

Abstract
Gene expression is a process central to any form of life. It involves multiple temporal and functional scales that extend from specific protein-DNA interactions to the coordinated regulation of multiple genes in response to intracellular and extracellular changes. This diversity in scales poses fundamental challenges to the use of traditional approaches to fully understand even the simplest gene expression systems. Recent advances in computational systems biophysics have provided promising avenues to reliably integrate the molecular detail of biophysical process into the system behavior. Here, we review recent advances in the description of gene regulation as a system of biophysical processes that extend from specific protein-DNA interactions to the combinatorial assembly of nucleoprotein complexes. There is now basic mechanistic understanding on how promoters controlled by multiple, local and distal, DNA binding sites for transcription factors can actively control transcriptional noise, cell-to-cell variability, and other properties of gene regulation, including precision and flexibility of the transcriptional responses.
Introduction
The process that leads to functional RNA and protein molecules from the information encoded in genes is known as gene expression. It starts with the binding of the RNA polymerase (RNAP) to the promoter, continues with transcription of the gene into RNA, and often concludes with translation into protein (1). This simple description is just the backbone of a much more complex set of events involving many processes that actively regulate, complement, affect, and critically refine these three steps (2).
The complexity of gene expression is already evident at the very early stages of the process. The RNAP rarely just binds to DNA and starts transcription. There are molecules, such as transcription factors (TFs), that enhance, stabilize, hinder, and prevent the binding of the RNAP to the promoter (3). This local layer of control is embedded in the underlying dynamic organization of the genome, which determines to a large extent the accessibility of the RNAP to the promoter and to the information content of sets of genes that are spatially in the same region (4–8). In addition, the RNAP is not a simple molecule but a multisubunit complex that, especially in eukaryotes, does not necessarily need to come preassembled to the promoter region or to be ready to start transcription upon binding (9,10). Along the way, there are molecular mechanisms that affect RNA stability and its information content, such as alternative splicing and RNA editing (1). To close up the loop, proteins and functional RNA are in charge of orchestrating all these processes, thus regulating their own synthesis.
This short review focuses on key, well-characterized guiding principles that allow the description of gene expression in terms of systems of biophysical processes and the application of these principles to actual systems, exemplified by the lac operon in prokaryotes and the retinoid X receptor in eukaryotes, which are both amenable to concise informative mechanistic descriptions. The goal is to accurately capture the effects of molecular interactions across scales up to the system behavior. To do so effectively, we will emphasize approaches that are scalable—namely, approaches that can be used with small and large systems, incorporate complex phenomena such as DNA looping, employ as few free parameters as possible, and the molecular parameters of which can be inferred from the experimental data and reused in modeling subsequent experiments.
There are two types of important situations that we will not consider explicitly because of space limitations. One type includes elementary mechanisms, such a cooperative interactions, which are described in virtually any biochemistry and molecular biology textbook (1), and which are applied to gene regulation exactly as described in the textbooks (11–14). The other type includes complex situations with missing key mechanistic information, such as eukaryotic enhancers (15,16), which would extend the discussion to cover many potential mechanisms that are compatible with the observed experimental data. The most effective avenue to modeling such complex problems so far has been to supplement known biophysical mechanisms with phenomenological rules and assumptions (17,18).
There are also many important aspects of gene expression that we will not be able to reach, including the effects of focused and dispersed transcription initiation (19), transcription elongation regulation (20,21), transcriptional traffic (22), and translation regulation by microRNAs (23), to mention just a few. The general principles reviewed here to a large extent also apply to those situations.
Modeling Gene Expression
A common starting point for most quantitative approaches to gene expression is a description based on reactions among molecular species (24–30). This description considers that there is a set of i different transcriptional states δi and that for each of these states there is a given transcription rate Γi that leads to mRNA, m. The simplest case with a single state would be a constitutive promoter with a constant transcription rate. The next step in complexity, a promoter with two states, already includes the potential for regulation, as for instance when a repressor turns off transcription upon binding the promoter. In general, the transitions between transcriptional states δi and δj with rates kij depend on the numbers of the different molecular species of the system. For each mRNA molecule, proteins p are produced at a rate Ω. Typically, mRNA and proteins are degraded at rates γm and γp, respectively. These reactions can be summarized as
| (1) |
The advantage of using such an approach is that it allows a direct connection of the description parameters with biophysical properties such as free energies of binding and DNA elastic properties.
The Deterministic Approach
When fluctuations are not relevant, either because they are small or because they can be averaged out (31), the set of expressions in Eq. 1 is usually written in terms of concentrations using traditional deterministic rate equations:
| (2) |
Here, Vc is the reaction volume; Pi = 〈δi〉 is the probability of having the system in the transcriptional state i; [m] = 〈m〉/Vc is the mRNA concentration; and [p] = 〈p〉/Vc is the average protein concentration, with angular brackets 〈…〉 representing averages.
The previous set of equations can be solved to obtain the steady-state protein content concentration [p]ss as
| (3) |
where pmax = ΓmaxΩ/γmγpVc is the maximum concentration, Γmax is the maximum transcriptional activity, and χi = Γi/Γmax is the normalized transcriptional activity. This result is extremely important because, besides collapsing the effects of many processes into a single parameter pmax, it directly connects microscopic probabilities of the transcriptional states with experimentally measurable quantities.
The deterministic approach, also known as mean-field approach, has been very useful to study systems with large numbers of molecules and negligible fluctuations. In the presence of fluctuations of small numbers of molecules, the average behavior of the system is still correctly described by the set of expressions in Eq. 2. The applicability of the deterministic approach, however, could break down with the additional presence of nonlinear terms. The reason is that the average of nonlinear terms cannot generally be expressed in terms of concentrations. For instance, the kinetics of dimerization of a protein p would involve the term 〈p2〉, which is not equivalent to 〈p〉2 = (Vc[p])2. In general, the validity of the deterministic approach should be carefully assessed on a case-by-case basis, taking into account that neither small numbers of molecules nor nonlinear terms by themselves always prevent its applicability, as illustrated by genetic nonlinear oscillators that can function in the deterministic regime even with just a few mRNA molecules per cell (32).
Control of Gene Expression
Control of gene expression is achieved through the dependence of the probability of the transcriptional states on the specific pattern of TFs that are assembled on DNA, as for instance, binding of an activator and absence of a repressor (2). In most instances, these interactions take place under quasi-equilibrium conditions and statistical thermodynamics can be used to express the probabilities of the states in terms of standard free energies and concentrations of the different regulatory molecules involved (33–35). The validity of the quasi-equilibrium assumption requires the binding kinetics, which by itself would be a completely reversible reaction, to be much faster than other cellular processes, such as cell growth, that could affect the binding process.
