 activity >90% and increased GLP-1 levels 4-fold following a 2 gm/kg glucose load but did not affect fasting GLP-1 levels. There was no difference in food intake or body weight between groups. At 56 days of age, oral glucose tolerance was improved in saxagliptin-versus vehicle-treated animals (AUC0-120 1340 ± 46 and 1501 ± 43 min·mmol/L, respectively, p<0.015). In contrast to the effect of a GLP-1 agonist in TG9 mice, saxagliptin had no effect on survival (80.7 ± 4.3 days) compared to vehicle-treated mice (79.6 ± 3.6 days, p = 0.46). Taken together, these data indicate that improvement in glucose tolerance is not sufficient to improve survival. Future efforts to confirm these findings in additional models of heart failure are warranted.
Keywords: cardiac function, insulin sensitivity, mouse model, glucose transport, incretin hormone
INTRODUCTION
Insulin resistance and cardiac dysfunction are intertwined in reciprocal causal relationships [1]. Diabetes increases the risk of developing overt heart failure 2–8 fold [2, 3] and is present in nearly 20% of patients with congestive heart failure. Furthermore, occult cardiomyopathy is prevalent in diabetic subjects [4]. Diabetic patients who develop heart failure have a greater than 2-fold increase in risk of death, compared to age and weight matched non-diabetic controls [5]. Conversely, idiopathic dilated cardiomyopathy patients develop secondary insulin resistance [6, 7]. Optimizing glucose homeostasis in heart failure patients, whether insulin resistance is primary or secondary to cardiac dysfunction, has gained greater attention [8, 9].
Glucagon like peptide-1 (GLP-1), a member of the pro-glucagon incretin family, mediates multiple physiologic effects that favorably affect whole body glucose homeostasis. These include augmentation of glucose-stimulated insulin secretion, suppression of glucagon secretion, preservation of beta cell mass, delay of gastric emptying, satiety, and improved peripheral glucose disposal [10]. In addition to these systemic metabolic effects, GLP-1 improves myocardial function in a canine model of tachycardia-induced cardiomyopathy [11]. In humans, GLP-1 can augment myocardial glucose uptake and cardiac function related to myocardial ischemia and dilated cardiomyopathy [12, 13].
The mechanism by which GLP-1 improves cardiac function could have clinical significance. Specifically, improvements in cardiac function may be due to direct effects of GLP-1 signaling in the heart or to improved systemic or tissue-specific glucose utilization. Multiple classes of drugs can effectively mediate the systemic but not necessarily the direct cardiac effects. Cardiac GLP-1 receptor expression suggests incretin signaling has a direct effect [14]. Studies in GLP-1R−/− mice indicate that GLP-1 receptor signaling affects both heart rate and contractile function [15]. Furthermore, GLP-1 improves cardiac preconditioning in isolated perfused rat hearts [16].
Endogenous GLP-1 consists of two active isoforms GLP-17-36 and GLP-17-37, both of which interact with the GLP-1 receptor. Dipeptidyl peptidase 4 (DPP4) rapidly degrades GLP-17-36 amide to the shorter 9-36 amide, which lacks insulinotropic effects. Several DPP4 inhibitors have therefore been developed for the treatment of type 2 diabetes [17]. DPP4 inhibitors produce many of the beneficial metabolic effects of GLP-1 agonists including preservation of beta cell mass, augmentation of insulin secretion and suppression of glucagon secretion. However, DPP4 inhibitors typically have a more modest anti-hyperglycemic effect than GLP-1 agonists and no effect on gastric emptying and satiety [17].
The ability of GLP-17-36 and GLP-19-36 to influence cardiac function in GLP-1R−/− mice suggests that some of the effects are mediated through GLP-1R independent mechanisms [1]. Given these observations, we sought to investigate the effect of DPP4 inhibition on glucose homeostasis and cardiac function in the failing heart using an established murine model of dilated cardiomyopathy. The TG9 mouse was generated by cardiac-specific overexpression of the cre recombinase protein under the α-mhc promoter [18]. Numerous features of the model system greatly assist efforts to understand the influence of incretin hormone biology on heart failure progression. TG9 mice develop progressive dilated cardiomyopathy in a highly predictable manner with many of the salient molecular and functional changes observed in human heart failure. This includes increased levels of brain naturetic peptide (BNP) and salutary effect of beta blockers and angiotensin converting enzyme (ACE) inhibitors. TG9 mice have normal left ventricular dimensions and systolic function at 6 weeks of age followed by progressive dilatation and worsening contractile function that leads to decompensated heart failure and death between 11–13 weeks of life [18]. The administration of the facilitative glucose transporter (GLUT) antagonist ritonavir to 10 week-old TG9 mice acutely exacerbates glucose intolerance and causes rapid cardiac decompensation [19], whereas the GLP-1 agonist exenatide improves glucose homeostasis, cardiac function and survival in this model [20]. We therefore hypothesized that if the effects of exenatide are mediated through improved systemic glucose homeostasis, the DPP4 inhibitor saxagliptin would produce similar beneficial effects on cardiac function and survival. Conversely, if the cardiac effects were mediated primarily through GLP-19-36, cardiac function would not improve despite incretin-induced changes in systemic glucose homeostasis. We report here the effect of saxagliptin on glucose tolerance, progression of cardiac dysfunction, and survival.
MATERIALS AND METHODS
Materials
Saxagliptin (Onglyza©, BMS-477118, or (1S,3S,5S)-2-[(2S)-2-Amino-2-(3-hydroxytricyclo(3.3.1.13,7]dec-1-yl)acetyl]-2-azabicyclo[3.1.0]hexane-3-carbonitrile, monohydrate) was supplied by Bristol-Myers Squibb (Princeton, NJ). GLUT4 antibody was custom produced by Invitrogen (Carlsbad, CA). GLUT1 antibody was a gift from Dr Mike Mueckler (Washington University, St Louis, MO). GAPDH monoclonal antibody was purchased from Abcam (Cambridge, MA). Anti human/rat/mouse monoclonal pan-AKT antibody and rabbit anti-phospho-AKT antibody were ordered from R&D Systems, Inc (Minneapolis, MN). AMPKα (F6) mouse antibody and Phospho-AMPKα (Thr 172) antibody were ordered from Cell Signaling (Danvers, MA). Secondary anti mouse and anti rabbit antibodies were ordered from LI-COR (Lincoln, NE). Unless noted, all other reagents were purchased from Sigma (St. Louis, MO).