The key quantity in the thermodynamic approach is the statistical weight, or Boltzmann factor, which is defined in terms of the free energy ΔGi of the state i as . Its main feature is its proportionality to the probability of the state i,
| (4) |
The normalization factor is known as the partition function and the term RT is, as usual, the gas constant, R, times the absolute temperature, T. This expression is particularly important because it encapsulates the dependence of the probabilities on the different molecular concentrations of regulatory molecules [pj] through
| (5) |
where the terms d(i,j) correspond to the number of molecules of the species j in the state i and is the corresponding standard free energy at 1 M concentration. Therefore, if the free energies, or alternatively the probabilities, of the different states are known for given values of the concentrations of regulatory molecules, it is possible to obtain the probabilities for any concentration using the previous two equations. These two equations can be combined into
| (6) |
which has been a cornerstone in quantitative modeling of gene expression since the beginning of the field (30,34). Its main advantage is that it only requires the values for each transcriptional state, which has traditionally been written in tabular form along with a description of the molecular configuration (34).
Combinatorial Complexity
The main advantage of using a free energy value for each transcriptional state may turn increasingly fast into a disadvantage when the number of components of the system increases. The reason is that there are potentially as many states as the number of possible ways of arranging the regulatory molecules on DNA, which grows exponentially with the number of components. The resulting combinatorial explosion in the number of states makes the straightforward application of Eq. 6 impracticable for systems with more than just a handful of components.
Several general approaches have been developed to tackle this exponentially large multiplicity in the number of states. They involve a diversity of methodologies that range from stochastic configuration sampling (36) to automatic generation of all the underlying equations (37). The complexity of the general problem makes each of these approaches work efficiently only on a particular type of problem, be it conformational changes, multi-site phosphorylation, or oligomerization (38–42).
In the case of gene regulation, it has been possible to capitalize on the unambiguous modular structure that macromolecular complexes typically have on DNA to capture this complexity in simple terms (43). The key idea is to describe the specific configuration, or state of the protein-DNA complex, through a set of M state variables, denoted by s = (s1,…sk,…sM), which indicate whether a particular molecular component or conformation is present (sk = 1) or absent (sk = 0) at a specific position within the complex (43). The main advantage is that the free energy ΔGi = ΔG(s) and transcription rates Γi = Γ(s) for each state can be specified as function of the state variables without explicitly enumerating all the states.
The Lac Operon
The Escherichia coli lac operon is the genetic system that regulates and produces the enzymes needed to metabolize lactose (44,45). Besides opening the doors to the field of gene regulation, the lac operon has provided an example of a sophisticated regulation mechanism where all the components are known in great detail (46–52).
The main player in the control of transcription is the tetrameric lac repressor. In the absence of allolactose, a derivative of lactose, the lac repressor can bind to the main operator to prevent the RNAP from binding to the promoter and transcribing the genes. Binding of allolactose to the repressor substantially reduces its specific binding for the operator and transcription is de-repressed. The effects of the lac repressor on transcription are characterized as negative control. There is also positive control through the catabolite activator protein (CAP), which acts as an activator of transcription when glucose is not present by stabilizing the binding of the RNAP to the promoter.
This account of positive and negative control does not offer the whole picture of the underlying complexity. There are also two additional auxiliary operators that bind the repressor without preventing transcription (Fig. 1 A). Early on, they were considered just remnants of evolution because they bind the repressor very weakly and because elimination of either one of them has only minor effects in transcription. It was later observed that the simultaneous elimination of both auxiliary operators reduces the repression level by ∼100 times (46–48). The reason for this astonishing effect is that the lac repressor can bind simultaneously two operators and loop the intervening DNA (Fig. 1 B). Thus, the main operator and at least one auxiliary operator are needed to form DNA loops that substantially increase the repressor's ability to bind the main operator. Without quantitative approaches, however, it is difficult to fully grasp how such weak auxiliary sites, as much as 300-times weaker in terms of binding affinity than the main operator, can help the binding so much.
Figure 1.
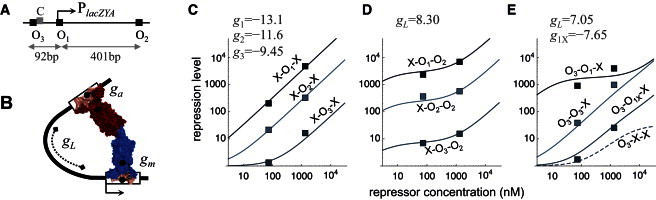
Gene expression in the lac operon for different operator configurations. (A) The relative positions of the main operator O1 and the auxiliary operators O2 and O3 are shown (solid rectangles) on the solid line representing DNA. The binding site for CAP is also shown (shaded rectangle). (B) A representation of the lac repressor is shown looping DNA (solid line) bound to the main and auxiliary operators (open rectangles). The contributions to the standard free energy of the looped DNA-repressor state from binding the main and auxiliary operators and from looping DNA are indicated by gm, ga, and gL, respectively. (C–E) The repression level computed from Eq. 9 (lines) and experimentally measured by Oehler et al. (46) (symbols) are shown for different combinations of operator replacements and deletions, including configurations with just the main operator, the O1-O2 loop, and the O3-O1 loop. The notation on each curve indicates the operator sequence at the positions of the operators O3-O1-O2. X corresponds to a complete deletion of O1, O2, or O3; O1X corresponds to a partial deletion of O1. The values of g1, g2, g3, and g1X show the standard free energy of binding (in kcal/mol) to the sequences of O1, O2, O3, and O1X, respectively. The value of gL used is shown (in kcal/mol) for each type of loop.
To illustrate the important effects of the presence of the auxiliary operators, we examine in detail the case with two operators, the main operator, Om, and an auxiliary operator, Oa. The main operator is located at the position of O1 and the auxiliary operator is located at the position of either O2 or O3 (Fig. 1 A).
The key piece of information that allowed capturing the effects of DNA looping in quantitative detail was shown to be the decomposition of the free energy of the looped protein–DNA complex, , into different modular contributions that take into account the binding to each operator and the looping contribution (49). Explicitly, = gm + ga + gL, where gm and ga are the standard free energy of binding to Om and Oa, respectively, and gL is the free energy of looping (Fig. 1 B).
This segmentation of the free energy allows for an efficient representation of all the transcriptional states in terms of state variables. These variables comprise sm and sa, which indicate whether (= 1) or not (= 0) a repressor is bound to the main and auxiliary operator, respectively, and sL, which indicates whether (= 1) or not (= 0) DNA forms the loop Om-Oa.
The free energy of the system in terms of these three state variables is given by
| (7) |
where [n] is the concentration of the lac repressor. The first two terms in the expression take into account the repressor binding to Om or Oa. The fourth term indicates that the presence of looping needs both operators occupied by a repressor; otherwise, the free energy would be infinite. Finally, the first three terms all together represent the binding of the repressor when the three state variables are equal to 1, which indicates that a single repressor is bound simultaneously to Oa and Om and that there is a looping contribution to the free energy.