Animal Procedures
All experiments were approved by the animal studies committee at Washington University School of Medicine. Mice were housed in the animal facility at Washington University under standard light/dark cycles and fed standard mouse chow diet and water ad libitum. For survival studies female TG9 mice were given saxagliptin (10 mg/kg/day) via oral gavage with litter-matched control receiving an equal volume of vehicle (0.5% Methylcellulose + 0.1% Tween 20).
Glucose tolerance
Following a 5-hour fast mice in the survival study group were subjected to a 2-hour glucose tolerance test. At time zero 20% dextrose (1mg/g) was administered via oral gavage and blood was sampled from a tail vein at 15 minutes intervals. Blood glucose was immediately determined using an Acenscia Contour glucometer (Bayer Health Care LLC). Serum insulin levels were determined by the DRTC immunoassay core facility at Washington University using a Singulex-based assay (Alameda, CA). Quantitative Insulin Sensitivity Check Index (QUICKI) was calculated as 1/[(log10I0)(log10G0)] where I0 = fasting serum insulin (μmol/l) and G0 = fasting serum glucose (mg/Dl) as previously described [21].
Myocardial glucose uptake
Tissue-specific glucose uptake was assessed by measuring 2-deoxyglucose incorporation in left ventricular myocardium under basal conditions following a 5-hour fast as previously described [19, 22, 23].
DPP4 activity
Following an overnight fast, 6-week old female TG9 mice were dosed with saxagliptin (10mg/kg) or vehicle. 75 minutes later, blood was drawn into chilled EDTA tubes via cardiac puncture. DPP4 activity was determined by measuring the cleavage of gly-pro-pNA as previously described [24]. Activity, the rate of release of free pNA from the pseudo-substrate, was assayed at 30°C, using a Spectramax 340 plate reader (Molecular Probes) with rate determining software. Absorbance at 390 nm was measured. Results were expressed as arbitrary units because standard pNA curves were not run for all experiments. The extent of DPP4 inhibition was determined from the reduction in the initial rate observed at each drug concentration with respect to untreated plasma.
GLP-1 levels
FVB, TG9 and C57 mice were dosed with saxagliptin (10mg/kg) or vehicle following a 4 hr fast. One hour later the mice were dosed with 2g/kg dextrose. Terminal bleeds were performed 10 minutes later. Blood was divided into 2 EDTA tubes; one with 5ul DPP4 inhibitor + 1μl Aprotinin, one blank and then assayed using the Meso Scale Active GLP-1 (7-36) assay kit (Mesoscale Diagnostics, Gaithersburg, MD).
RNA isolation and quantification
Total RNA was isolated from the left ventricular myocardium using Trizol® Plus RNA Purification System (Invitrogen, Carlsbad, CA). One microgram of total RNA was then treated with DNase I (Invitrogen, Carlsbad, CA) and reverse transcribed using the SuperScript® III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). Quantitative RT-PCR was performed using Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) on a Stratagene MX3005 qPCR thermal cycler. Each reaction was run in triplicate using previously validated primers for BNP (sense: 5′-TCACCGCTGGGAGGTCACTC-3′, antisense: 5′-GTGAGGCCTTGGTCCTTCAAG-3′) [20], and GLP1R (sense: 5′-TCTTCATCCGTGTCATCTGC-3′, antisense: 5′-CGTCCATCACAAAGGCAAAG-3′. β-Actin (sense: 5′-GATTACTGCTCTGGCTCCTAG-3′, antisense: 5′-GACTCATCGTACTCCTGCTTG-3′) and HPRT (sense: 5′-CCCCAAAATGGTTAAGGTTGC-3′, and antisense: 5′-AACAAAGTCTGGCCTGTATCC-3′) as internal controls, respectively. The relative changes in BNP and GLP1R expression levels were calculated by the ΔΔCt method.
Protein expression
Left ventricular myocardium was harvested from the mice immediately following euthanasia and snap frozen in liquid nitrogen. Lysates were prepared by homogenization in buffer containing 1% Triton X100 in PBS, Sigma protease inhibitor cocktail, sodium vanadate 1mM, sodium fluoride 50mM, and sodium pyrophosphate 10mM. Lysates were kept on ice for 15 minutes and cleared by centrifugation at 1500g for 20 minutes at 4C. Protein concentration was determined by the Bradford method [Bio-Rad, Hercules, CA]. Western blot analysis was then performed on 10 μg of total protein per lane using GLUT1 (1:1000) or GLUT4 (1:1000) rabbit polyclonal antibody recognizing the C terminus of the transporter, pan-AKT (1:5000), Phospho AKT (1:2000), AMPKα (1:1000) and phospho-AMPKα (1:1000). A mouse monoclonal GAPDH antibody (1:5000) was used as the loading control. Protein bands were quantified using Odyssey infrared imaging system version 3.0 (LI-COR Biosciences, Lincoln, NE).
Echocardiography
Echocardiography was performed on conscious 75-day old TG9 mice treated with vehicle or saxagliptin starting at 6 weeks of age using a Vevo 2100 Imaging System (VisualSonics, Toronto, Canada) equipped with a 30 MHz linear-array transducer. Parasternal long-axis views were obtained with a handheld technique. Quantitative image analysis was performed using a speckle-tracking algorithm implemented in the Vevo 2100 system (VevoStrain, VisualSonics, Toronto, Canada). In brief, suitable 2-D cine-loops were selected from digitally acquired echocardiographic images based on adequate visualization of the endo-and epicardial border and absence of image artifacts. Three consecutive cardiac cycles were selected for analysis based on image quality. Semi-automated tracing of the endocardial and epicardial borders were performed and verified over all 3 cardiac cycles and then corrected as needed to achieve good quality tracking throughout each cine loop. Tracked images were then processed in a frame-by-frame manner to derive the following parameters: LV volumes (end-diastolic and end-systolic, calculated by the disk summation method); rate-of-change of LV volumes (peak positive and peak-negative); stroke volume (end-diastolic volume minus end-systolic volume), ejection fraction (stroke volume divided by end-diastolic volume multiplied by 100); LV mass (volume of the epicardial shell minus volume of the endocardial shell multiplied by the specific gravity of muscle); global radial myocardial strain (averaged peak segmental radial strain).