The normalized transcriptional activity is expressed in terms of state variables as
| (8) |
This expression specifies that there is no transcription when the repressor is bound to Om. When Om is free, transcription occurs at a maximum rate if Oa is free, and at rate χa if the repressor occupies Oa.
The advantage of using state variables is that Eqs. 7 and 8 completely specify the transcriptional properties of the lac operon. The steady-state protein production is computed directly from [p]ss = pmaxΣsχ(s)P(s), where the sum can be performed by hand or automatically using software like CplexA (53,54). The resulting repression level is given succinctly by
| (9) |
This expression is important because it connects macroscopically measurable quantities, such as protein content in a cell population, with microscopic binding parameters. The value of pmax can be obtained from measurements for strains without repressor, which transcribe the lac genes at a maximum rate, and it is customary to report just the ratio pmax/[p]ss, which is known as repression level. In the case of the lac operon, all the parameters needed for modeling can be inferred from the experimentally available data.
For instance, when the auxiliary operator is deleted, or more precisely when it is mutated so that the binding is very low (ga → ∞), the repression level reduces to
| (10) |
From this expression and the data of experimental setups that used the sequence of O1, O2, and O3 as a main operator, it is possible to obtain the free energy of binding for each operator (Fig. 1 C), which can be reused in subsequent modeling.
In the case of the O1-O2 loop for different sequences of the main operator, the only additional parameter needed to accurately reproduce the experimental data is the free energy of looping gL (Fig. 1 D). In this case, binding of the repressor to the auxiliary operator O2 does not affect transcription and χa = 1.
In the case of the O3-O1 loop, binding of the repressor to the auxiliary operator prevents CAP from activating transcription and the transcription rate is reduced to χa = 0.03. In this case as well, just a single additional parameter for the free energy of looping gL is needed to reproduce most of the experimental data (Fig. 1 E). It turns out, however, that the deletion of O1 in the strain labeled O3-O1X-X is not complete and the site is still able to form the O3-O1X loop even though its binding is reduced by 5.5 kcal/mol, a factor 10,000 in terms of binding affinity. This moderate decrease in binding can be inferred from the position-weight matrix score of the specific sequence of the incomplete deletion (55). The computed repression level for the complete deletion of O1, strain O3-X-X, is shown in Fig. 1 E as a discontinuous line that is clearly below the incomplete deletion.
The Retinoid X Receptor
Gene expression in eukaryotes is substantially more involved than in prokaryotes (2,3,56). Just the core of the eukaryotic transcriptional machinery itself involves a wide variety of components with oscillatory patterns of macromolecular assembly and phosphorylation (9,57). In addition, there are many additional layers of control that extend from the accessibility and assembly of the transcriptional machinery at the promoter to the intracellular transport and regulation of mRNA and proteins. Despite all these differences, it has been argued that there are many general principles that apply to both prokaryotes and eukaryotes (2). We use the retinoid X receptor (RXR) to illustrate how the main ideas and methodology used in the lac operon can also be applied to this complex eukaryotic system.
RXR is a nuclear receptor that is responsible for regulating a large number of genes. It exerts its function by binding to DNA as homodimer, homotetramer, or obligatory heterodimerization partner for other nuclear receptors (58).
Similarly to the lac operon, RXR can bind multiple sites simultaneously as a tetramer by looping the intervening DNA (Fig. 2 A). A distinct feature, however, is that in the case of RXR, tetramers and dimers coexist in the cell and their relative populations are regulated by the RXR cognate ligands, which prevent the formation of tetramers besides imparting RXR the ability to recruit coactivators of transcription.
Figure 2.
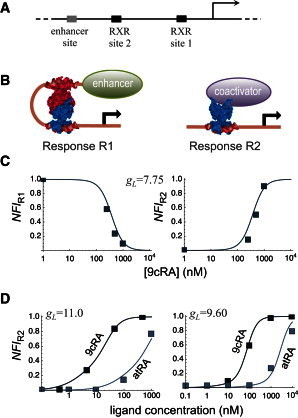
RXR-mediated transcriptional responses to 9cRA and atRA ligands. (A) A prototypical arrangement of binding sites for RXR and an enhancer element are shown (solid and shaded rectangles, respectively) on a solid line representing DNA. (B) In response R1, an RXR tetramer loops DNA (represented as a continuous line) to bring an enhancer close to the promoter region. In response R2, an RXR dimer recruits a coactivator to the promoter region. (C and D) The normalized fold induction for responses R1 and R2 (lines) was computed from Eqs. 15 with the experimental values Klig = 8 nM for 9cRA (97) or Klig = 350 nM for atRA (98), and Ktd = 4.4 nM (99). The value of the free energy of looping gL (shown in kcal/mol) depends on the specific promoter and cell line. (C) Response to 9cRA for a promoter incorporating two RXR binding sites with (left) and without (right) a distal enhancer, which considers responses R1 and R2, respectively. Experimental gene expression data (symbols) was obtained from Yasmin et al. (62). (D) Responses to 9cRA and atRA for promoters without a distal enhancer (response R2). Experimental gene expression data (symbols) correspond to reporter plasmids ADH-CRBPII-LUC (left) and TK-CRBPII-LUC (right) from Heyman et al. (70).
The first step in the signaling cascade for sensing the ligand concentration is regulation of the relative abundance of the oligomerization states of the RXR, which include tetramers, n4, dimers, n2, and nontetramerizing dimers, n2∗. The effects of the ligand are quantitated in general through the modulator function f([l]) = [n2∗]/[n2], which describes the partitioning into the tetramerizing and nontetramerizing dimers by the ligand l. In this system, the canonical ligand is the hormone 9cRA (9-cis-retinoic acid), a derivative of Vitamin A, which binds each RXR monomeric subunit independently of its oligomerization state (59) and prevents dimers with their two subunits occupied from tetramerizing (60). Therefore, considering [n2∗] as the concentration of dimers with two ligands bound and [n2] as the concentration of dimers with one or zero ligand leads to , where Klig is the ligand-RXR dissociation constant and [l] is the concentration of the ligand (61). This process determines dimer and tetramer concentrations, which are related to each other through [n2]2/[n4] = Ktd, where Ktd is the tetramer-dimer dissociation constant.
Control of gene expression results from the dependence of the transcriptional response on the type of oligomeric species that are assembled on DNA (62). There are two differentiated types of responses (Fig. 2 B): The first type, referred to as response R1, involves a tetramer that simultaneously binds two nonadjacent DNA sites. Upon binding, the tetramer can bring a distal enhancer close to the promoter region by looping DNA and control transcription. In this case, dimers do not elicit transcriptional responses. In general, promoting and preventing DNA looping has been found to be a fundamental mechanism for controlling the effects of distal enhancers (63,64). The second type, denoted response R2, relies on differentiated recruitment abilities by different oligomerization states. Specifically, dimers can recruit a coactivator by binding of a region that is secluded in the tetramer (61).