RESULTS
Saxagliptin increases GLP-1 levels and inhibits DPP4 activity in TG9 Mice
To determine the effect of augmentation of endogenous GLP-1 on heart failure progression, TG9 mice were treated with the DPP4 inhibitor saxagliptin (10 mg/kg/d) administered by oral gavage once daily beginning at 42 days (6 weeks) of age, just prior to the onset of cardiac dysfunction. The pharmacokinetics of saxagliptin in rodents has been previously studied [25], and this dosing regimen has been shown to produce sustained effects on glucose tolerance [26]. Saxagliptin produced the expected inhibition of DPP4 activity in the blood 75 minutes after administration (Figure 1A). Compared to vehicle-treated mice, cleavage of the reporter peptide was reduced by 80%, (p<0.05). Furthermore, following an oral glucose load GLP-1 levels were augmented more than 4-fold in saxagliptin-treated TG9 mice as well as non-transgenic FVB and C57Bl/6J animals compared to vehicle-treated littermate controls (Figure 1B).
Figure 1. Saxagliptin Effect on GLP-1 and DPP4.
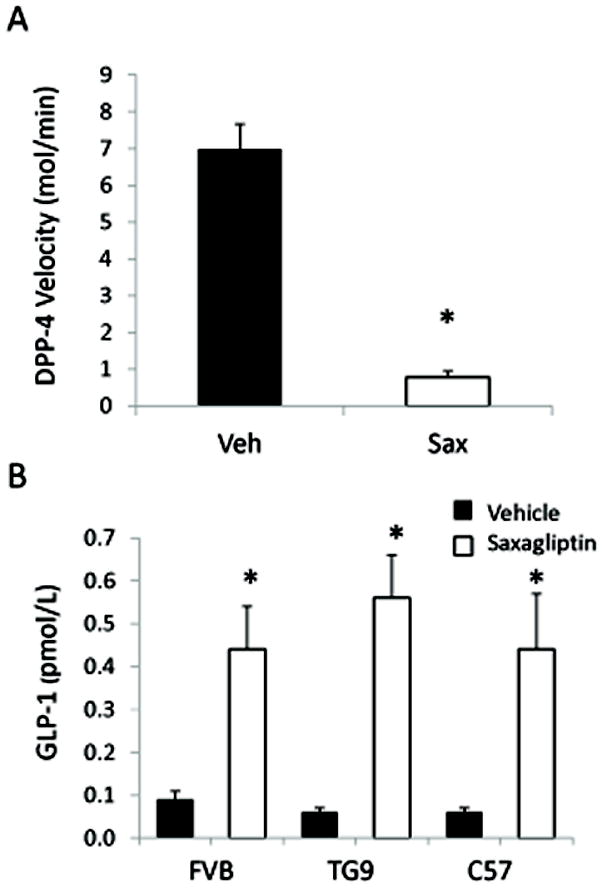
A. DPP-4 Activity was determined in female TG9 mice dosed with 10mg/kg saxagliptin [Sax] or vehicle [Veh] after an overnight fast. Terminal blood samples were taken 75 min later. B. Serum GLP-1 levels were determined on age-matched C57Bl/6J, FVB and TG9 mice dosed with 10mg/kg saxagliptin [□] or vehicle [■], following a 4 hr fast. After one hour, mice were given dextrose [2 gm/kg] by oral gavage. Serum was sampled 10 minutes later. *p < 0.05, student’s t-test.
Glucose transporter expression and function
To assess effects of saxagliptin treatment on whole body glucose homeostasis, glucose tolerance tests were performed at baseline (6 weeks) and then again after 2 and 4 weeks of treatment with either drug or vehicle alone (8 and 10 weeks of age, respectively). Blood glucose levels and responses to GTT did not differ between the two groups at baseline (Figure 2). At 8 and 10 weeks of age, the saxagliptin treatment group exhibited a significantly improved response to glucose challenge compared to vehicle-treated littermate control animals. However, relative insulin sensitivity as determined by the quantitative insulin sensitivity check index (QUICKI) [27] was the same in mice treated with vehicle (0.346 ± 0.023) and saxagliptin (0.343 ± 0.017, p=0.9).
Figure 2. Effect of saxagliptin on glucose tolerance in TG9 mice.
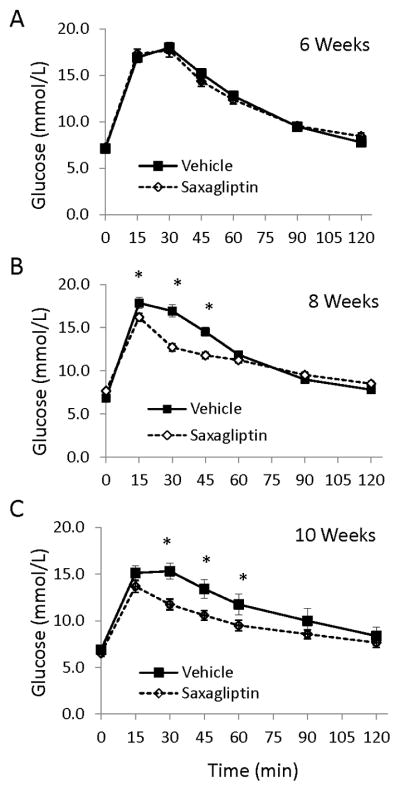
Oral glucose tolerance tests [2 gm/kg] were performed on age-matched female TG9 mice following a 5 hour fast. A. Baseline responses at 6 weeks of age. B. Following 2 weeks of treatment with either vehicle or saxagliptin [Age 8 weeks]. C. Following 4 weeks of treatment with either vehicle or saxagliptin [Age 10 weeks]. Data is shown as the mean +/− SEM, [n = 26–30]. *p < 0.05, ANOVA.
In prior studies, we determined that changes in peripheral glucose disposal in TG9 mice induced by exenatide correlate with increased expression of cardiac GLUT4 protein [20]. In contrast to these earlier findings, we did not observe a difference in either GLUT1 and GLUT4 protein levels in saxagliptin treated mice compared to vehicle-treated littermate controls (Figure 3). Consistent with this finding, no difference was observed in glucose uptake into left ventricular myocardium in saxagliptin-treated TG9 mice compared to vehicle treated littermate control animals (Figure 4).
Figure 3. Western blot analysis of GLUT protein levels in left ventricular myocardium harvested from 75-day-old TG9 mice.
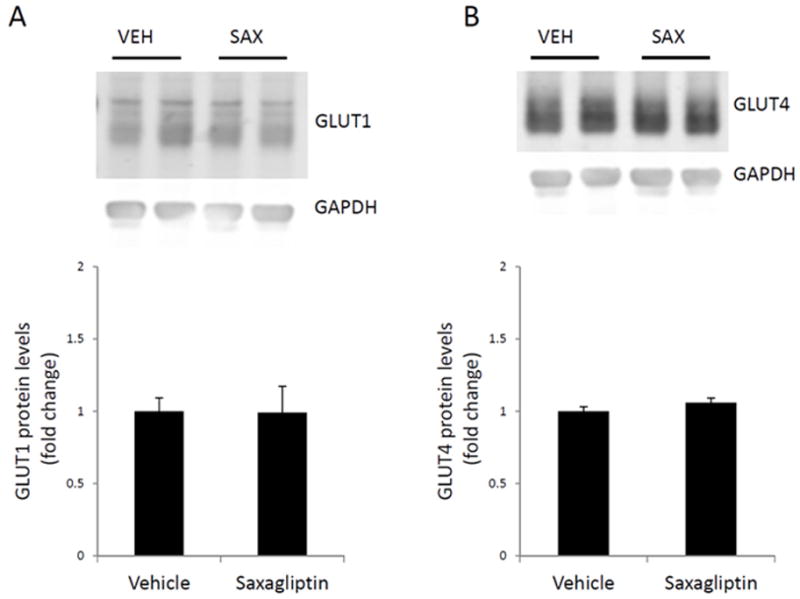
GLUT1 [A] and GLUT4 [B] protein levels were assessed using western blotting and normalized to the internal control, GAPDH, for loading variability. Fold change of GLUT expression in the saxagliptin-treated compared to the vehicle-treated mice is represented in the bar graphs. A representative western blot is shown. GLUT1 and GLUT4 levels are unchanged in left ventricular myocardium following sustained treatment with saxagliptin. Values are expressed as the mean ± SEM. n = 6 mice per group.
Figure 4. Effect of saxagliptin on left ventricular myocardial glucose uptake.
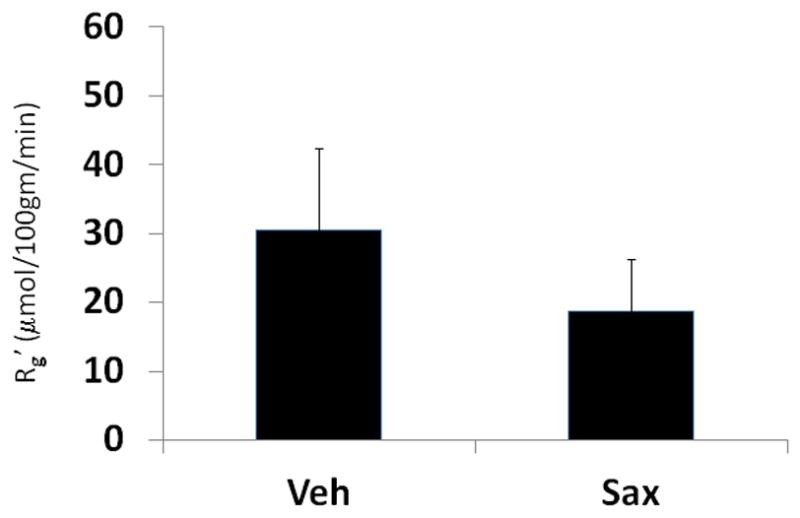
Female TG9 mice treated with vehicle [Veh] or 10 mg/kg saxagliptin [Sax] daily by oral gavage starting at 42 days of age. Glucose transport activity was determined by [3H]-2-deoxyglucose incorporation following a 5-h fast [n=8 per group]. Rg′, tissue glucose metabolic index.
Activation of AKT and AMPK
To assess changes in the metabolic status of the heart in response to saxagliptin therapy, we measured activation of the serine/threonine kinase AKT (Protein Kinase B). Total and phophorylated levels of AKT in left ventricular myocardium were not different between saxagliptin- and vehicle-treated TG9 mice (Figure 5A). Similarly, no difference was observed in activation of AMP-activated protein kinase (AMPK) in the two treatment groups (Figure 5B).
Figure 5. Western blot analysis of AKT and AMPK phosphorylation in left ventricular myocardium harvested from 75-day-old TG9 mice.