The different configurations for binding of RXR to two DNA sites are described by the state variables s1t and s2t that indicate whether (= 1) or not (= 0) a tetramer is bound to site 1 and 2, respectively; sL that indicates whether (= 1) or not (= 0) DNA forms the loop between these two sites; and two additional state variables s1d and s2d that indicate whether (= 1) or not (= 0) a dimer is bound to site 1 and 2, respectively.
The free energy of the system in terms of these state variables is given by
| (11) |
Here, g1 and g2 are the standard free energies of binding to sites 1 and 2, respectively, which are assumed to be the same for all three oligomeric species, and gL is the free energy of looping. The first four terms of this expression are equivalent to those for the lac operon in Eq. 7 because it is the same type of tetrameric binding to two sites. The fifth and sixth terms represent the binding of a dimeric species to sites 1 and 2, respectively. The last term indicates that dimers and tetramers cannot be bound simultaneously to the same site by assigning an infinite free energy to those states.
The normalized transcriptional activities for responses R1 and R2 are expressed in terms of state variables as
| (12) |
where χref does not depend on the ligand concentration and is the normalized basal activity of the promoter in absence of any activation. The explicit forms of χd = (1 – (1 + [l]/Klig)−2)(1 – χref) and χdd = (1 – (1 + [l]/Klig)−4)(1 – χref) indicate that at least one of the ligand-binding sites of one dimer and of a pair of dimers, respectively, needs to be occupied by the ligand for the coactivator to be recruited.
It is straightforward to obtain analytic expressions of the transcriptional activity from Eqs. 11 and 12 using software packages like CplexA (53), but it is more illustrative for the purposes of this review to focus on the functional regime, which guarantees that there is response to changes in the ligand concentration.
The functional regime considers two properties. The first one is that the total RXR concentration is sufficiently high for it to significantly bind DNA. The second one is that the concentration of tetramers is low enough for them not to completely saturate the binding. The reason is that for typical values of gL, tetramers bind more strongly to two DNA sites simultaneously than dimers do to a single DNA site, as in the case of the lac operon (43,49,65). Under these conditions the representative states, described by s = (s1t, s2t, sL, s1d, s2d), are those with a tetramer bound to the two sites simultaneously, s = (1,1,1,0,0), and with one dimer bound to each of the two sites, s = (0,0,0,1,1). The corresponding statistical weights for these states, the only ones needed in this case, are
and
respectively.
The key implication of this regime is that the steady-state protein production, computed from
simplifies in such a way that the transcriptional responses are governed by the reduced expressions
| (13) |
where
| (14) |
is the probability of the state s = (1,1,1,0,0).
The particular form of Pt is exceptionally remarkable because it imparts precision and flexibility to the transcriptional responses—two properties that are the cornerstone of natural gene expression systems but that have proved to be highly elusive because of their seemingly antagonistic character (66). Precision ensures that the transcriptional response is consistently triggered at a given ligand concentration irrespective of the particular total RXR concentration, which cancels out in the reduced equations that govern the system behavior. Flexibility, on the other hand, allows the precise triggering point to be altered both at the individual promoter level through gL (67,68) and at a genomewide scale through f([l]) and Ktd.
To compare with the experimental data, the most convenient approach is to use the normalized fold induction (NFI), which is defined as NFI = (FI – 1)/(FImax – 1), where FI is the fold induction and FImax is its maximum value. The value of FI is obtained experimentally as the actual expression of a gene over its baseline expression and in mathematical terms as FI = [p]ss/(pmaxχref). In terms of the NFI, the results do not depend on parameters related to the baseline and maximum expression levels and it becomes possible to effectively compare experiments on different promoters and cell lines. The explicit form of the NFI for responses R1 and R2 is
| (15) |
respectively. Importantly, the only parameters needed to characterize the shape of the response in the functional regime are Klig and Ktd, which have been measured experimentally, and gL, which can be inferred by adjusting its value to reproduce the experimental data.
A fully predictive framework without free parameters has been obtained with this approach because it collapses most of the intracellular complexity into just one unknown parameter gL. Therefore, once this parameter is known for a particular experimental setup (specific cell type, cellular conditions, and promoter), it can be used to predict other responses just from thermodynamic principles.
One possibility is to use the value of gL inferred for one type of response to predict the other one. There is experimental data that tested in the same cell type and promoter both types of transcriptional responses, one mediated by an enhancer (response R1) and the other, by a coactivator (response R2). The results of the model indicate that just a single value of gL is needed to reproduce with high accuracy the experimental data in both cases (Fig. 2 C).
Another possibility is to use the value of gL inferred for one ligand to predict the response to other ligands. The all-trans-retinoic acid (atRA) was tested early on as a potential candidate for the RXR cognate ligand, and it was observed that binding was present but very weak (69,70). The values of gL inferred for 9cRA responses can be used to closely match the experimental transcription data in response to atRA without any free parameter by simply changing the value of the ligand-binding constant, Klig, to the corresponding one for atRA (Fig. 2 D).
Combinatorial Assembly of Nucleoprotein Complexes
There are many situations in which the DNA loop is formed not by a single protein, as in the lac operon and RXR, but by a protein complex that is assembled on DNA as the loop forms. The term “combinatorial assembly” is used because there are many potential complexes that can arise from the combinations of binding to multiple sites, even when just a single TF is involved. An illustrative example is present in the regulation of phage λ. It has two operators located 2.4 kb away from one another and each operator contains a tandem of three sites where phage λ cI repressors can bind as dimers. In this case, two dimers bound to an operator can form an octamer with two dimers bound to another operator by looping the intervening DNA (43,71,72). Another example is the interaction of TFs bound at distal enhancers with the transcriptional complexes bound at the promoter (63). To study this type of problem, it is crucial to properly take into account that proteins bound to distal DNA regions can interact with each other only if DNA looping is present.
Interactions mediated by DNA looping would lead to terms with products of three or more state variables in the free energy (43). An illustrative example is
where the state variables sU,i, sD,j, and sL indicate whether (= 1) or not (= 0) a protein is bound to site i at the upstream DNA region, a protein is bound to site j at the downstream DNA region, and DNA looping is present, respectively. The quantities ei,j account for the interactions between proteins bound at different DNA regions and gL is the free energy of looping. The formation of the DNA loop would be energetically favorable only when a sufficient number of interactions can be achieved between the two DNA regions. In the case of phage λ, only octamers and dodecamers are able to form the looped complex among the many possible combinations of binding (43,71,72). In turn, the presence of DNA looping can enhance DNA binding through the interactions that can be established between the two DNA regions, which can lead to highly cooperative phenomena in the formation of the nucleoprotein complex (42).
Stochastic Kinetics
The lac operon and RXR have been used so far in this review to demonstrate how biophysical principles can be used to efficiently capture the system behavior when noise in the form of random fluctuations is not relevant. The very same principles can be extended to take into account the inherent stochastic nature of the underlying processes in a wide range of situations. An efficient avenue to do so is to consider the dynamics of the macromolecular complexes that control gene expression through the stochastic dynamics of the state variables (43,73).
The dynamics of the macromolecular complex can be described in terms of components that can change in a transition. For the widespread case in which only one component can change at a given time (either the component i gets into or out of the complex), one can define on (kion) and off (kioff) rates for the association-like and dissociation-like rates, respectively, which in general depend on the pretransition and posttransition states of the complex.
The explicit dynamics can be obtained by considering the change in state variables as reactions given by
| (16) |
These reactions change the variable si to 1 when it is 0 and to 0 when it is 1, representing that the element gets into or out of the complex. Typically, the on-rate does not depend as strongly on the state of the complex as the off-rate. The on-rate is essentially the rate of transferring the component from solution to the complex. The off-rate, in contrast, depends exponentially on the free energy change.
The principle of detailed balance (33) can be used to obtain the off-rates from the on-rates:
| (17) |
The remarkable property of this expression is that reactions with known rates can be used to infer the rates of more complex reactions from the equilibrium properties (for instance, to infer dissociation rates for different binding sites from a single association rate (43)). In general, the association rate could also depend on the state of the complex and its free energy, as for instance if the presence of a TF facilitates the association of another TF. If this dependence is included in the on-rate, Eq. 17 can also be applied straightforwardly to obtain the off-rate.
The stochastic dynamics of the resulting networks of reactions and transitions can then be obtained with kinetic Monte Carlo simulations using well-established algorithms (26,74,75).
Noise and Fluctuations in the Lac Operon
Stochastic effects in the lac operon have been known to be important since the late 1950s (76), predating the discovery of gene regulation (45). The most salient example is the all-or-none induction process (76), which was measured at the single-cell level with a resolution of a few molecules of the gene products per cell (77). This effect has its roots in the amplification of the inherent stochastic fluctuations of transcription and translation processes (78–81) close to the boundary that separates the induced from noninduced states of the lac operon (24,82).
The underlying molecular mechanisms and parameters have been shown to shape transcriptional noise to a large extent (43,49,83–86). To illustrate these effects in the lac operon, we discuss, first, regulation through just the main operator. The use of state variables leads to a single reaction that describes both the binding and unbinding of the repressor to the main operator:
| (18) |
Here, the on-rate is given by [n]ka, where ka is the association rate constant, and the off-rate, , is obtained from the detailed balance principle. The transcription rate is described by
| (19) |
and mRNA degradation, protein production, and protein degradation are described by the stochastic counterpart of the expressions in Eq. 1. The time courses of the number of proteins produced from this promoter show relatively small fluctuations for the experimental values of the parameters (Fig. 3 A). The downside of having just a binding site for regulation is that repression is relatively weak and a substantial number of proteins are produced.
Figure 3.
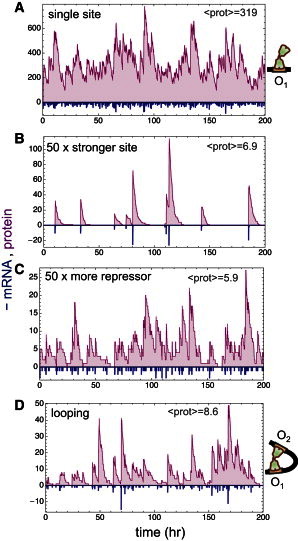
Transcriptional noise in the lac operon. (A–C) Time courses of the number of protein and mRNA (shown as negative values) produced from a promoter with just the main operator described by Eqs. 18 and 19 and the stochastic implementation of the expressions in Eq. 1. The values of the common parameters for all three panels are Γmax = 0.5 s−1, Ω = 0.01 s−1, γm = 3.3 × 10−3 s−1, γp = 9.2 × 10−5 s−1, and ka = 2.2 × 10−6 M−1 s−1. The values of the remaining parameters are: (A) gm = −13.1 kcal/mol and [n] = 15 nM for wild-type Om and wild-type repressor concentration; (B) gm = −13.1 – RT ln 50 kcal/mol and [n] = 15 nM for 50-times stronger Om and wild-type repressor concentration; and (C) gm = −13.1 kcal/mol and [n] = 750 nM for wild-type Om and 50-times more repressor. (D) Time courses of the number of protein and mRNA (shown as negative values) produced from a promoter with the operators O1 and O2 described by Eqs. 19–21 and the stochastic implementation of the expressions in Eq. 1. The values of the parameters are the same as in panel A with the addition of ga = −11.6 kcal/mol and gL = 8.30 kcal/mol. The illustrations to the right of panels A and D represent respectively a lac repressor bound to the main operator and a lac repressor bound simultaneously to the main and auxiliary operators looping the intervening DNA.
To increase repression, two simple alternatives exist. The first one is to consider a stronger site. For a site 50-times stronger than the wild-type main operator, protein production would be close to the value expected for the lac operon with the three operators. In this case, the average protein production is reduced ∼50 times, as expected from the deterministic theory, but fluctuations increase dramatically (Fig. 3 B). There are infrequent mRNA bursts that lead to large protein amounts that decay in a few hours and long periods of time without any protein at all. The second alternative is to include more repressors. For a repressor concentration 50-times higher than in wild-type, the average protein production is reduced ∼50 times and the fluctuations remain relatively small (Fig. 3 C). In this case, mRNA production happens in smaller quantities but more frequently. The physiological downside is that the repressor production would have to be 50-times higher than in wild-type and if that happens for all the proteins of the cell, E. coli would have to be 50-times more crowded.
A more efficient alternative to increase repression is to use DNA looping, which has been chosen by evolution not only in the lac operon but also in a large variety of systems. The computational approach in this case is slightly more involved because it has to take into account that an operator can be bound by a repressor in solution or by a repressor bound to the other operator thus forming a DNA loop (Fig. 1 B). For binding to the main operator, these two processes are represented by
| (20) |
| (21) |
For the binding to the auxiliary operator, the reactions have the same representation except that the terms sm, sa, and gm are replaced by sa, sm, and ga, respectively.
The stochastic kinetics of the regulation through the O1-O2 loop shows a small average number of proteins with low fluctuations, thus behaving in a manner very similar to a single operator with 50-times more repressor (Fig. 3 D). Therefore, DNA looping in this case allows the system to achieve the same behavior as it would with 50-times more repressors.
Intuitively, both looping and high repressor concentration lead to lower noise than a single strong site because of the characteristic timescales involved. In the strong site case, there are long periods of time with maximum transcriptional activity and long periods without any activity, which results in the number of proteins fluctuating strongly between high and low values. In the cases of looping and high repressor concentration, the off-rate of the repressor from the main operator is 50-times larger than for the strong site and the average on-rate increases accordingly to keep the same repression level. Therefore, the switching between transcriptional states is very fast and mRNA production is in the form of short and frequent bursts. This lack of long periods of time with either full or null production gives a narrower distribution of the number of proteins. Explicitly, the coefficient of variation of protein (mRNA) content shown in Fig. 3 for the strong site, the high repressor concentration, and the DNA looping cases is 2.3 (12.9), 0.81 (4.8), and 0.95 (5.4), respectively.