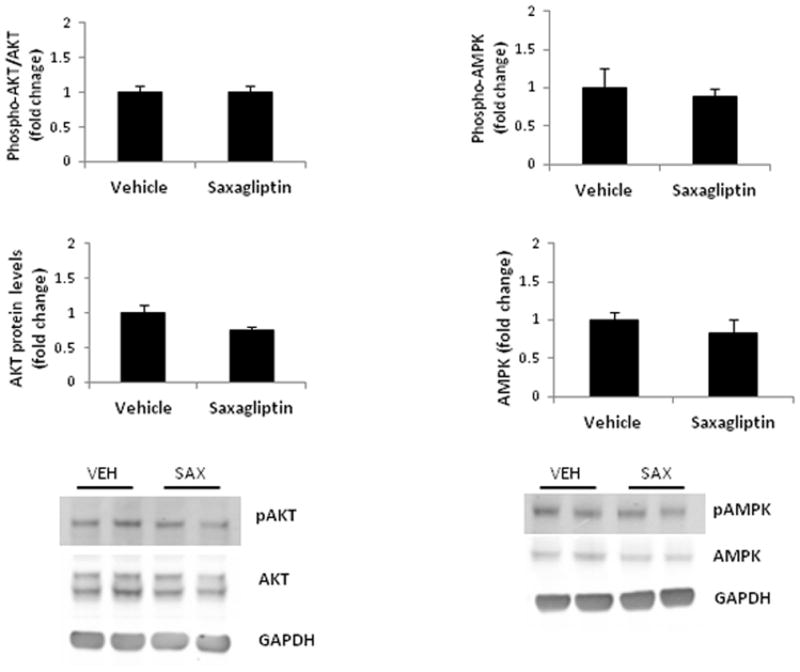
Total protein and phosphorylation levels of AKT [A] and AMPK [B] were assessed using western blotting and normalized to the internal control, GAPDH, for loading variability. Phosphorylation levels of [A] AKT and [B] AMPK in saxagliptin- compared to vehicle-treated animals is represented in the upper bar graph. Fold change of AKT and AMPK expression in the same mice is represented in the lower bar graph. A representative western blot is shown. Values are expressed as the mean ± SEM. n = 6 mice per group.
GLP-1 Receptor Expression
To determine whether saxagliptin treatment influenced the expression of the GLP1 receptor in left ventricular myocardium, mRNA was quantified by real-time PCR analysis in 75-day old TG9 mice. There was no difference observed in message level between saxagliptin- and vehicle-treated animals (Figure 6A).
Figure 6. RT-PCR analysis of BNP and GLP1R transcript levels in left ventricular myocardium harvested from 75-day-old FVB and TG9 mice.
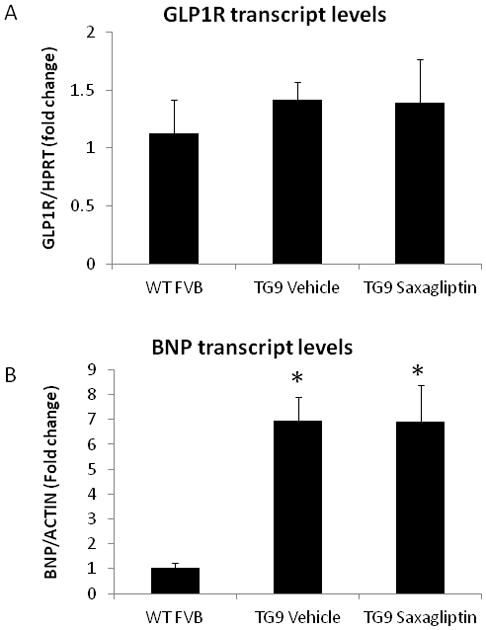
Mice were treated with saxagliptin or vehicle starting at 42 days. Comparative real time PCR was performed to examine the relative mRNA expression of (A) GLP1R and (B) BNP transcripts in the left ventricles of saxagliptin- versus vehicle-treated TG9 animals relative to message from age-matched FVB controls. *, p<0.01 compared to FVB control mice (BNP only). Real time PCR shows no change in the relative expression levels of BNP or GLP1R transcripts between the two TG9 groups [n= 4 mice/group].
Cardiac function
To assess the effect of saxagliptin on heart failure progression in the TG9 mouse, BNP mRNA expression in the left ventricular myocardium of 75-day old mice was quantified by real-time PCR. BNP expression was elevated approximately 7-fold in TG9 animals compared to age-matched non-transgenic controls. Saxagliptin treatment had no effect on BNP levels (Figure 6B).
Cardiac contractile function was assessed by echocardiography in 75-day old TG9 mice. Left ventricular dimensions and systolic function were significantly worse than non-transgenic mice, consistent with previously published data [4]. No difference was observed in any echocardiographic parameter between saxagliptin-treated mice and vehicle-treated littermate controls (Table 1).
Table 1.
Echocardiography of TG9 mice
| Parameter | Vehicle [n=7] | Saxagliptin [n=7] | p-value |
|---|---|---|---|
| Body weight [g] | |||
| Age 42 days | 19.8 ± 0.37 | 20.0 ± 0.37 | 0.69 |
| Age 70 days | 21.5 ± 0.73 | 21.4 ± 0.2 | 0.90 |
| Heart rate [beats per minute] | 522 ± 49 | 450 ± 18 | 0.19 |
| EDV [μL] | 64 ± 4.1 | 67 ± 3.2 | 0.59 |
| ESV[μL] | 37 ± 3.4 | 40 ± 2.7 | 0.48 |
| EF [%] | 43 ± 1.9 | 41 ± 1.6 | 0.38 |
| dV/dt-s | 0.77 ± 0.02 | 0.75 ± 0.02 | 0.50 |
| dV/dt-d | 1.31 ± 0.06 | 1.27 ± 0.06 | 0.63 |
| LVMd [mg] | 84 ± 4.3 | 86 ± 3.6 | 0.82 |
| e-rad [%] | 18 ± 1.5 | 17 ± 1.5 | 0.77 |
| R-TPk [ms] | 64 ± 4.4 | 65 ± 1.5 | 0.93 |
| e-long [%] | 11 ± 0.7 | 10 ± 0.8 | 0.15 |
| L-TPK [ms] | 64 ± 3.3 | 66 ± 1.6 | 0.64 |
Transthoracic echocardiography performed on conscious 75-day old TG9 mice. Saxagliptin [or vehicle] was administered by oral gavage at a dose of 10 mg/kg/day starting at 42 days of age. LV, left ventricle. EDV, end-diastolic LV volume. ESV = end-systolic LV volume. EF = ejection fraction [(EDV−ESV)/EDV]. dV/dt-d = peak rate of LV volume increase in diastole. dV/dt-s = peak rate of LV volume decrease in systole. e-rad = average peak radial strain. R-TPK = average time to peak of radial strain. elong = average peak longitudinal strain. L-TPk = average time to peak of longitudinal strain. Data is shown at the average +/− SEM
Survival analysis
The stereotypical rate of progression of heart failure in TG9 mice provides a means to assess survival as an endpoint with statistical confidence. Untreated TG9 mice typically die within a 2–3 day window between 11–13 weeks of age. In the current study, the mean survival of saxagliptin-treated mice was 80.7 +/− 4.3 days. This was indistinguishable from vehicle-treated littermate control animals (79.6 +/− 3.6 days, p = 0.46; Figure 7). Thus, despite the ability of saxagliptin to improve glucose tolerance, DPP4 inhibition had no effect on heart failure progression and time to death in TG9 mice.