Discussion
Gene expression relies on intricate molecular mechanisms to function in extraordinarily diverse intra- and extracellular environments. Biophysical approaches have provided new avenues to unravel how these different levels of molecular complexity contribute to the observed behavior. The results reviewed here show that the underlying complexity of biological systems is not just an accident of evolution but has a functional role.
Explicitly, the lac operon exemplifies how escalating complexity from one to two operators introduces stronger repression while preserving low transcriptional noise, which is not possible with a stronger single binding site.
In the case of the RXR, the additional complexity embedded in the control of its oligomeric state by the cognate ligand and its ability to bind simultaneously single and multiple DNA sites has been shown to impart precision and flexibility, two seemingly antagonistic properties, to the sensing of cellular signals.
This type of regulated oligomerization has also been observed explicitly in other transcription factors that can bind multiple DNA sites simultaneously, such as the tumor suppressor p53 (87), the nuclear factor κB (NF-κB) (88,89), the signal transducers and activators of transcription (STATs) (90), and the octamer-binding proteins (Oct) (91,92). In these systems, the properties of self-assembly, and the partitioning into low- and high-order oligomeric species, are strongly regulated and modulated by several types of signals, such as ligand binding (60), protein binding (93,94), acetylation (95), and phosphorylation (92,96).
The combined presence of flexibility and precision in the control of gene expression, as explicitly shown for RXR, allows a single TF to simultaneously regulate multiple genes with promoter-tailored dose-response curves that consistently maintain their diverse shapes for a broad range of the TF concentration changes.
Thus, the complexity of multiple repeated distal DNA binding sites both in prokaryotes and eukaryotes, far from being just a remnant of evolution or a backup system as often assumed, can confer fundamental properties that are not present in simpler setups.
Acknowledgments
This work was supported by the El Ministerio de Economía y Competitividad under grants No. FIS2009-10352 and No. FIS2012-38105 (to J.M.G.V.) and the University of California at Davis (to L.S.).
Contributor Information
Jose M.G. Vilar, Email: j.vilar@ikerbasque.org.
Leonor Saiz, Email: lsaiz@ucdavis.edu.
References
- 1.Alberts B., Johnson A., Walter P. Garland Science; New York: 2008. Molecular Biology of the Cell. [Google Scholar]
- 2.Ptashne M., Gann A. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2002. Genes and Signals. [Google Scholar]
- 3.Levine M., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 4.Hermsen R., ten Wolde P.R., Teichmann S. Chance and necessity in chromosomal gene distributions. Trends Genet. 2008;24:216–219. doi: 10.1016/j.tig.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Teif V.B., Rippe K. Calculating transcription factor binding maps for chromatin. Brief. Bioinform. 2012;13:187–201. doi: 10.1093/bib/bbr037. [DOI] [PubMed] [Google Scholar]
- 6.Li Q., Barkess G., Qian H. Chromatin looping and the probability of transcription. Trends Genet. 2006;22:197–202. doi: 10.1016/j.tig.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Fudenberg G., Mirny L.A. Higher-order chromatin structure: bridging physics and biology. Curr. Opin. Genet. Dev. 2012;22:115–124. doi: 10.1016/j.gde.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naumova N., Dekker J. Integrating one-dimensional and three-dimensional maps of genomes. J. Cell Sci. 2010;123:1979–1988. doi: 10.1242/jcs.051631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Métivier R., Penot G., Gannon F. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 10.Djordjevic M., Bundschuh R. Formation of the open complex by bacterial RNA polymerase—a quantitative model. Biophys. J. 2008;94:4233–4248. doi: 10.1529/biophysj.107.116970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ay A., Arnosti D.N. Mathematical modeling of gene expression: a guide for the perplexed biologist. Crit. Rev. Biochem. Mol. Biol. 2011;46:137–151. doi: 10.3109/10409238.2011.556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Jong H. Modeling and simulation of genetic regulatory systems: a literature review. J. Comput. Biol. 2002;9:67–103. doi: 10.1089/10665270252833208. [DOI] [PubMed] [Google Scholar]
- 13.Garcia H.G., Sanchez A., Phillips R. Transcription by the numbers redux: experiments and calculations that surprise. Trends Cell Biol. 2010;20:723–733. doi: 10.1016/j.tcb.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlebach G., Shamir R. Modeling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008;9:770–780. doi: 10.1038/nrm2503. [DOI] [PubMed] [Google Scholar]
- 15.Struffi P., Corado M., Small S. Combinatorial activation and concentration-dependent repression of the Drosophila even skipped stripe 3+7 enhancer. Development. 2011;138:4291–4299. doi: 10.1242/dev.065987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry M.W., Boettiger A.N., Levine M. Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc. Natl. Acad. Sci. USA. 2011;108:13570–13575. doi: 10.1073/pnas.1109873108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segal E., Raveh-Sadka T., Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 18.He X., Samee M.A., Sinha S. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLOS Comput. Biol. 2010;6:e1000935. doi: 10.1371/journal.pcbi.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juven-Gershon T., Kadonaga J.T. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol. 2010;339:225–229. doi: 10.1016/j.ydbio.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boettiger A.N., Ralph P.L., Evans S.N. Transcriptional regulation: effects of promoter proximal pausing on speed, synchrony and reliability. PLOS Comput. Biol. 2011;7:e1001136. doi: 10.1371/journal.pcbi.1001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine M. Paused RNA polymerase II as a developmental checkpoint. Cell. 2011;145:502–511. doi: 10.1016/j.cell.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klumpp S., Hwa T. Stochasticity and traffic jams in the transcription of ribosomal RNA: intriguing role of termination and antitermination. Proc. Natl. Acad. Sci. USA. 2008;105:18159–18164. doi: 10.1073/pnas.0806084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eulalio A., Huntzinger E., Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132:9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 24.Vilar J.M.G., Guet C.C., Leibler S. Modeling network dynamics: the lac operon, a case study. J. Cell Biol. 2003;161:471–476. doi: 10.1083/jcb.200301125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulsson J. Models of stochastic gene expression. Phys. Life Rev. 2005;2:157–175. [Google Scholar]
- 26.McAdams H.H., Arkin A. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA. 1997;94:814–819. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morelli M.J., Allen R.J., Wolde P.R. Effects of macromolecular crowding on genetic networks. Biophys. J. 2011;101:2882–2891. doi: 10.1016/j.bpj.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teif V.B. Predicting gene-regulation functions: lessons from temperate bacteriophages. Biophys. J. 2010;98:1247–1256. doi: 10.1016/j.bpj.2009.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cottrell D., Swain P.S., Tupper P.F. Stochastic branching-diffusion models for gene expression. Proc. Natl. Acad. Sci. USA. 2012;109:9699–9704. doi: 10.1073/pnas.1201103109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saiz L. The physics of protein-DNA interaction networks in the control of gene expression. J. Phys. Condens. Matter. 2012;24:193102. doi: 10.1088/0953-8984/24/19/193102. [DOI] [PubMed] [Google Scholar]
- 31.Gillespie D.T. Deterministic limit of stochastic chemical kinetics. J. Phys. Chem. B. 2009;113:1640–1644. doi: 10.1021/jp806431b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilar J.M.G., Kueh H.Y., Leibler S. Mechanisms of noise-resistance in genetic oscillators. Proc. Natl. Acad. Sci. USA. 2002;99:5988–5992. doi: 10.1073/pnas.092133899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill T.L. Addison-Wesley; Reading, MA: 1960. An Introduction to Statistical Thermodynamics. [Google Scholar]
- 34.Ackers G.K., Johnson A.D., Shea M.A. Quantitative model for gene regulation by λphage repressor. Proc. Natl. Acad. Sci. USA. 1982;79:1129–1133. doi: 10.1073/pnas.79.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Segal E., Widom J. From DNA sequence to transcriptional behavior: a quantitative approach. Nat. Rev. Genet. 2009;10:443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Novère N., Shimizu T.S. STOCHSIM: modeling of stochastic biomolecular processes. Bioinformatics. 2001;17:575–576. doi: 10.1093/bioinformatics/17.6.575. [DOI] [PubMed] [Google Scholar]
- 37.Hlavacek W.S., Faeder J.R., Fontana W. Rules for modeling signal-transduction systems. Sci. STKE. 2006;2006:re6. doi: 10.1126/stke.3442006re6. [DOI] [PubMed] [Google Scholar]
- 38.Borisov N.M., Markevich N.I., Kholodenko B.N. Trading the micro-world of combinatorial complexity for the macro-world of protein interaction domains. Biosystems. 2006;83:152–166. doi: 10.1016/j.biosystems.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bray D., Lay S. Computer-based analysis of the binding steps in protein complex formation. Proc. Natl. Acad. Sci. USA. 1997;94:13493–13498. doi: 10.1073/pnas.94.25.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ollivier J.F., Shahrezaei V., Swain P.S. Scalable rule-based modeling of allosteric proteins and biochemical networks. PLOS Comput. Biol. 2010;6:e1000975. doi: 10.1371/journal.pcbi.1000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deeds E.J., Bachman J.A., Fontana W. Optimizing ring assembly reveals the strength of weak interactions. Proc. Natl. Acad. Sci. USA. 2012;109:2348–2353. doi: 10.1073/pnas.1113095109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vilar J.M.G., Saiz L. Multiprotein DNA looping. Phys. Rev. Lett. 2006;96:238103. doi: 10.1103/PhysRevLett.96.238103. [DOI] [PubMed] [Google Scholar]
- 43.Saiz L., Vilar J.M.G. Stochastic dynamics of macromolecular-assembly networks. Mol. Syst. Biol. 2006;2:2006.0024. doi: 10.1038/msb4100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Müller-Hill B. Walter de Gruyter; Berlin, Germany: 1996. The Lac Operon: a Short History of a Genetic Paradigm. [Google Scholar]
- 45.Jacob F., Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 46.Oehler S., Amouyal M., Müller-Hill B. Quality and position of the three lac operators of E. coli define efficiency of repression. EMBO J. 1994;13:3348–3355. doi: 10.1002/j.1460-2075.1994.tb06637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oehler S., Eismann E.R., Müller-Hill B. The three operators of the lac operon cooperate in repression. EMBO J. 1990;9:973–979. doi: 10.1002/j.1460-2075.1990.tb08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mossing M.C., Record M.T., Jr. Upstream operators enhance repression of the lac promoter. Science. 1986;233:889–892. doi: 10.1126/science.3090685. [DOI] [PubMed] [Google Scholar]
- 49.Vilar J.M.G., Leibler S. DNA looping and physical constraints on transcription regulation. J. Mol. Biol. 2003;331:981–989. doi: 10.1016/s0022-2836(03)00764-2. [DOI] [PubMed] [Google Scholar]
- 50.Saiz L., Vilar J.M.G. Ab initio thermodynamic modeling of distal multisite transcription regulation. Nucleic Acids Res. 2008;36:726–731. doi: 10.1093/nar/gkm1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhlman T., Zhang Z., Hwa T. Combinatorial transcriptional control of the lactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA. 2007;104:6043–6048. doi: 10.1073/pnas.0606717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Narang A. Effect of DNA looping on the induction kinetics of the lac operon. J. Theor. Biol. 2007;247:695–712. doi: 10.1016/j.jtbi.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 53.Vilar J.M.G., Saiz L. CPLEXA: a MATHEMATICA package to study macromolecular-assembly control of gene expression. Bioinformatics. 2010;26:2060–2061. doi: 10.1093/bioinformatics/btq328. [DOI] [PubMed] [Google Scholar]
- 54.Vilar, J. M. G., and L. Saiz. 2010. CPLEXA. http://sourceforge.net/projects/cplexa.