Figure 7. Kaplan-Meier survival curves.
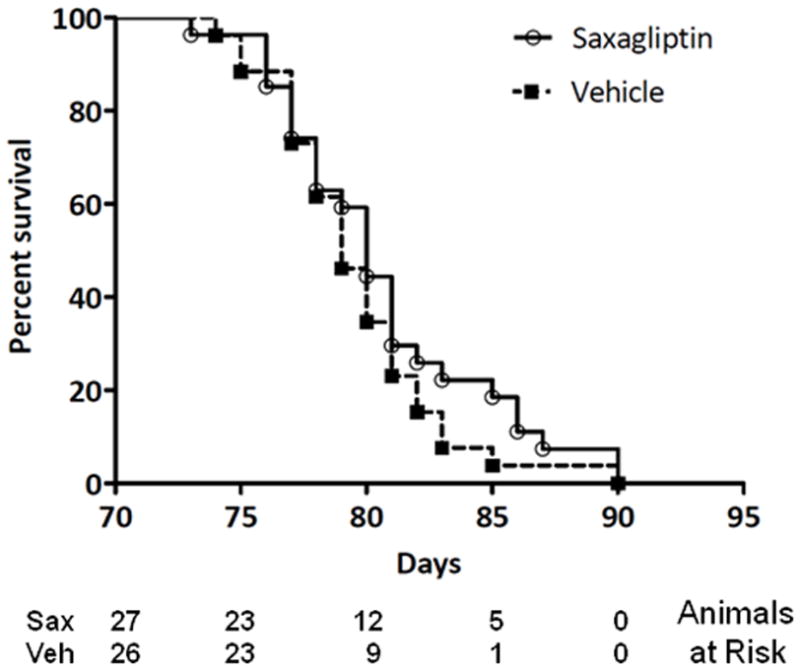
Female TG9 mice were treated with 10 mg/kg/day of saxagliptin or vehicle orally beginning at 42 days of life and continuing until the time of death. ○, Saxagliptin treated mice [n = 27]; ■, vehicle [n = 26]. P =0.3, Log Rank Test.
DISCUSSION
With growing interest in developing more effective strategies for the prevention and treatment of congestive heart failure, particularly in patients with type 2 diabetes, GLP-1 agonists have emerged as promising pharmacologic agents [28]. The beneficial effects on systemic glucose homeostasis and body weight in combination with a low risk of hypoglycemia have garnered incretin mimetics significant attention in this patient population. Beyond the influence of incretin hormones on glucose tolerance, exogenously administered GLP-1 and GLP-1R agonists have been shown to have beneficial effects on cardiac function and survival in numerous animal models, including rodents [29, 30]. Whether DDP4 inhibition has similar benefits on cardiac function and survival is less well understood. DDP4 has other substrates in addition to GLP-1, so DPP4 inhibition could have effects beyond improvement in glucose homeostasis that pertain to cardiac function in the setting of concomitant injury or stress [31]. The ability of DPP4 to cleave and inactivate BNP, for instance, has led to the hope that inhibition of this enzymatic activity may prove beneficial in patients with congestive heart failure [32]. DPP4 blockade has recently been shown to improve cardiac and renal function in a porcine model of tachycardia-induced cardiomyopathy [33]. Genetic and pharmacologic ablation of DPP4 activity in a rodent model of acute ischemia/reperfusion injury has also shown beneficial effects on glucose homeostasis and cardiovascular function [34]. Understanding of the ability of DPP4 inhibitors to influence cardiac function and survival in patients with heart failure has significant implications for recommendations and clinical use of these agents. Beyond direct clinical applicability, delineation of the role of incretin cleavage products in augmenting myocardial contractility is necessary to determine more precisely mechanisms for these effects.
Despite the strong rationale for exploring DDP4 blockade as a means for improving cardiac function, improvement in heart failure progression did not occur with augmentation of endogenous GLP-1 levels in mice with dilated cardiomyopathy. A similar lack of benefit of the DPP4 inhibitor vildagliptin on cardiac function has recently been reported in a rodent myocardial infarction model [35]. Unlike the effect of saxagliptin in TG9 mice, vildagliptin did not alter glucose tolerance. The current data demonstrate that in the TG9 mouse improved systemic glucose tolerance alone is not sufficient to produce a detectable increase in cardiac function or survival. Several aspects of the current data could account for this observation. Since saxagliptin did not influence cardiac insulin signaling and glucose uptake, it remains possible that altered myocardial glucose delivery and utilization in the setting of heart failure contributes to decreased contractile function. This would be consistent with the data showing reduced cardiac function and survival following pharmacologic glucose transport blockade [19]. Exenatide produces greater improvement in glucose tolerance, improves myocardial glucose uptake and contractile function, and prolonged survival in TG9 mice [20] and each of these factors may contribute to the difference in cardioprotective effects of saxagliptin.
The improved glucose tolerance observed in the saxagliptin-treated mice likely reflects augmented insulin secretion due to DPP4-mediated increases in endogenous GLP-1 levels. While we did observe a significant increase in glucose-stimulated GLP-1 levels with saxagliptin, the absolute incretin hormone activity was less than what can be achieved by exogenous GLP-1 or GLP-1 agonist administration. Higher GLP-1 levels may be necessary to enhance glucose uptake in the myocardium than in skeletal muscle. Supra-physiologic action of GLP-1 on myocardial tissue, whether direct or indirect, could account for the observed differences between the effect of exenatide and saxagliptin on GLUT4 expression [20]. Alternatively, supraphysiologic GLP-1 levels may be required to activate cardioprotective mechanisms unrelated to myocardial glucose uptake [36]. Since DDP4 activity was nearly completely abolished by saxagliptin treatment, increasing the dose of drug or treatment with additional agents within this class are unlikely to produce any further increases in endogenous GLP-1. Consistent with prior reports that the DPP4 cleavage product GLP-1 9-36 influences cardiac function independent of GLP-1R [37], it is also possible that this peptide contributes to the cardiac effects of augmented incretin hormone administration. While protein levels may not directly correlate with mRNA levels, the failure to observe a change in GLP-1R expression in saxagliptin indicates that this does not contribute to the observed difference from our previous study on the effect of exenatide on cardiac function and survival in this cardiomyopathy model.
The potential of saxagliptin to alter the levels of DPP4 substrates other than GLP-1 may have contributed to the lack of survival benefit observed in this study [38]. If such mechanisms exist, it is likely that the relative influence of these insulinotropic-independent signals will vary among different mechanisms for cardiac injury and may at least partially explain conflicting reports of the effects of DPP4 inhibition on cardiac function. Thus, until the effects of DPP4 inhibition are more fully elucidated, it may be premature to rule out a potential benefit. Nevertheless, emerging data indicate that there are fundamental differences between the effects of GLP-1 agonist and DPP4 inhibitor therapy [39]. Our data suggest the therapeutic differences extend to the setting of primary dilated cardiomyopathy.
Acknowledgments
Grants: This study was supported in part by research grants from Bristol Myers Squibb, the National Institutes of Health (DK064572 and HL092798) and the Washington University NORC P30DK056341. Insulin assays were performed in the Diabetes Research and Training Center core laboratory funded by NIH grant DK 020579. Echocardiography was performed in the Mouse Phenotyping Core Facility at Washington University.
Footnotes
Author Contributions: Conceived and designed experiments: PWH, PYJ, AKV. Performed experiments: AKV, LAF, MAP. Wrote paper: AKV, PWH. Reviewed and edited manuscript: LAF, AKV, PYJ.
References
- 1.Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294(3):334–41. doi: 10.1001/jama.294.3.334. [DOI] [PubMed] [Google Scholar]
- 2.Nichols GA, Hillier TA, Erbey JR, Brown JB. Congestive heart failure in type 2 diabetes: prevalence, incidence, and risk factors. Diabetes Care. 2001;24(9):1614–9. doi: 10.2337/diacare.24.9.1614. [DOI] [PubMed] [Google Scholar]
- 3.Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care. 2003;26(8):2433–41. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- 4.Fang ZY, Schull-Meade R, Leano R, Mottram PM, Prins JB, Marwick TH. Screening for heart disease in diabetic subjects. Am Heart J. 2005;149(2):349–54. doi: 10.1016/j.ahj.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 5.Gustafsson I, Brendorp B, Seibaek M, Burchardt H, Hildebrandt P, Kober L, Torp-Pedersen C. Influence of diabetes and diabetes-gender interaction on the risk of death in patients hospitalized with congestive heart failure. J Am Coll Cardiol. 2004;43(5):771–7. doi: 10.1016/j.jacc.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 6.Witteles RM, Tang WH, Jamali AH, Chu JW, Reaven GM, Fowler MB. Insulin resistance in idiopathic dilated cardiomyopathy: a possible etiologic link. Journal of the American College of Cardiology. 2004;44(1):78–81. doi: 10.1016/j.jacc.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 7.Kim J, Nakatani S, Hashimura K, Komamura K, Kanzaki H, Asakura M, et al. Abnormal glucose tolerance contributes to the progression of chronic heart failure in patients with dilated cardiomyopathy. Hypertens Res. 2006;29(10):775–82. doi: 10.1291/hypres.29.775. [DOI] [PubMed] [Google Scholar]
- 8.Schulze PC, Biolo A, Gopal D, Shahzad K, Balog J, Fish M, et al. Dynamics in Insulin Resistance and Plasma Levels of Adipokines in Patients With Acute Decompensated and Chronic Stable Heart Failure. Journal of Cardiac Failure. 2011;17(12):1004–1011. doi: 10.1016/j.cardfail.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falcão-Pires I, Leite-Moreira A. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Failure Reviews. :1–20. doi: 10.1007/s10741-011-9257-z. [DOI] [PubMed] [Google Scholar]
- 10.Holst JJ, Deacon CF, Vilsboll T, Krarup T, Madsbad S. Glucagon-like peptide-1, glucose homeostasis and diabetes. Trends Mol Med. 2008;14(4):161–8. doi: 10.1016/j.molmed.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, et al. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation. 2004;110(8):955–61. doi: 10.1161/01.CIR.0000139339.85840.DD. [DOI] [PubMed] [Google Scholar]
- 12.Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. 2006;12(9):694–9. doi: 10.1016/j.cardfail.2006.08.211. [DOI] [PubMed] [Google Scholar]
- 13.Sokos GG, Bolukoglu H, German J, Hentosz T, Magovern GJ, Jr, Maher TD, et al. Effect of glucagon-like peptide-1 (GLP-1) on glycemic control and left ventricular function in patients undergoing coronary artery bypass grafting. Am J Cardiol. 2007;100(5):824–9. doi: 10.1016/j.amjcard.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Nauck MA. Glucagon-like peptide 1 (GLP-1): a potent gut hormone with a possible therapeutic perspective. Acta Diabetol. 1998;35(3):117–29. doi: 10.1007/s005920050116. [DOI] [PubMed] [Google Scholar]
- 15.Gros R, You X, Baggio LL, Kabir MG, Sadi AM, Mungrue IN, et al. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology. 2003;144(6):2242–52. doi: 10.1210/en.2003-0007. [DOI] [PubMed] [Google Scholar]
- 16.Bose AK, Mocanu MM, Carr RD, Yellon DM. Glucagon like peptide-1 is protective against myocardial ischemia/reperfusion injury when given either as a preconditioning mimetic or at reperfusion in an isolated rat heart model. Cardiovasc Drugs Ther. 2005;19(1):9–11. doi: 10.1007/s10557-005-6892-4. [DOI] [PubMed] [Google Scholar]
- 17.Gallwitz B. GLP-1 agonists and dipeptidyl-peptidase IV inhibitors. Handb Exp Pharmacol. 2011;(203):53–74. doi: 10.1007/978-3-642-17214-4_3. [DOI] [PubMed] [Google Scholar]