- 55.Vilar J.M.G. Accurate prediction of gene expression by integration of DNA sequence statistics with detailed modeling of transcription regulation. Biophys. J. 2010;99:2408–2413. doi: 10.1016/j.bpj.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simicevic J., Deplancke B. DNA-centered approaches to characterize regulatory protein-DNA interaction complexes. Mol. Biosys. 2010;6:462–468. doi: 10.1039/b916137f. [DOI] [PubMed] [Google Scholar]
- 57.Lemaire V., Lee C.F., Glass L. Sequential recruitment and combinatorial assembling of multiprotein complexes in transcriptional activation. Phys. Rev. Lett. 2006;96:198102. doi: 10.1103/PhysRevLett.96.198102. [DOI] [PubMed] [Google Scholar]
- 58.Altucci L., Gronemeyer H. The promise of retinoids to fight against cancer. Nat. Rev. Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 59.Kersten S., Dawson M.I., Noy N. Individual subunits of heterodimers comprised of retinoic acid and retinoid X receptors interact with their ligands independently. Biochemistry. 1996;35:3816–3824. doi: 10.1021/bi952737k. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z.P., Iyer J., Gronemeyer H. Ligand- and DNA-induced dissociation of RXR tetramers. J. Mol. Biol. 1998;275:55–65. doi: 10.1006/jmbi.1997.1413. [DOI] [PubMed] [Google Scholar]
- 61.Vilar J.M.G., Saiz L. Control of gene expression by modulated self-assembly. Nucleic Acids Res. 2011;39:6854–6863. doi: 10.1093/nar/gkr272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yasmin R., Yeung K.T., Noy N. DNA-looping by RXR tetramers permits transcriptional regulation “at a distance”. J. Mol. Biol. 2004;343:327–338. doi: 10.1016/j.jmb.2004.08.070. [DOI] [PubMed] [Google Scholar]
- 63.Nolis I.K., McKay D.J., Thanos D. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. USA. 2009;106:20222–20227. doi: 10.1073/pnas.0902454106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chopra V.S., Kong N., Levine M. Transcriptional repression via antilooping in the Drosophila embryo. Proc. Natl. Acad. Sci. USA. 2012;109:9460–9464. doi: 10.1073/pnas.1102625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vilar J.M.G., Saiz L. DNA looping in gene regulation: from the assembly of macromolecular complexes to the control of transcriptional noise. Curr. Opin. Genet. Dev. 2005;15:136–144. doi: 10.1016/j.gde.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 66.Ptashne M., Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 67.Saiz L., Rubi J.M., Vilar J.M.G. Inferring the in vivo looping properties of DNA. Proc. Natl. Acad. Sci. USA. 2005;102:17642–17645. doi: 10.1073/pnas.0505693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jackson P., Mastrangelo I., Barrett J. Synergistic transcriptional activation of the MCK promoter by p53: tetramers link separated DNA response elements by DNA looping. Oncogene. 1998;16:283–292. doi: 10.1038/sj.onc.1201491. [DOI] [PubMed] [Google Scholar]
- 69.Levin A.A., Sturzenbecker L.J., Lovey A. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR α. Nature. 1992;355:359–361. doi: 10.1038/355359a0. [DOI] [PubMed] [Google Scholar]
- 70.Heyman R.A., Mangelsdorf D.J., Thaller C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 71.Ptashne M. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. A Genetic Switch: Phage λ Revisited. [Google Scholar]
- 72.Révet B., von Wilcken-Bergmann B., Müller-Hill B. Four dimers of λ repressor bound to two suitably spaced pairs of λ operators form octamers and DNA loops over large distances. Curr. Biol. 1999;9:151–154. doi: 10.1016/s0960-9822(99)80069-4. [DOI] [PubMed] [Google Scholar]
- 73.Saiz L., Vilar J.M.G. Protein-protein/DNA interaction networks: versatile macromolecular structures for the control of gene expression. IET Syst. Biol. 2008;2:247–255. doi: 10.1049/iet-syb:20080091. [DOI] [PubMed] [Google Scholar]
- 74.Gillespie D.T. General method for numerically simulating stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976;22:403–434. [Google Scholar]
- 75.Bortz A.B., Kalos M.H., Lebowitz J.L. New algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975;17:10–18. [Google Scholar]
- 76.Novick A., Weiner M. Enzyme induction as an all-or-none phenomenon. Proc. Natl. Acad. Sci. USA. 1957;43:553–566. doi: 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maloney P.C., Rotman B. Distribution of suboptimally induces -D-galactosidase in Escherichia coli. The enzyme content of individual cells. J. Mol. Biol. 1973;73:77–91. doi: 10.1016/0022-2836(73)90160-5. [DOI] [PubMed] [Google Scholar]
- 78.Golding I., Paulsson J., Cox E.C. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123:1025–1036. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 79.Paulsson J. Summing up the noise in gene networks. Nature. 2004;427:415–418. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- 80.Elowitz M.B., Levine A.J., Swain P.S. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 81.Guet C.C., Bruneaux L., Cluzel P. Minimally invasive determination of mRNA concentration in single living bacteria. Nucleic Acids Res. 2008;36:e73. doi: 10.1093/nar/gkn329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ozbudak E.M., Thattai M., van Oudenaarden A. Multistability in the lactose utilization network of Escherichia coli. Nature. 2004;427:737–740. doi: 10.1038/nature02298. [DOI] [PubMed] [Google Scholar]
- 83.Blake W.J., Kærn M., Collins J.J. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- 84.Coulon A., Gandrillon O., Beslon G. On the spontaneous stochastic dynamics of a single gene: complexity of the molecular interplay at the promoter. BMC Syst. Biol. 2010;4:2. doi: 10.1186/1752-0509-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Elf J., Li G.W., Xie X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hammar P., Leroy P., Elf J. The lac repressor displays facilitated diffusion in living cells. Science. 2012;336:1595–1598. doi: 10.1126/science.1221648. [DOI] [PubMed] [Google Scholar]
- 87.Wang P., Reed M., Tegtmeyer P. p53 domains: structure, oligomerization, and transformation. Mol. Cell. Biol. 1994;14:5182–5191. doi: 10.1128/mcb.14.8.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Phelps C.B., Sengchanthalangsy L.L., Ghosh G. Mechanism of κB DNA binding by Rel/NF-κB dimers. J. Biol. Chem. 2000;275:24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 89.Sengchanthalangsy L.L., Datta S., Ghosh G. Characterization of the dimer interface of transcription factor NFκB p50 homodimer. J. Mol. Biol. 1999;289:1029–1040. doi: 10.1006/jmbi.1999.2823. [DOI] [PubMed] [Google Scholar]
- 90.Zhang X., Darnell J.E., Jr. Functional importance of Stat3 tetramerization in activation of the α2-macroglobulin gene. J. Biol. Chem. 2001;276:33576–33581. doi: 10.1074/jbc.M104978200. [DOI] [PubMed] [Google Scholar]
- 91.Tomilin A., Reményi A., Schöler H.R. Synergism with the coactivator OBF-1 (OCA-B, BOB-1) is mediated by a specific POU dimer configuration. Cell. 2000;103:853–864. doi: 10.1016/s0092-8674(00)00189-6. [DOI] [PubMed] [Google Scholar]
- 92.Kang J., Gemberling M., Tantin D. A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev. 2009;23:208–222. doi: 10.1101/gad.1750709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Dieck J., Fernandez-Fernandez M.R., Fersht A.R. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J. Biol. Chem. 2009;284:13804–13811. doi: 10.1074/jbc.M901351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hanson S., Kim E., Deppert W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 95.Li A.G., Piluso L.G., Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell. 2006;23:575–587. doi: 10.1016/j.molcel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 96.Wenta N., Strauss H., Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc. Natl. Acad. Sci. USA. 2008;105:9238–9243. doi: 10.1073/pnas.0802130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bissonnette R.P., Brunner T., Heyman R.A. 9-cis retinoic acid inhibition of activation-induced apoptosis is mediated via regulation of Fas ligand and requires retinoic acid receptor and retinoid X receptor activation. Mol. Cell. Biol. 1995;15:5576–5585. doi: 10.1128/mcb.15.10.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vuligonda V., Lin Y., Chandraratna R.A.S. Synthesis of highly potent RXR-specific retinoids: the use of a cyclopropyl group as a double bond isostere. Bioorg. Med. Chem. Lett. 1996;6:213–218. [Google Scholar]
- 99.Kersten S., Kelleher D., Noy N. Retinoid X receptor α forms tetramers in solution. Proc. Natl. Acad. Sci. USA. 1995;92:8645–8649. doi: 10.1073/pnas.92.19.8645. [DOI] [PMC free article] [PubMed] [Google Scholar]