- 18.Buerger A, Rozhitskaya O, Sherwood MC, Dorfman AL, Bisping E, Abel ED, et al. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J Card Fail. 2006;12(5):392–8. doi: 10.1016/j.cardfail.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Hruz PW, Yan Q, Struthers H, Jay PY. HIV protease inhibitors that block GLUT4 precipitate acute, decompensated heart failure in a mouse model of dilated cardiomyopathy. FASEB J. 2008;22(7):2161–7. doi: 10.1096/fj.07-102269. [DOI] [PubMed] [Google Scholar]
- 20.Vyas AK, Yang KC, Woo D, Tzekov A, Kovacs A, Jay PY, Hruz PW. Exenatide improves glucose homeostasis and prolongs survival in a murine model of dilated cardiomyopathy. PLoS One. 2011;6(2):e17178. doi: 10.1371/journal.pone.0017178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, Quon MJ. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85(7):2402–10. doi: 10.1210/jcem.85.7.6661. [DOI] [PubMed] [Google Scholar]
- 22.Hom FG, Goodner CJ, Berrie MA. A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes. 1984;33(2):141–52. doi: 10.2337/diab.33.2.141. [DOI] [PubMed] [Google Scholar]
- 23.Smith SA, Young P, Cawthorne MA. Quantification in vivo of the effects of insulin on glucose utilization in individual tissues of warm- and cold-acclimated rats. The Biochemical Journal. 1986;237(3):789–95. doi: 10.1042/bj2370789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergman AJ, Stevens C, Zhou Y, Yi B, Laethem M, De Smet M, et al. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Ther. 2006;28(1):55–72. doi: 10.1016/j.clinthera.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Fura A, Khanna A, Vyas V, Koplowitz B, Chang SY, Caporuscio C, et al. Pharmacokinetics of the dipeptidyl peptidase 4 inhibitor saxagliptin in rats, dogs, and monkeys and clinical projections. Drug Metab Dispos. 2009;37(6):1164–71. doi: 10.1124/dmd.108.026088. [DOI] [PubMed] [Google Scholar]
- 26.Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, et al. Discovery and preclinical profile of Saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48(15):5025–37. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- 27.Lee S, Muniyappa R, Yan X, Chen H, Yue LQ, Hong EG, et al. Comparison between surrogate indexes of insulin sensitivity and resistance and hyperinsulinemic euglycemic clamp estimates in mice. American Journal of Physiology - Endocrinology And Metabolism. 2008;294(2):E261–E270. doi: 10.1152/ajpendo.00676.2007. [DOI] [PubMed] [Google Scholar]
- 28.Khan MA, Deaton C, Rutter MK, Neyses L, Mamas MA. Incretins as a novel therapeutic strategy in patients with diabetes and heart failure. Heart Fail Rev. 2012 doi: 10.1007/s10741-012-9318-y. [DOI] [PubMed] [Google Scholar]
- 29.Michael HD. Cardiovascular Effects of Glucagonlike peptide–1 Agonists. The American Journal of Cardiology. 2011;108(3, Supplement):33B–41B. doi: 10.1016/j.amjcard.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 30.Liu Q, Anderson C, Broyde A, Polizzi C, Fernandez R, Baron A, Parkes DG. Glucagon-like peptide-1 and the exenatide analogue AC3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovasc Diabetol. 2010;9:76. doi: 10.1186/1475-2840-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fadini GP, Avogaro A. Cardiovascular effects of DPP-4 inhibition: Beyond GLP-1. Vascular Pharmacology. 55(1–3):10–16. doi: 10.1016/j.vph.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Vanderheyden M, Bartunek J, Goethals M, Verstreken S, Lambeir AM, De Meester I, Scharpé S. Dipeptidyl-peptidase IV and B-type natriuretic peptide. From bench to bedside. Clinical Chemistry and Laboratory Medicine. 2009;47(3):248–252. doi: 10.1515/CCLM.2009.065. [DOI] [PubMed] [Google Scholar]
- 33.Gomez N, Touihri K, Matheeussen V, Mendes Da Costa A, Mahmoudabady M, Mathieu M, et al. Dipeptidyl peptidase IV inhibition improves cardiorenal function in overpacing-induced heart failure. European Journal of Heart Failure. 2012;14(1):14–21. doi: 10.1093/eurjhf/hfr146. [DOI] [PubMed] [Google Scholar]
- 34.Sauvé M, Ban K, Momen MA, Zhou YQ, Henkelman RM, Husain M, Drucker DJ. Genetic Deletion or Pharmacological Inhibition of Dipeptidyl Peptidase-4 Improves Cardiovascular Outcomes After Myocardial Infarction in Mice. Diabetes. 2010;59(4):1063–1073. doi: 10.2337/db09-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin M, Sillje HH, Meissner M, van Gilst WH, de Boer RA. Early and late effects of the DPP-4 inhibitor vildagliptin in a rat model of post-myocardial infarction heart failure. Cardiovasc Diabetol. 2011;10:85. doi: 10.1186/1475-2840-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon JS, Lee HW. Understanding the cardiovascular effects of incretin. Diabetes Metab J. 2011;35(5):437–43. doi: 10.4093/dmj.2011.35.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonne DP, Engstrom T, Treiman M. Protective effects of GLP-1 analogues exendin-4 and GLP-1(9-36) amide against ischemia-reperfusion injury in rat heart. Regul Pept. 2008;146(1–3):243–9. doi: 10.1016/j.regpep.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Brandt I, Lambeir AM, Ketelslegers JM, Vanderheyden M, Scharpé S, De Meester I. Dipeptidyl-Peptidase IV Converts Intact B-Type Natriuretic Peptide into Its des-SerPro Form. Clinical Chemistry. 2006;52(1):82–87. doi: 10.1373/clinchem.2005.057638. [DOI] [PubMed] [Google Scholar]
- 39.Morales J. The Pharmacologic Basis for Clinical Differences Among GLP-1 Receptor Agonists and DPP-4 Inhibitors. Postgraduate Medicine. 2011;123(6):189–201. doi: 10.3810/pgm.2011.11.2508. [DOI] [PubMed] [Google Scholar]